

Abstract
Loss of E-cadherin–mediated cell–cell contacts can elicit a signaling pathway that leads to acquisition of an invasive phenotype. Here, we show that at the receiving end of this pathway is the proto-oncogene c-Jun, a member of the activator protein-1 family of transcription factors that play a key role in stimulation of cell proliferation and tumor promotion. Cell separation or abrogation of E-cadherin–mediated cell–cell contacts both cause a dramatic increase in accumulation of the c-Jun protein. Unlike growth factors that enhance the expression of c-Jun by activating the transcription of the c-jun gene, the cell contact-dependent increase in c-Jun accumulation is not accompanied by a corresponding increase in c-Jun mRNA or c-Jun protein stability but rather in the translatability of the c-Jun transcript. Consistently, the increase in c-Jun accumulation is not dependent on activation of the mitogen-activated protein kinase or β-catenin pathways but is mediated by signals triggered by the restructured cytoskeleton. Depolymerization of the cytoskeleton can mimic the effect of cell separation and cause a dramatic increase in c-Jun accumulation, whereas Taxol inhibits the cell contact-dependent increase. This novel mechanism of c-Jun regulation seems to underlie the robust overexpression of c-Jun in tumor cells of patients with colon carcinoma.
INTRODUCTION
Contact between neighboring cells plays an important role in the process of embryonic development and malignant transformation. Formation of cell–cell contacts is an integral component of morphogenetic programs controlling structural and functional properties of the developing tissue, whereas loss of cell–cell contacts is considered a key step in the progression of tumors toward the invasive phase. The ability of cell–cell contacts to affect physiological events within the cell is well illustrated by the functional role of the adhesion molecule E-cadherin. E-cadherin is a transmembrane glycoprotein that is primarily expressed in epithelia and is a key player in inducing cell polarity and in organizing an epithelium through the establishment of calcium-dependent homophilic interactions at sites of cell–cell contacts. In most cancers of epithelial origin, E-cadherin–mediated cell–cell adhesion is lost concomitantly with progression toward tumor malignancy (Birchmeier and Behrens, 1994; Hirohashi, 1998). Loss of E-cadherin promotes the progression from adenoma to carcinoma, whereas reexpression of E-cadherin results in reversion from an invasive mesenchymal phenotype to a benign epithelial phenotype of cultured tumor cells (Behrens et al., 1989; Frixen et al., 1991; Vleminckx et al., 1991; Birchmeier and Behrens, 1994; Perl et al., 1998). The molecular mechanisms that underlie the loss of E-cadherin–mediated cell–cell contacts have been extensively studied in the past (Hirohashi, 1998; Thiery, 2002; Cavallaro and Christofori, 2004), but the mechanisms by which changes in cell contacts trigger a transition in the cellular phenotype remained largely unknown.
Studies by us and by others have identified the proto-oncogene c-Jun as a potential target for regulation by cell–cell contacts. Cells cultured at low density have been shown to accumulate a high level of c-Jun, whereas at high density or in the intact tissue the accumulation of c-Jun is low (Reisfeld and Vardimon, 1994; Lallemand et al., 1998). The c-Jun protein is a transcription factor that forms a variety of dimeric complexes, collectively termed activator protein (AP)-1 and can positively regulate cell proliferation and tumor progression. The c-Jun protein stimulates cell cycle progression through two main mechanisms: 1) induction of genes coding for components of the cell cycle clock machinery, such as cyclin D1, and 2) repression of tumor suppressor genes, such as p53 (Shaulian and Karin, 2002). In addition, the c-Jun protein activates several events required for tumor progression, including the expression of matrix metalloproteinases (MMPs), proteolitic enzymes that facilitate growth, invasion, and metastasis of cancer cells (Chakraborti et al., 2003). Reduction of c-Jun/AP-1 activity, using dominant-negative c-Jun (TAM67) or conditional inactivation of the c-jun gene, causes cell arrest (Hennigan and Stambrook, 2001), interferes with tumor development (Young et al., 1999; Eferl et al., 2003), suppress the invasive ability in keratinocytes (Dong et al., 1997) and fibroblasts (Lamb et al., 1997), and blocks papilloma-to-carcinoma conversion (Cooper et al., 2003). Given the physiological properties of the c-Jun protein, the induction of c-Jun expression by loss of cell–cell contact might represent a key step in the transition to an invasive phenotype.
Two major signaling pathways have been implicated in the control of c-Jun expression. 1) The β-catenin signaling pathway—this pathway elevates the expression of c-Jun by inducing the transcription of the c-jun gene. The transcriptional increase is mediated by a T cell factor (TCF)/β catenin binding site in the regulatory region of the c-jun gene and is dependent on transcriptional cooperation between TCF4, c-Jun, and β-catenin (Nateri et al., 2005). 2) The mitogen-activated protein kinases (MAPKs), in particular c-Jun NH2-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and p38—these pathways are activated by a diverse array of extracellular stimuli, including peptide growth factors, various cellular stresses, and UV irradiation, and they cause a rapid and transient increase in c-Jun protein accumulation, mainly by activating the transcription of the c-jun gene. The induction of c-jun transcription is mediated by JNK and p38, which phosphorylate the transcription factors c-Jun, ATF2, and MEF2C and thereby activate the transcription of the c-jun gene (Shaulian and Karin, 2002). The MAPK pathways can also contribute to the stability of the c-Jun protein. Phosphorylation by JNK protects c-Jun from ubiquitination and subsequent degradation (Musti et al., 1997), whereas ERK increases c-Jun stability via inactivation of glycogen synthase kinase 3 (Lopez-Bergami et al., 2007). Recent studies have shown that expression of c-Jun can also be elevated by restructuring of the cytoskeleton. Overexpression of cofilin 1, an actin depolymerization protein (Ruegg et al., 2004), or addition of cytoskeleton-disrupting agents to intact tissue or to cells at high density (Oren et al., 1999; Polak et al., 2006) can both cause a marked and sustained increase in c-Jun protein accumulation. The cytoskeletal-dependent increase in c-Jun accumulation is not accompanied by an increase in c-Jun mRNA or in the half-life of the c-Jun protein but rather in the translatability of the c-Jun transcript (Polak et al., 2006). β-Catenin, MAPK, and the cytoskeletal network have been all shown to be functionally linked to cell adhesion complexes (Cavallaro and Christofori, 2004) and therefore might be directly involved in mediating cell contact control of c-Jun expression. Here, we show that loss of cell–cell contacts by cell separation or by functional inhibition of E-cadherin can both cause a marked increase in c-Jun protein accumulation and that this increase is not transcriptionally but rather translationally controlled. Consistently, the increase in c-Jun accumulation is not dependent on activation of the β-catenin or MAPK signaling pathways but rather on signals elicited by the restructured cytoskeleton. This mechanism might be relevant to the control of c-Jun in human carcinomas, as in colon carcinoma presented here.
MATERIALS AND METHODS
Reagents and Plasmids
Nocodazole (Noc), sodium orthovanadate (VOOH), and 12-O-tetradecanoylphorbol-13 acetate (TPA) were purchased from Sigma Chemie (Deisenhofen, Germany). PD98059 was purchased from A.G. Scientific (San Diego, CA), and epidermal growth factor (EGF) was purchased from R&D Systems (Minneapolis, MN). The expression vector for dominant-negative E-cadherin, pEc1M-W156A (Laur et al., 2002), was kindly provided by Dr. S. Troyanovsky (Washington University, St. Louis, MO). pTOPFLASH (contains multimerized TCF-binding sites linked to luciferase reporter) and its mutated version pFOPFLASH (Caspi et al., 2008) were kindly provided by Dr. R. Rosin-Arbesfeld (Tel Aviv University, Tel Aviv, Israel). The reporter constructs 5XcollTRE-TATA-Luc (TRE-TATA), 5Xjun2TRE-TATA-Luc (Jun2-TATA), and TATA-Luc (TATA) (van Dam et al., 1998) were kindly provided by Dr. P. Angel (DKFZ, Karlsruhe, Germany). The expression vector for dominant-negative c-Jun, pEGP-TAM67 (TAM67) (Hennigan and Stambrook, 2001), was kindly provided by Dr. R. F Hennigan (University of Cincinnati, Cincinnati, OH). pTK-Renilla was generated by removing the cytomegalovirus (CMV) promoter from the plasmid pRL-CMV (Promega, Madison, WI) and replacing it with the TK promoter.
Cell Culture, Transfection Procedure, and Luciferase Assay
Cells were seeded at low (0.5 × 106cells/10 cm2) or high (4.5 × 106cells/10 cm2) density in DMEM supplemented with 10% fetal bovine serum. Confluent cultures were treated with drugs at the following final concentrations: Noc (30 μg/ml), EGF (20 ng/ml), TPA (100 ng/ml), and VOOH (1 mM). To generate clones with stable expression of dominant-negative E-cadherin (E-Cad DN), HaCaT cells were transfected with the pEc1M-W156A plasmid, and clones with altered and unaltered morphology were selected in the presence of Geneticin (G418; 500 μg/ml; Invitrogen, Carlsbad, CA). Confluent cultures were transiently transfected with 3 μg of DNA/6 × 105cells, by using jetPEI (Polyplus transfection) according to the manufacturer's instructions. In all experiments, pTK-Renilla (0.5 μg of DNA/6 × 105cells) was cotransfected to control for transfection efficacy. The total amount of plasmid DNA was adjusted with empty vectors. Forty-eight hours after transfection, the cells were washed with phosphate-buffered saline and lysed in passive lysis buffer (Promega). Luciferase activity was assayed using a dual-luciferase reporter assay system (Promega) according to the manufacturer's instructions and recorded by a luminometer (LKB, Rockville, MD).
Cell and Tissue Staining and Fluorescence Imaging
To visualize the actin cytoskeleton, cells grown in six-well plates on glass coverslips were washed with buffer B (2 mM CaCl2 in Tris-buffered saline [TBS]), fixed with 4% paraformaldehyde for 30 min, and washed twice with buffer B. Cells were permeabilized with 0.2% Triton X-100 (Sigma Chemie) for 10 min at room temperature. Nonspecific binding was blocked by incubation of the permeabilized cells for 1 h with buffer A (2 mM CaCl2, 2% bovine serum albumin [BSA] in TBS). The cells were then incubated for 1 h with buffer B containing 0.5% BSA and 0.2 U/ml Alexa Fluor 568 phalloidin (A12380; Invitrogen). After three washes with buffer A, the cells were counterstained with 0.1 μg/ml Hoechst dye 33258 (Sigma Chemie). Confocal imaging was performed using a 510 META confocal laser scanning microscope (Carl Zeiss, Jena, Germany).
For c-Jun staining, cells were fixed with methanol for 15 min and washed twice with PBS. Cells were permeabilized with 0.1% Triton X-100 (Sigma Chemie) for 5 min at room temperature and washed twice with buffer B. Nonspecific binding was blocked by incubation for 1 h with blocking buffer (PBS containing 1% BSA). The cells were stained with rabbit anti-c-Jun antibody (sc-1694; Santa Cruz Biotechnology, Santa Cruz, CA) and fluorescein isothiocyanate-conjugated anti-rabbit (1:40; Dako Deutschland, Hamburg, Germany), washed again, and finally sealed with VECTASHIELD mounting medium (Vector Laboratories, Burlingame, CA) including 1 mg/ml 4,6-diamidino-2-phenylindole (DAPI) (Sigma Chemie). Images were collected by fluorescence microscopy. Tissue sections of normal colon and colon carcinoma were screened for c-Jun and E-cadherin protein expression by immunohistochemistry. The tissues were deparaffinized, rehydrated, and subsequently incubated with primary rabbit anti-c-Jun antibody (Santa Cruz Biotechnology) and mouse anti-E-cadherin antibody overnight at 4°C. The secondary antibody (biotin-labeled anti-rabbit/anti-mouse; Dako Deutschland) was incubated for 30 min at room temperature, followed by incubation with streptavidin-POD (Dako Deutschland) for 30 min. Antibody binding was visualized using AEC-solution (Dako Deutschland). Finally, the tissues were counterstained by hemalaun solution (Dako Deutschland). The evaluation of the staining was performed semiquantitatively by light microscopy.
Protein Preparation and Western Blot Analysis
Cellular protein extracts were prepared by sonication of the cells in passive lysis buffer (Promega) containing a cocktail of protease inhibitors (Roche Diagnostics, Mannheim, Germany), followed by centrifugation at 20,000 × g for 15 min at 4°C. Protein amounts were determined using the Bradford reagent (Bio-Rad, Hemel Hempstead, United Kingdom). Western blot analysis was performed using standard procedures. Briefly, equal amounts (40 μg/lane) of protein were separated on 10% SDS-polyacrylamide gel electrophoresis (PAGE) gels, electroblotted onto nitrocellulose, and incubated with the following mouse monoclonal antibodies (Abs): anti-c-Jun (J31920; BD Biosciences Transduction Laboratories, Erembodegem, Belgium), phospho-c-Jun or p38 (sc-822 or sc-7972, respectively; Santa Cruz Biotechnology), phospho-ERK (M8159; Sigma Chemie), and anti-myc (clone 9E10); or the following rabbit polyclonal Abs: anti-JNK, phospho-p38, or c-Fos (sc-571, sc-17852, or sc-52; Santa Cruz Biotechnology), phospho-JNK (9251; Cell Signaling Technology, Danvers, MA), or ERK (M5670; Sigma Chemie). The corresponding horseradish peroxidase-conjugated secondary Abs (Valeant Pharmaceuticals, Costa Mesa, CA, or Jackson ImmunoResearch Laboratories, West Grove, PA) were used, and cross-reactivity was visualized by the enhanced chemoluminescence procedure (Pierce Chemical, Rockford, IL).
RNA Preparation, Northern Blotting, and Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Cellular RNA was prepared with the EZ-RNA reagent (Biological Industries, Kibbutz Beit Ha-Emek, Israel) according to the manufacturer's instructions. For Northern blot analysis, RNA was denatured by heating at 68°C for 10 min in 2.2 M formaldehyde/50% formamide, and equal amounts (30 μg/lane) were fractionated by electrophoresis in 1.2% agarose gels containing 2.2 M formaldehyde. The fractionated RNA was transferred to a nitrocellulose filter and hybridized with specific DNA probes, labeled with 32P by the random primer DNA labeling mix (Biological Industries). The probes used for hybridization were a 1.1-kb PstI fragment of human GAPDH, and a 1.8-kb EcoRI fragment of mouse c-Jun. The levels of hybridization were determined by autoradiography. For real-time RT-PCR analysis, RNA was digested with RNase-free DNase (MBI Fermentas, Hanover, MD) to remove residual DNA and purified with phenol-chloroform. First-strand cDNA synthesis was preformed using the Verso cDNA synthesis kit (AB-1453; Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. Quantitative real-time PCR was performed on a LightCycler (Takara Bio USA, Madison, WI). cDNA template (2 μl), 0.5 μl (20 mM) of forward and reverse primers, and 10 μl of SYBR Premix Ex Taq in a total of 20 μl were applied to the following PCR program: 30 s at 95°C (initial denaturation); 20°C/s temperature transition rate up to 95°C for 15 s, 3 s at 68°C, 5 s at 72°C, 86°C acquisition mode single, repeated for 40 times (amplification). The PCR reaction was evaluated by melting curve analysis and checking the PCR products on 2% agarose gels. β-Actin was amplified to ensure cDNA integrity and to normalize expression.
The following oligonuleotide primers were used: 5′-AAGTAAGAGTGCGGGAGGCA3′ (forward) and 5′-GGGCATCGTCATAGAAGGTCG-3′ (reverse) for the c-jun transcript and 5′-CTACGTCGCCCTGGACTTCGAGC-3′ (forward) and 5′-GATGGAGCCGCCGATCCACACGG-3′ (reverse) for the β-actin transcript.
Pulse-Chase Analysis
Pulse-chase analysis was performed as described previously (Polak et al., 2006). Briefly, A12/1 cells were pulse labeled with 250 μCi of [35S]methionine and [35S]cysteine (PerkinElmer Life and Analytical Sciences, Boston, MA) per milliliter, in methionine-free medium for 60 min, and chased in medium containing 2 mM unlabeled methionine for the indicated periods. Total cell extracts were prepared as described above. For immunoprecipitation, cell extracts were precleared by incubation overnight at 4°C with preimmune serum bound to protein A/G-Sepharose (Santa Cruz Biotechnology). Cleared extracts were immunoprecipitated with protein A/G-Sepharose bound to mouse anti-c-Jun antibodies overnight at 4°C. Immunoprecipitates were washed twice with buffer I (10 mM Tris, pH 8, 150 mM NaCl) and once with 20 mM Tris, pH 8.0, before the addition of sample buffer, and separated by SDS-PAGE (on 10% polyacrylamide gels), transferred to nitrocellulose, and analyzed by autoradiography. Bends intensities were determined using the ImageJ software (National Institutes of Health, Bethesda, MD). The identity of the c-Jun protein was verified by Western blotting.
Measurement of Translation Efficiency
To measure the rate of de novo c-Jun synthesis, HaCaT cells that express (A12/1) or do not express (E8/2) the dominant-negative E-cadherin or cells treated or untreated with EGF were pulse labeled with 250 μCi of [35S]methionine and [35S]cysteine per milliliter in methionine-free medium for 30 min. Total cell extracts were immediately prepared, and the c-Jun protein was immunoprecipitated and analyzed as described above. In parallel, total RNA was prepared from duplicated cell cultures, and the cellular amount of c-Jun mRNA was determined by real-time RT-PCR, as described above. Translation efficiency was calculated as the ratio between the rate of de novo synthesis of the c-Jun protein and the cellular amount of the c-Jun mRNA.
RESULTS
Loss of Cell-Cell Contacts Results in a Marked Increase in c-Jun Protein Accumulation
Our previous studies have shown that dissociation of intact retinal tissue into separate cells results in a marked increase in accumulation of the c-Jun protein (Reisfeld and Vardimon, 1994). To determine whether the increase in c-Jun accumulation represents a general pattern of response, we examined the influence of cell separation on c-Jun expression in chicken brain and liver tissues and in the spontaneously immortalized cell lines of mouse fibroblasts (NIH 3T3) and human keratinocytes (HaCaT). Protein extracts were prepared from intact tissues (T), from tissues that were dissociated into separate cells and cultured monodispersed (M) (Figure 1A), and from NIH 3T3 or HaCaT cells that were cultured at low or high density (Figure 1, B and C). As control, high-density cultures of NIH 3T3 or HaCaT cells were treated with known activators of c-Jun expression, TPA or EGF, respectively. Western blot analysis revealed that in all cases, cells that were cultured at low density accumulated a high amount of c-Jun, whereas at high density or in the intact tissue the cellular amount of c-Jun was low. The increase in c-Jun accumulation could not be attributed to a general, nonspecific effect on protein expression, because cell separation did not alter the cellular amounts of c-Fos, c-Myc, or ERK (Figure 1, B and C). In addition, immunostaining of a wounded HaCaT culture with anti-c-Jun antibodies revealed that cells at the edge of the wound, which form no or only partial cell contacts, became c-Jun positive, whereas most cells at the confluent area were c-Jun negative (Figure 1D). This suggests that the cell contact effect is not mediated by a freely diffusible factor but rather by the close juxtaposition between cells.
Figure 1.

Expression of c-Jun is dependent on cell-cell contacts. (A) Protein samples from intact tissue (T) or monolayer culture (M) of chicken retina, brain, or liver were analyzed by Western blotting using anti-c-Jun or anti-ERK Abs. NIH 3T3 (B) or HaCaT (C) cells were plated at low or high cell density and harvested after 18h of culture. As control, TPA (B) or EGF (C) was added to cells plated at high density for the last 1 h of culture. Protein samples were analyzed by Western blotting using anti-c-Jun, c-Fos, c-Myc, or ERK Abs. (D) HaCaT cells were grown to confluence on coverslips and then wounded using a pipette tip. Cells were allowed to grow for additional 16 h and then stained with DAPI to visualize nuclei and with anti-c-Jun Ab.
Loss of E-cadherin–mediated cell–cell contacts is considered a key step in tumor progression. To examine whether this step is also accompanied by an increase in c-Jun expression, we immunostained tumor sections from patients with colon carcinoma with antibodies against c-Jun and E-cadherin. The staining revealed that although in normal colon tissue E-cadherin was localized at the membrane and the cells expressed no or only a low level of c-Jun, colon carcinoma cells in all patients examined lost E-cadherin expression at the membrane and exhibited strong and selective expression of c-Jun in the nucleus (Figure 2A). To examine whether loss of the E-cadherin–mediated cell–cell contacts may be a direct cause for the marked increase in c-Jun expression, we stably transfected the E-cadherin–expressing HaCaT cells with the myc-tagged dominant-negative E-cadherin construct, EcMW156A (E-cad DN) (Laur et al., 2002). This construct abrogates cell adhesion due to a tryptophan-to-alanine amino acid substitution (W156A) in the first cadherin-like repeat, a substitution that prevents the formation of transdimers. Clones that exhibited altered (A12/1, A10/1, E12/1) or unaltered (E8/2) epithelial morphology were selected and assayed for the expression of the dominant-negative E-cadherin construct (via the use of an anti-myc antibody), c-Jun or ERK, as an internal control. Western blot analysis revealed that expression of dominant-negative E-cadherin was restricted to clones with altered morphology and that in these clones the expression of c-Jun was markedly elevated (Figure 2B). Thus, disruption of cell–cell contacts by cell separation or by functional inhibition of the adhesion molecule E-cadherin both caused a marked increase in accumulation of the c-Jun protein.
Figure 2.

Abrogation of E-cadherin–dependant cells adhesion results in elevation of c-Jun expression. (A) Immunohistochemical staining of c-Jun and E-cadherin in tissue sections of human normal colon (left) or colon carcinoma (right). (B) The E-cadherin dominant-negative (E-cad DN) construct pEc1M-W156A (illustrated schematically in the top panel), was stably transfected into HaCaT cells. Clones with altered (A12/1, A10/1, E12/1) or unaltered (E8/2) epithelial morphology were selected. Representative phase-contrast images of the selected clones are shown. Protein samples were analyzed by Western blotting using anti-myc, c-Jun, or ERK Abs.
The Increase in c-Jun Accumulation Is Posttranscriptionally Controlled
Loss of E-cadherin–mediated cell–cell contacts has been shown to activate, in some systems, the β-catenin signaling pathway (Conacci-Sorrell et al., 2003), a pathway that can elevate the transcription of the c-jun gene (Nateri et al., 2005). We examined therefore whether the cell–cell contact-dependent increase in c-Jun protein accumulation was transcriptionally regulated. Northern blot analysis revealed that the increase in c-Jun protein accumulation in separate cells (Figure 3A) or in cells that express dominant-negative E-cadherin (Figure 3B) was not accompanied by a corresponding increase in accumulation of the c-Jun transcript. This was in clear contrast to the concomitant increase in c-Jun mRNA and protein observed in cells treated with EGF, a known activator of c-Jun transcription. Consistent with this finding, transfection of β-catenin-LEF/TCF-responsive reporter plasmid showed that expression of dominant negative E-cadherin did not result in activation of β-catenin signaling in HaCaT cells (Figure 3C). Thus, although in EGF-treated cells the increase in c-Jun protein accumulation mainly reflects an increase in gene transcription, accumulation of c-Jun in cells that have lost cell–cell contacts is posttranscriptionally controlled. Interestingly, inspection of the Cancer Genomic Anatomy Project data (cgap.nci.nih.gov), which contains collections of human cancer transcriptome profiles, revealed that normal colon and colon carcinoma cells accumulate a similar level of the c-Jun transcript. This suggests that similarly to the in vitro situation, the increase in c-Jun accumulation in vivo, in human colon carcinoma cells that lost E-cadherin–mediated cell contact, is posttranscriptionally controlled.
Figure 3.

Cell contact dependent increase in c-Jun accumulation is posttranscriptionally controlled. Protein and RNA extracts were prepared from NIH3T3 and HaCaT cells that were plated at low or high cell density and harvested after 18 h of culture (A) or from confluent cultures of HaCaT, E8/2 or A12/1 cells (B). As a control, EGF was added to cells plated at high density for the last 30 min of culture. Protein samples were analyzed by Western blotting using anti-c-Jun or ERK Abs. RNA samples were analyzed by Northern blotting using a 32P-labeled probe specific for the c-jun or the GAPDH gene. (C) E8/2 or A12/1 cells were transfected with the reporter plasmids pTOPFLASH or pFOPFLASH together with pTK-Renilla. β-Catenin transcriptional activity was defined as ratio of pTOPFLASH/pFOPFLASH luciferase activities normalized to Renilla luciferase level detected in each transfection. The results (means ± SD) of three separate experiments are shown.
The c-Jun protein is known to autoregulate its own transcription, via AP-1 sites in the regulatory region of the gene (Shaulian and Karin, 2002). The finding that the increase in c-Jun protein accumulation is not accompanied by an increase in c-Jun mRNA raised the possibility that the accumulated protein is transcriptionally inactive. The transactivation activity of c-Jun is augmented by N-terminal phosphorylation at serines 63 and 73 (Minden et al., 1994). Analysis using anti-phospho-c-Jun antibodies revealed that the accumulated c-Jun protein in A12/1cells is phosphorylated, albeit at a lower level (40%) than that in the EGF-treated HaCaT cells (Figure 4A). To examine the transactivating capability of c-Jun directly, A12/1 cells were transfected with reporter constructs that contain a minimal TATA box promoter attached to five copies of the AP-1 sequence from the regulatory region of MMP1 (TRE-TATA) or of the c-jun gene (Jun2-TATA). A reporter construct that lacks the AP1 sequence was used as control. Analysis of reporter gene expression revealed that expression of the two AP-1–containing constructs differed significantly. Although expression of Jun2-TATA was low, similar to that of the control construct, expression of TRE-TATA was 17-fold higher (Figure 4B). This high level declined considerably upon cotransfection of TAM67, a dominant-negative form of the c-Jun protein (Figure 4C). It should be noted that although expression of Jun2-TATA was low in HaCaT cells that express dominant-negative E-cadherin, it was elevated in HaCaT cells that were treated with EGF (data not shown). These findings suggest that the accumulated c-Jun is transcriptionally active but incapable of activating its own promoter.
Figure 4.
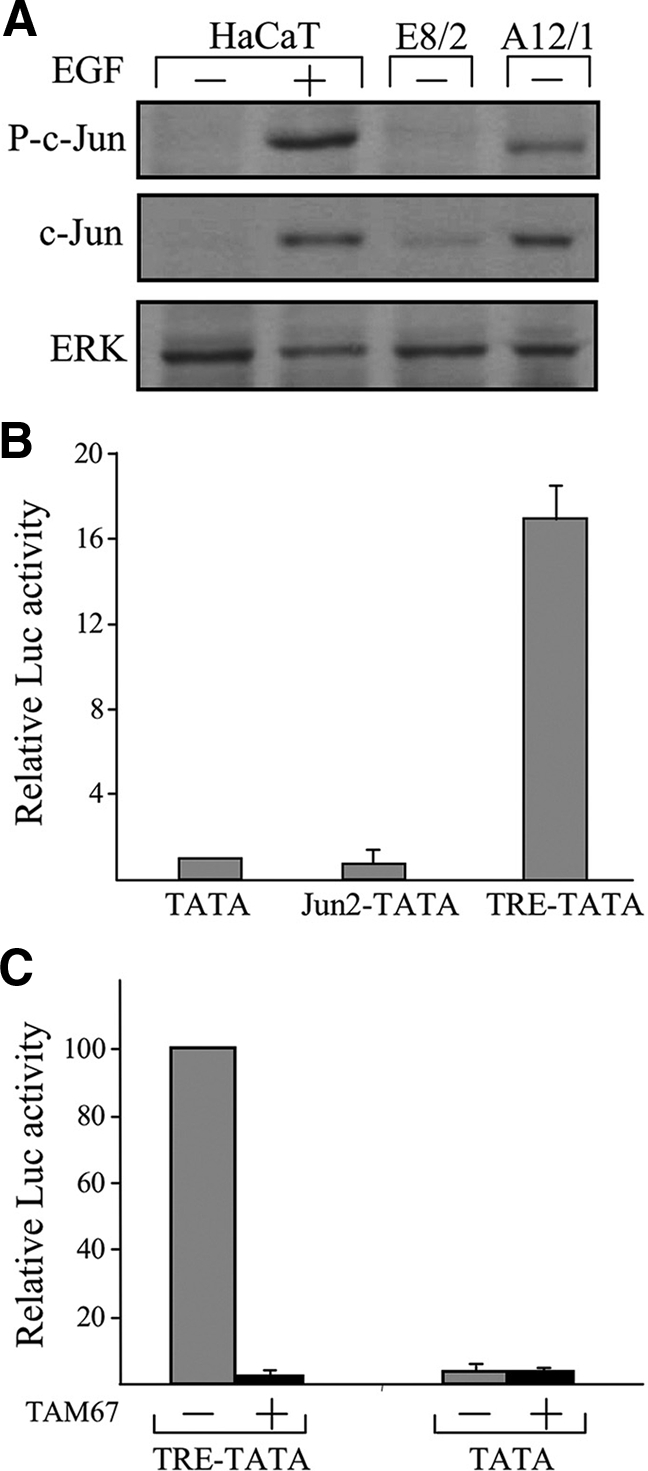
The accumulated c-Jun protein is transcriptionally active. (A) Protein samples from confluent cultures of E8/2, A12/1, or HaCaT cells, untreated (−) or treated (+) for 1 h with EGF, were analyzed by Western blotting using anti-phospho-c-Jun (P-c-Jun), c-Jun, or ERK Abs. (B) A12/1 cells were transfected with the reporter constructs TATA, TRE-TATA, or Jun2-TATA together with pTK-Renilla. In each experiment, luciferase activity obtained in the presence of the reporter construct TATA was given the arbitrary value of 1 and used to normalize all other results. The data shown are the means ±SD of three separate experiments. (C) A12/1 cells were transfected with the reporter constructs TATA or TRE-TATA together with the dominant-negative c-Jun expression vector TAM67 (+) or with an empty vector and with TK-Renilla. In each experiment, luciferase activity obtained in presence of the control vector and the reporter construct TRE-TATA was given the arbitrary value of 100 and used to normalize all other results. The data shown are the means ± SD of two separate experiments.
Loss of Cell–Cell Contacts Enhances the Translation of the c-Jun Transcript
The possibility that cell contact dependent increase in c-Jun accumulation is due to an increase in the stability of the c-Jun protein was examined next. The half-life of c-Jun has been shown to be elevated upon activation of the MAPK pathways, in particular JNK and ERK (Musti et al., 1997; Lopez-Bergami et al., 2007). To examine whether the MAPK pathways are activated in the E-cadherin dominant-negative cells (A12/1), we used specific antibodies that recognize phosphorylated (and therefore activated) JNK, ERK, and p38. HaCaT cells treated with EGF or VOOH, a general phosphatase inhibitor, were used as positive control. The results disclosed that loss of E-cadherin–mediated cell contacts did not activate the MAPK pathways (Figure 5A). We occasionally observed a small increase in ERK phosphorylation, but inhibition of the ERK pathway by the specific mitogen-activated protein kinase kinase (MEK) inhibitor PD98059 did not prevent the accumulation of c-Jun (Figure 5B). Consistent with this finding, expression of c-Fos, a known target of the MAPK pathway, was not altered upon cell separation (Figure 1, B and C) or abrogation of E-cadherin–mediated cell adhesion (Figure 5A). Thus, cell contact-dependent increase in c-Jun accumulation was not dependent on activation of the MAPK pathways. Pulse-chase analysis indeed revealed that the increase in c-Jun protein accumulation was not due to an increase in the stability of the c-Jun protein. From the pulse-chase results shown in Figure 5C, the half-life of c-Jun in A12/1 cells was estimated at 70 min, shorter than the reported half-life of endogenous c-Jun in EGF-treated (130 min) or untreated (90 min) cells (Lamph et al., 1988; Treier et al., 1994; Musti et al., 1997). Given that changes in cell–cell contacts did not alter the transcription or stability of c-Jun, the possibility arose that the increase in c-Jun protein accumulation is translationally controlled. We measured the rate of de novo c-Jun synthesis in HaCaT cells that express (A12/1) or do not express (E8/2) the dominant-negative E-cadherin or in cells treated with EGF. Protein extracts from cells labeled metabolically with [35S]Met/Cys were immunoprecipitated with anti-c-Jun antibodies (Figure 6A). In parallel, the cellular amount of c-Jun mRNA was determined by real-time RT-PCR (Figure 6B). Translation efficiency was calculated as the ratio between the rate of de novo synthesis of the c-Jun protein and the cellular amount of the c-Jun mRNA (Figure 6C). The results demonstrated that although translation efficiency of c-Jun was not altered by EGF, it was elevated by more than fivefold upon expression of dominant-negative E-cadherin.
Figure 5.

Accumulation of the c-Jun protein is not dependent on MAPK activity and is not due to an increase in c-Jun stability. (A) Protein samples from confluent cultures of E8/2, A12/1, or HaCaT cells, untreated (−) or treated for 1 h with EGF or for 15 min with VOOH were analyzed by Western blotting using the indicted antibodies. (B) Protein samples from confluent cultures of E8/2 or A12/1 cells, untreated (−) or treated for the indicated times with 50 μM of the MEK inhibitor PD98059, were analyzed by Western blotting using anti-c-Jun, phospho-ERK (P-ERK), or ERK Abs, as indicated. (C) To estimate the half-life of c-Jun, A12/1 cells were metabolically pulse labeled with [35S]methionine/[35S]cysteine and chased for the indicated times. The intensities of the immunoprecipitated c-Jun bands were determined by scanning and calculated using the ImageJ software. The c-Jun signal intensities are expressed as a percentage of that present at the end of the labeling pulse. The data shown are of two independent experiments.
Figure 6.

The cell contact-dependent increase in c-Jun accumulation is translationally controlled. (A) Confluent cultures of E8/2, A12/1, or HaCaT cells treated (+) or untreated (−) with EGF were metabolically labeled with [35S]methionine/[35S]cysteine for 30 min. Total cell extracts were prepared, fractionated by electrophoresis before or after immunoprecipitation with anti-c-Jun Abs, transferred to a nitrocellulose filter, and visualized by autoradiography. The identity of the c-Jun protein was verified by Western blot analysis (data not shown). To determine the rate of de novo c-Jun synthesis, the blot was scanned and the intensity of the c-Jun band was calculated using the ImageJ software. (B) Parallel cultures were used to prepare total RNA extracts. RNA samples were quantitatively analyzed for c-Jun and β-actin mRNA by real-time RT-PCR. The data were quantified and are represented as ratios of c-Jun to β-actin mRNA. The ratio of c-Jun to β-actin mRNA in untreated HaCaT cells was assigned the arbitrary value of 1 and used to normalize all other results. The data shown are the means ± SD of two independent experiments. (C) Translation efficiency was calculated as the ratio between the rate of de novo synthesis of the c-Jun protein and the cellular amount of the c-Jun mRNA. The calculated ratio in untreated HaCaT cells was assigned the arbitrary value of 1 and used to normalize all other results. The data shown are the means ± SD of two separate experiments.
Cell Contact-dependent Increase in c-Jun Expression Is Mediated by the Cytoskeleton Network
Previous studies have demonstrated that depolymerization of the cytoskeleton causes an increase in c-Jun accumulation (Oren et al., 1999; Ruegg et al., 2004) and that this increase is not transcriptionally but rather translationally controlled (Polak et al., 2006). Considering that the cytoskeletal network is linked to adherent junctions and is restructured upon changes in cell contacts, we examined whether the cytoskeletal network might be involved in transmitting the cell contact signals to the translation machinery of c-Jun. We first examined whether depolymerization of the cytoskeleton in HaCaT cells can mimic the effect of cell separation and cause an increase in accumulation of the c-Jun protein. HaCaT cells were treated with the microtubule-disrupting agents nocodazole, vinblastine, or colchicine, and c-Jun expression was assayed. The results showed that each of these agents caused a marked increase in c-Jun protein accumulation and that here, too, the increase in c-Jun protein accumulation was not accompanied by a corresponding increase in c-Jun mRNA (Figure 7A). Staining with phalloidin revealed that treatment with nocodazole promoted, as expected, the formation of stress fibers and that similar changes occurred upon loss of E-cadherin (Figure 7B, a–c). Formation of stress fibers could also be observed in wounded HaCaT culture, in cells at the edge of the wound, but not in cells at the confluent area (Figure 7B, d–f). To examine whether restructuring of the cytoskeleton is an essential step in the cell contact signaling pathway, we used the microtubule-stabilizing agent Taxol. Confluent HaCaT cultures were treated with or without Taxol and subsequently wounded by scratching. Immunostaining with anti-c-Jun antibodies revealed that in contrast to untreated cultures, in cultures that were pretreated with Taxol, cells at the edge of the wound, which exhibited no or only partial cell–cell contact, remained c-Jun negative (Figure 7C).
Figure 7.

The cytoskeletal network is functionally involved in cell-contact control of c-Jun expression. (A) Confluent cultures of HaCaT cells were untreated (−) or treated with nocodazole (Noc), colchicine (Colchi), or vinblastine (Vin) for 18 h or with EGF for 1 h. Protein and RNA extracts were prepared. Protein samples were analyzed by Western blotting using anti-c-Jun or ERK Abs. RNA samples were analyzed by Northern blotting using a 32P-labeled probe specific for the c-Jun or the GAPDH gene. (B) Confluent cultures of HaCaT cells, untreated (a), treated with nocodazole for 16 h (b), stably transfected with dominant-negative E-cadherin (c), or wounded by using a pipette tip and cultured for additional 16 h (d–f) were fixed and stained with phalloidin to visualize the actin cytoskeleton and with Hoechst 33258 to visualize the nuclei. (C) HaCaT cells were pretreated with the indicated amounts of Taxol or with dimethyl sulfoxide (DMSO, vehicle control) for 6 h and then wounded using a pipette tip. Cells were allowed to grow for additional 16 h and then stained with DAPI to visualize nuclei and with anti-c-Jun Abs.
DISCUSSION
Loss of E-cadherin contributes to tumor progression not only by altering the adhesion status of the cell but also by stimulating the expression of genes that are integral components of the malignant process. Here, we showed that loss of E-cadherin–mediated cell adhesion triggers a signaling pathway that elicits robust overexpression of the proto-oncogene c-Jun, a critical regulator of genes that define the invasive phenotype. Several signaling pathways may be used for transmitting cell contact signals. A most attractive candidate is the β-catenin pathway, because β-catenin exhibits a dual function as a structural component in adherent junctions and a regulatory protein in the transcription machinery of the c-jun gene (Nateri et al., 2005). However, the β-catenin–LEF/TCF-dependent transactivation assay revealed that functional inhibition of E-cadherin in HaCaT cells did not activate the β-catenin signaling, and consistently Northern blot and real time RT-PCR analysis showed that the cell contact-dependent increase in c-Jun accumulation was posttranscriptionally controlled. Abrogation of E-cadherin–mediated cell adhesion activates the β-catenin signaling pathway in some experimental systems, such as in SW480 cells (Conacci-Sorrell et al., 2003), but not in others (Comijn et al., 2001). SW480 cells, unlike HaCaT cells, express a truncated form of the adenomatous polyposis coli (APC) protein, which is part of a complex that targets β-catenin for degradation. The presence of functional APC molecules in HaCaT cells might prevent the accumulation of β-catenin and inhibit thereby the TCF/β-catenin–mediated transcription activity. The finding that the increase in c-Jun protein accumulation is posttranscriptionally controlled was also supported by transfection experiments that assayed the expression of reporter constructs that contain the AP-1 sequence from the c-Jun or the MMP1 promoter. The results indicated that the accumulated c-Jun protein is transcriptionally active but incapable of activating its own promoter. The transcription activity of c-Jun is executed by forming AP-1 complexes that consist of homo- or heterodimers with members of the Jun, Fos, and ATF protein subfamilies. These c-Jun/AP-1 complexes display subtle but important variations in DNA binding specificity, and their formation depends on the relative abundance of each of the Jun, Fos, and ATF proteins in the cell (van Dam et al., 1998; Eferl and Wagner, 2003). Thus, the observed differences in transactivation of reporters that contain the AP-1 sequence from the c-Jun or the MMP1 promoter suggest that in cells expressing the dominant-negative E-cadherin, the cellular context facilitates the formation of c-Jun/AP1 complexes that can interact with the AP-1 sequence of the MMP1 promoter (and thereby activate the expression of proteolytic enzymes that contribute to the invasive capability of cancer cells), but not with that of c-Jun. Cell–cell contact signals may also be transmitted by the MAPK pathways, through the engagement of tyrosine kinase receptors, such as the EGF receptor (Takahashi and Suzuki, 1996; Pece and Gutkind, 2000). These pathways are known to be triggered by various extracellular signals and to cause an increase in both c-jun gene transcription and c-Jun protein stability. Here, we showed that loss of E-cadherin–mediated cell contacts did not significantly alter the phosphorylation and therefore the activation of JNK, ERK, and p38 and consistently that the sustained increase in c-Jun protein accumulation was not due to an increase in transcription or stability of the c-Jun protein but rather due to an increase in the translatability of the c-Jun transcript. We measured the rate of de novo c-Jun synthesis in HaCaT cells that express (A12/1) or do not express (E8/2) the dominant-negative E-cadherin or in cells treated with EGF. Our results clearly demonstrated that unlike EGF, which causes an increase in c-Jun protein accumulation by stimulating the transcription of the c-jun gene, the cell contact-dependent increase was translationally controlled.
How are cell–cell contact signals transmitted to the translation machinery of c-Jun? Recent studies have demonstrated that depolymerization of the actin or microtubule networks can both cause a marked increase in c-Jun protein accumulation (Oren et al., 1999; Ruegg et al., 2004) and that this increase is not accompanied by a corresponding increase in c-Jun mRNA accumulation or c-Jun protein stability but rather in the translation efficiency of the c-Jun transcript (Polak et al., 2006). The increase in c-Jun translation is mediated by the untranslated regions (UTRs) of the c-Jun transcript, in particular by the 5′ UTR, and is not dependent on activation of the MAPK pathways (Polak et al., 2006). Given that E-cadherin is linked via its cytoplasmic domain to the actin and microtubule networks and that loss of cell–cell contacts results in restructuring of the cytoskeleton (Cavallaro and Christofori, 2004), we examined whether cell contact control of c-Jun translation is mediated by the cytoskeletal network. Our results demonstrated that 1) depolymerization of the microtubules in HaCaT cells mimicked the effect of cell separation and caused a marked increase in c-Jun protein accumulation; 2) loss of cell contacts in HaCaT cells resulted in restructuring of the cytoskeleton; and 3) Taxol, a microtubule-stabilizing agent, prevented the accumulation of c-Jun in cells that lost cell contacts. Based on these findings we suggest that the cytoskeletal network plays a critical role in mediating the cell contact dependent control of c-Jun translation.
Several studies point at connection between the translation machinery and the components of the cytoskeleton (Howe and Hershey, 1984; Suprenant, 1993; Blower et al., 2007). All major cytoskeletal structures, including microtubules and microfilaments, have been implicated in the targeting and transport of mRNA molecules directly, via specific localization signals in the mRNA molecules or through components of the translation machinery. The microtubule network has also been shown to associate with clusters of ribosomes (Hamill et al., 1994) and recently to be physically connected to processing bodies (P-bodies), subcellular ribonucleoprotein granules that have been implicated in translational control (Sweet et al., 2007). Although the mechanism that underlies the cytoskeletal control of c-Jun translation has yet to be unraveled, it seems to involve the untranslated regions of the c-Jun transcript (Polak et al., 2006). These regions are exceptionally long (the 5′ and 3′ UTRs of the human transcript are 974 and 1364 nucleotides, respectively), are conserved across species, and have the potential of forming stable secondary structures. It is possible that the c-Jun 5′ UTR forms a physical barrier that blocks the translational machinery and that this inhibition is alleviated upon binding of a specific protein complex, whose assembly is influenced by cytoskeletal dynamics. This mechanism might harness an internal ribosome entry site, which has been identified in chicken c-Jun 5′ UTR (Sehgal et al., 2000) and is known to stimulate initiation of translation during the G2/M phase of the cell cycle, when cap-dependent protein synthesis is inhibited (Bonneau and Sonenberg, 1987). Such a mechanism might be activated during epithelial-mesenchymal transition, a phenomenon which involves loss of E-cadherin and massive remodeling of the cytoskeleton and occurs during embryonic development and malignant progression of carcinoma (Thiery, 2002). Indeed, analysis of patients with colon carcinoma revealed that loss of membrane E-cadherin in tumor cells is accompanied by a robust increase in accumulation of the c-Jun protein but apparently not of c-Jun mRNA (cgap.nci.nih.gov). Further elucidation of the mechanism that underlies the cell contact and/or cytoskeletal control of c-Jun translation may provide novel molecular targets for inhibition of the invasive phenotype.
ACKNOWLEDGMENTS
We thank Drs. S. Troyanovsky, R. Rosin-Arbesfeld, P. Angel, and R. F. Hennigan for plasmids. This research was supported by the Israel Cancer Association, the Israel Ministry of Health, and the Israel Science Foundation (grant 425/08).
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-12-1196) on February 4, 2009.
REFERENCES
- Behrens J., Mareel M. M., Van Roy F. M., Birchmeier W. Dissecting tumor cell invasion: epithelial cells acquire invasive properties after the loss of uvomorulin-mediated cell-cell adhesion. J. Cell Biol. 1989;108:2435–2447. doi: 10.1083/jcb.108.6.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchmeier W., Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim. Biophys. Acta. 1994;1198:11–26. doi: 10.1016/0304-419x(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Blower M. D., Feric E., Weis K., Heald R. Genome-wide analysis demonstrates conserved localization of messenger RNAs to mitotic microtubules. J. Cell Biol. 2007;179:1365–1373. doi: 10.1083/jcb.200705163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneau A. M., Sonenberg N. Involvement of the 24-kDa cap-binding protein in regulation of protein synthesis in mitosis. J. Biol. Chem. 1987;262:11134–11139. [PubMed] [Google Scholar]
- Caspi M., Zilberberg A., Eldar-Finkelman H., Rosin-Arbesfeld R. Nuclear GSK-3beta inhibits the canonical Wnt signalling pathway in a beta-catenin phosphorylation-independent manner. Oncogene. 2008;27:3546–3555. doi: 10.1038/sj.onc.1211026. [DOI] [PubMed] [Google Scholar]
- Cavallaro U., Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer. 2004;4:118–132. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- Chakraborti S., Mandal M., Das S., Mandal A., Chakraborti T. Regulation of matrix metalloproteinases: an overview. Mol. Cell Biochem. 2003;253:269–285. doi: 10.1023/a:1026028303196. [DOI] [PubMed] [Google Scholar]
- Comijn J., Berx G., Vermassen P., Verschueren K., van Grunsven L., Bruyneel E., Mareel M., Huylebroeck D., van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell. 2001;7:1267–1278. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- Conacci-Sorrell M., Simcha I., Ben-Yedidia T., Blechman J., Savagner P., Ben-Ze'ev A. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of beta-catenin signaling, Slug, and MAPK. J. Cell Biol. 2003;163:847–857. doi: 10.1083/jcb.200308162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S. J., MacGowan J., Ranger-Moore J., Young M. R., Colburn N. H., Bowden G. T. Expression of dominant negative c-jun inhibits ultraviolet B-induced squamous cell carcinoma number and size in an SKH-1 hairless mouse model. Mol. Cancer Res. 2003;1:848–854. [PubMed] [Google Scholar]
- Dong Z., Crawford H. C., Lavrovsky V., Taub D., Watts R., Matrisian L. M., Colburn N. H. A dominant negative mutant of jun blocking 12-O-tetradecanoylphorbol-13-acetate-induced invasion in mouse keratinocytes. Mol. Carcinog. 1997;19:204–212. doi: 10.1002/(sici)1098-2744(199707)19:3<204::aid-mc8>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Eferl R., Ricci R., Kenner L., Zenz R., David J. P., Rath M., Wagner E. F. Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell. 2003;112:181–192. doi: 10.1016/s0092-8674(03)00042-4. [DOI] [PubMed] [Google Scholar]
- Eferl R., Wagner E. F. AP-1, a double-edged sword in tumorigenesis. Nat. Rev. Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- Frixen U. H., Behrens J., Sachs M., Eberle G., Voss B., Warda A., Lochner D., Birchmeier W. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J. Cell Biol. 1991;113:173–185. doi: 10.1083/jcb.113.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill D., Davis J., Drawbridge J., Suprenant K. A. Polyribosome targeting to microtubules: enrichment of specific mRNAs in a reconstituted microtubule preparation from sea urchin embryos. J. Cell Biol. 1994;127:973–984. doi: 10.1083/jcb.127.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennigan R. F., Stambrook P. J. Dominant negative c-jun inhibits activation of the cyclin D1 and cyclin E kinase complexes. Mol. Biol. Cell. 2001;12:2352–2363. doi: 10.1091/mbc.12.8.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirohashi S. Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am J. Pathol. 1998;153:333–339. doi: 10.1016/S0002-9440(10)65575-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe J. G., Hershey J. W. Translational initiation factor and ribosome association with the cytoskeletal framework fraction from HeLa cells. Cell. 1984;37:85–93. doi: 10.1016/0092-8674(84)90303-9. [DOI] [PubMed] [Google Scholar]
- Lallemand D., Ham J., Garbay S., Bakiri L., Traincard F., Jeannequin O., Pfarr C. M., Yaniv M. Stress-activated protein kinases are negatively regulated by cell density. EMBO J. 1998;17:5615–5626. doi: 10.1093/emboj/17.19.5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb R. F., Hennigan R. F., Turnbull K., Katsanakis K. D., MacKenzie E. D., Birnie G. D., Ozanne B. W. AP-1-mediated invasion requires increased expression of the hyaluronan receptor CD44. Mol. Cell. Biol. 1997;17:963–976. doi: 10.1128/mcb.17.2.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamph W. W., Wamsley P., Sassone-Corsi P., Verma I. M. Induction of proto-oncogene JUN/AP-1 by serum and TPA. Nature. 1988;334:629–631. doi: 10.1038/334629a0. [DOI] [PubMed] [Google Scholar]
- Laur O. Y., Klingelhofer J., Troyanovsky R. B., Troyanovsky S. M. Both the dimerization and immunochemical properties of E-cadherin EC1 domain depend on Trp(156) residue. Arch. Biochem. Biophys. 2002;400:141–147. doi: 10.1006/abbi.2002.2774. [DOI] [PubMed] [Google Scholar]
- Lopez-Bergami P., et al. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell. 2007;11:447–460. doi: 10.1016/j.ccr.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minden A., Lin A., Smeal T., Derijard B., Cobb M., Davis R., Karin M. c-Jun N-terminal phosphorylation correlates with activation of the JNK subgroup but not the ERK subgroup of mitogen-activated protein kinases. Mol. Cell. Biol. 1994;14:6683–6688. doi: 10.1128/mcb.14.10.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musti A. M., Treier M., Bohmann D. Reduced ubiquitin-dependent degradation of c-Jun after phosphorylation by MAP kinases. Science. 1997;275:400–402. doi: 10.1126/science.275.5298.400. [DOI] [PubMed] [Google Scholar]
- Nateri A. S., Spencer-Dene B., Behrens A. Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature. 2005;437:281–285. doi: 10.1038/nature03914. [DOI] [PubMed] [Google Scholar]
- Oren A., Herschkovitz A., Ben-Dror I., Holdengreber V., Ben-Shaul Y., Seger R., Vardimon L. The cytoskeletal network controls c-Jun expression and glucocorticoid receptor transcriptional activity in an antagonistic and cell-type-specific manner. Mol. Cell. Biol. 1999;19:1742–1750. doi: 10.1128/mcb.19.3.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pece S., Gutkind J. S. Signaling from E-cadherins to the MAPK pathway by the recruitment and activation of epidermal growth factor receptors upon cell-cell contact formation. J. Biol. Chem. 2000;275:41227–41233. doi: 10.1074/jbc.M006578200. [DOI] [PubMed] [Google Scholar]
- Perl A. K., Wilgenbus P., Dahl U., Semb H., Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190–193. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- Polak P., Oren A., Ben-Dror I., Steinberg D., Sapoznik S., Arditi-Duvdevany A., Vardimon L. The cytoskeletal network controls c-Jun translation in a UTR-dependent manner. Oncogene. 2006;25:665–676. doi: 10.1038/sj.onc.1209114. [DOI] [PubMed] [Google Scholar]
- Reisfeld S., Vardimon L. Cell to cell contacts control the transcription activity of the glucocorticoid receptor. Mol. Endocrinol. 1994;8:1224–1233. doi: 10.1210/mend.8.9.7838155. [DOI] [PubMed] [Google Scholar]
- Ruegg J., Holsboer F., Turck C., Rein T. Cofilin 1 is revealed as an inhibitor of glucocorticoid receptor by analysis of hormone-resistant cells. Mol. Cell. Biol. 2004;24:9371–9382. doi: 10.1128/MCB.24.21.9371-9382.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal A., Briggs J., Rinehart-Kim J., Basso J., Bos T. J. The chicken c-Jun 5′ untranslated region directs translation by internal initiation. Oncogene. 2000;19:2836–2845. doi: 10.1038/sj.onc.1203601. [DOI] [PubMed] [Google Scholar]
- Shaulian E., Karin M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- Suprenant K. A. Microtubules, ribosomes, and RNA: evidence for cytoplasmic localization and translational regulation. Cell Motil. Cytoskeleton. 1993;25:1–9. doi: 10.1002/cm.970250102. [DOI] [PubMed] [Google Scholar]
- Sweet T. J., Boyer B., Hu W., Baker K. E., Coller J. Microtubule disruption stimulates P-body formation. RNA. 2007;13:493–502. doi: 10.1261/rna.355807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Suzuki K. Density-dependent inhibition of growth involves prevention of EGF receptor activation by E-cadherin-mediated cell-cell adhesion. Exp. Cell Res. 1996;226:214–222. doi: 10.1006/excr.1996.0221. [DOI] [PubMed] [Google Scholar]
- Thiery J. P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- Treier M., Staszewski L. M., Bohmann D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell. 1994;78:787–798. doi: 10.1016/s0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- van Dam H., Huguier S., Kooistra K., Baguet J., Vial E., van der Eb A. J., Herrlich P., Angel P., Castellazzi M. Autocrine growth and anchorage independence: two complementing Jun-controlled genetic programs of cellular transformation. Genes Dev. 1998;12:1227–1239. doi: 10.1101/gad.12.8.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vleminckx K., Vakaet L., Jr, Mareel M., Fiers W., van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 1991;66:107–119. doi: 10.1016/0092-8674(91)90143-m. [DOI] [PubMed] [Google Scholar]
- Young M. R., Li J. J., Rincon M., Flavell R. A., Sathyanarayana B. K., Hunziker R., Colburn N. Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proc. Natl. Acad. Sci. USA. 1999;96:9827–9832. doi: 10.1073/pnas.96.17.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]