

Abstract
Using small angle X-ray scattering (SAXS) and tryptophan fluorescence spectroscopy, we have identified multiple compact denatured states of a series of T4 lysozyme mutants that are stabilized by high pressures. Recent studies imply that the mechanism of pressure denaturation is the penetration of water into the protein rather than the transfer of hydrophobic residues into water. To investigate water penetration and the volume change associated with pressure denaturation, we studied the solution behavior of four T4 lysozyme mutants having different cavity volumes at low and neutral pH up to a pressure of 400 MPa (0.1 MPa = 0.9869 atm). At low pH, L99A T4 lysozyme expanded from a compact folded state to a partially unfolded state with a corresponding change in radius of gyration from 17 to 32 Å. The volume change upon denaturation correlated well with the total cavity volume, indicating that all of the molecule's major cavities are hydrated with pressure. As a direct comparison to high-pressure crystal structures of L99A T4 lysozyme solved at neutral pH [Collins, M. D., Hummer, G., Quillin, M. L., Matthews, B. W., and Gruner, S. M. (2005), PNAS 102, 16668-16671], pressure denaturation of L99A and the structurally similar L99G/E108V mutant was studied at neutral pH. The pressure-denatured state at neutral pH is even more compact than at low pH, and the small volume changes associated with denaturation suggest that the preferential filling of large cavities is responsible for the compactness of the pressure-denatured state. These results confirm that pressure denaturation is characteristically distinct from thermal or chemical denaturation.
The characterization of non-native states stabilized under a variety of conditions is important towards understanding how proteins fold into their biologically functional, native structures (2). It is widely believed that the dominant driving force in protein folding is the hydrophobic effect and that denaturation can be described as the transfer of hydrophobic residues to water. Although the hydrophobic compound transfer model (3-6) largely succeeds in explaining the thermodynamic stability of proteins as a function of temperature, it does not explain denaturation with pressure (7). In particular, this model fails to explain the magnitude and pressure dependence of the volume difference between the native and denatured states. Recent simulation studies and experimental work suggest that this failure is due to fundamental differences between the temperature- and pressure-denatured states of a protein (1, 8-12). These studies suggest that unlike thermally and chemically denatured states, the pressure-denatured state is one in which water penetration into the protein is favorable, and that a significant contribution to the volume reduction with pressure is the hydration of internal cavities or packing defects.
The literature raises specific questions that require further study. Is pressure denaturation consistent with the water penetration model? How does the volume change upon denaturation correlate with cavity volume? Can other hydration mechanisms be distinguished from cavity filling? What constitutes a pressure-denatured state? We attempt to answer these questions by characterizing the pressure-denatured states of several mutants of the protein T4 lysozyme with varying cavity sizes.
T4 lysozyme is a small globular protein, 164 amino acid residues in length, with a molecular weight of 18.6 kDa (Figure 1 (f)) (13). Over 300 X-ray structures of T4 lysozyme mutants have been deposited in the Protein Data Bank, and the thermal stabilities of many of these mutants have been measured (14). Pressure-induced water filling of an enlarged hydrophobic cavity in the L99A mutant of the pseudo-wild-type T4 lysozyme (Figure 1 (b), cavity 6) has been observed by X-ray crystallography (1). Molecular dynamics simulations suggested that four water molecules cooperatively fill this cavity as the applied hydrostatic pressure is increased (1). No water molecules were observed in the corresponding cavity of the pseudo-wild-type, WT* (Figure 1 (a), cavity 6) (1).
Figure 1.
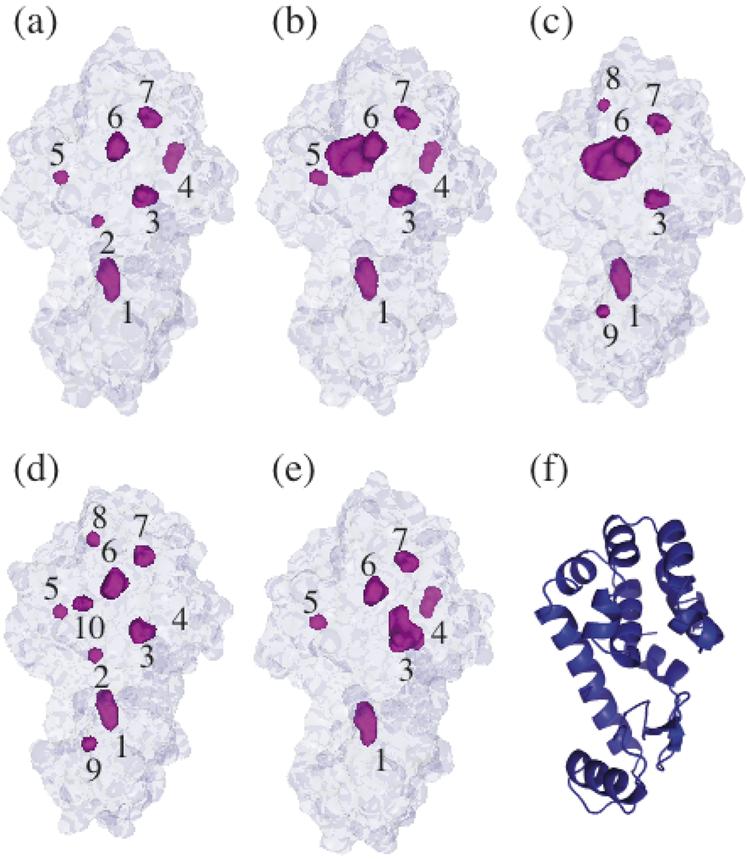
T4 lysozyme structures shown from the same perspective. The external surface and buried cavities (shown in magenta) of (a) WT* (1L63), (b) L99A (1L90), (c) L99G/E108V (1QUH), (d) A98L (1QS5), and (e) V149G (1G0P) identified with a 1.2 Å probe in MSMS (34) with all internal solvent molecules removed. Cavities are identified by numbers 1 - 10 (refer to Table 2). (f) Cartoon representation of WT* T4 lysozyme. The C-terminal lobe is on the top side.
In this study, the solution behavior of L99A and three additional T4 lysozyme mutants (Figure 1) were compared using high-pressure small-angle X-ray scattering (SAXS) and tryptophan fluorescence spectroscopy. The three additional mutants were identified from approximately 300 T4 lysozyme mutants available as satisfying the criteria of having similar thermal stabilities as L99A, approximately 5 kcal/mol less than that of WT* (13, 15-18), yet having differing internal cavity volumes. These features were important for this study in order observe complete denaturation transitions of all the mutants under identical solvent conditions and to determine the effect of internal cavity volume on the volume change of pressure denaturation. Two of the investigated mutants, L99A and L99G/E108V, are structurally very similar to WT* except for differing volumes of the hydrophobic cavities at the mutation site where the 99th residue, leucine, L, has been replaced by alanine, A, or glycine, G (15, 16). The V149G mutation enlarges an existing solvent-bound polar cavity at the mutation site (17). A98L is destabilized relative to WT* by a strain-inducing small-to-large amino acid substitution (18). Small angle X-ray scattering (SAXS) was used to characterize the pressure-induced change in radius of gyration and overall shape of L99A T4 lysozyme. Thermodynamic analysis was performed on tryptophan fluorescence data taken at various solvent conditions on all mutants. A fluorescence quenching study of a seleno-methionine variant of L99A provided additional information on global structural changes due to high pressure.
The results provide insight into the relationship between the thermodynamic and structural characteristics of T4 lysozyme. In particular, we characterize the contributions of cavity volume of the native state and electrostriction to the volume change of pressure denaturation. Low-resolution models of native and pressure-denatured solution structures have been determined. Multiple, compact pressure-denatured states are identified with differing degrees of hydration. These results confirm that pressure denaturation is characteristically distinct from thermal and chemical denaturation. It is shown that the study of pressure effects provides further understanding of the energy landscape and the structure-stability relationship of proteins.
Experimental Procedures
Sample and Buffer Preparation
In this work the cysteine-free pseudo-wild-type, C54T/C97A T4 lysozyme (13), is referred to as WT*. All mutants used in this study contain the mutations C54T and C97A. The compositions of cell culture media used in these procedures are listed in supporting material 1.
The L99A, L99G/E108V, and A98L mutants were expressed and purified by a modified version of the protocol described by Poteete, et al. (19) (supporting material 2). The V149G mutant was isolated using a modified version of the inclusion body protocol described by Vetter et al. (20) (supporting material 3). A seleno-methionine containing variant of the L99A mutant (Se-Met L99A) was prepared in a methionine deficient, seleno-methionine rich growth medium using an adaptation of the procedure by Van Duyne et al. (21) using non methionine-auxotrophic cells (22) (See supporting material 4).
High concentration buffers with low volume changes of ionization were chosen to stabilize the pH as a function of pressure (23, 24). At pH 3.0, 50 mM glycine buffer (Cat. 17-1323-01; GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA) and at pH 7.0, 50 mM Tris-HCl buffer (T-3253; Sigma, St. Louis, MO, USA) were used, unless otherwise noted. The NaCl (Cat. 7581; Mallinckrodt Baker, Phillipsburg, NJ, USA) concentrations used were 20 and 100 mM. Buffers were sterilized with 0.22 μm cellulose acetate filters (Catalog Number 431175; Corning Inc, Corning NY, USA), stored at 4 °C, and used within two weeks of preparation.
High-Pressure Small Angle X-ray Scattering
SAXS samples were prepared up to 48 hours in advance of experiments by dialyzing against buffers in micro-dialysis buttons (Cat. HR3-362; Hampton Research, Aliso Viejo, CA, USA) closed with a 10 kDa molecular weight cut-off dialysis membrane (Cat. 68100; Pierce Biotech, Rockford, IL, USA). Protein solution concentrations were adjusted to 4 to 25 g/L by UV absorption measurement (Nanodrop ND1000; Nanodrop Technologies, Wilmington, DE, USA) and centrifugal re-concentration when necessary (Microcon YM-10 centrifugal concentrator; Millipore, Billerica, MA, USA). The final dialysate solutions were used for sample dilutions and background scattering measurements. Samples were stored at 4 °C or on ice. Experiments were performed at room temperature.
The custom built high-pressure SAXS cell, equipment, and data reduction procedures were previously described (25). X-ray experiments were performed at the G1 station at the Cornell High Energy Synchrotron Source (CHESS, Cornell University, Ithaca, NY, USA). Data on 10 g/L L99A T4 lysozyme in 50 mM glycine, 100 mM NaCl, pH 3.0 (q range of 0.019 - 0.328 Å) and 16 g/L WT* T4 lysozyme in 50 mM glycine, 150 mM NaCl, pH 3.0 (q range of 0.025 - 0.295 Å) were collected with a detector distance of 1.25 m and a 12 keV 250 μm square beam. The salt concentration was adjusted at each protein concentration to minimize protein interactions (25). The momentum transfer, q, is defined as q = 4π/λ sin θ, where 2θ is the scattering angle and λ is the X-ray wavelength. The samples were contained in custom acrylic inner cells (ALine Inc., Redondo Beach, CA, USA) (25) with 7.5 μm Kapton film windows (Spectromembrane No. 3022; Chemplex Industries, Palm City, FL, USA). Data were collected on a home-built 1024 × 1024 pixel CCD detector, and the transmission intensity was measured with a home-made PIN diode beamstop. At each pressure, protein samples were equilibrated for 10 to 60 minutes. Buffer scattering was measured at each pressure. Measurements were repeated on fresh samples to identify radiation damage. Exposures exhibiting radiation damage were not used for analysis.
The scattering profile of the protein, I(q), was produced by subtracting the transmission-normalized scattering profile of the buffer taken at the sample pressure from that of the protein solution without further scaling. A Guinier approximation can be applied to the low q region of the scattering profiles.
| (1) |
The radius of gyration, Rg, and zero-angle scattering intensity, I(0), are determined from a linear fit to the Guinier plot, log(I(q)) vs. q, in the approximate region where the condition, qRg < 1.3, holds (26). I(0) is a function of the electron density contrast between the protein and solvent, Δρ, the excluded volume of the protein, Vp, and the structure factor, S(q), which is a function of protein-protein interactions (27).
| (2) |
The radius of gyration can also be determined from the pair distance distribution function, P(r) (27).
| (3) |
P(r) was calculated with program GNOM, which implements the indirect Fourier transform method (27, 28) using the experimental I(q) as the input. This method is sensitive to low q data and therefore, the choice of the maximum dimension of the protein, Dmax, where P(r) approaches zero. Thus, it was only applied to data where Dmax could be unambiguously determined. Low-resolution models of protein structures were generated from the I(q) produced by GNOM using the ab initio reconstruction program GASBOR (29). The scattering profile of the denatured state was also analyzed with the Ensemble Optimization Method (EOM) package developed for modeling flexible proteins in solution (30).
High-Pressure Fluorescence Spectroscopy
Three tryptophan residues are present in the C-terminal lobe of T4 lysozyme (Trp126, Trp138, and Trp158). Samples for tryptophan fluorescence spectroscopy were prepared up to 48 hours in advance of experiments by dialyzing 100 - 200 μl protein solutions in micro-dialysis floats (Cat. PI-69560; Thermo Scientific Pierce Slide-A-Lyzer, Thermo Fisher Scientific, Waltham, MA, USA). The dialyzed protein solutions were further diluted to a final volume of 2.4 - 2.5 ml, resulting in protein concentrations of < 3 μM. No contaminants from the dialysis step were detected spectroscopically.
A high-pressure fluorescence cell (HP Cell; ISS, Champaign, IL, USA) equipped with sapphire windows was used with a scanning fluorimeter (Chronos; ISS, Champaign, IL, USA). The pressurizing medium in the high-pressure cell, isopropanol, was separated from the pressurizing medium (Fluorinert FC-77; 3M, St. Paul, MN, USA) in the high-pressure pump (Cat. No. 37-6.75-60; High Pressure Equipment Co., Erie, PA, USA) by a piston in a home-built stainless steel high-pressure reservoir. Samples were contained in a square bottle with a cylindrical neck formed by fusing a short 3 mm inner diameter quartz tube to the top of a 10 mm path length square quartz cell (Cat. No. CQ110; VitroCom, Mountain Lakes, NJ, USA). The sample-filled bottle was sealed with a soft bulb made from polyolefin heat shrink tubing (Cat. No. 03F3745; Newark Electronics, Chicago, IL, USA) and positioned in the high-pressure cell with a home-made Delrin cuvette holder. The large flat windows of this cuvette minimize scattering of the incident beam, while the soft bulb closure acts as a deformable pressure-transmitting membrane.
Samples were excited at 295 nm with a bandwidth of 8 nm to minimize tyrosine excitation, and emission from 310 to 550 nm was collected at 90° with a bandwidth of 4 nm. Vinci software (ISS, Champaign, IL, USA) was used for instrument control and data acquisition. Protein solutions were equilibrated at each pressure for 20 to 90 minutes. Buffer reference spectra were pressure independent and featureless, with the exception of a small pressure independent water Raman peak around 330 nm. For this reason only one representative reference spectrum was taken at 50 MPa for each protein sample. Sample temperature was maintained with a water circulation pump (Neslab, Thermo Scientific, Waltham, MA, USA).
Data were analyzed with MATLAB (The MathWorks, Natick, MA, USA). The collected emission spectra were background-subtracted. Intensity corrections were performed with calibration data collected by ISS (Champaign, IL, USA) with 4 nm excitation and emission bandwidths. The center of spectral mass, <λ>, of each spectrum was calculated in the region 310 to 550 nm with the following equation (31).
| (4) |
where Ii is the fluorescence intensity at the wavenumber, νi, and N is the number of data points collected in the spectrum. The weighted average <ν> was then converted to wavelength, <λ>.
For two-state denaturation, the thermodynamic stability of the protein is the free energy difference between the native (N) and denatured (D) states.
| (5) |
Assuming that the protein solution is an ideal solution, ΔG at a given pressure can be expressed as a function of the observable quantity, the center of spectral mass.
| (6) |
where Keq is the equilibrium constant, R is the gas constant (83.1447 mL-bar/mol-K), T is the experimental temperature, and <λ>N and <λ>D are the spectral centers of mass of the native and denatured states. <λ>N and <λ>D were inferred by fitting experimental data. No baseline corrections to the spectral centers of mass were necessary.
The dominant contribution to the pressure-induced change in protein stability is the volume change (32, 33). Thus, a first-order approximation was used to model the denaturation of T4 lysozyme. The pressure stability with respect to a reference pressure, P0, can be expressed by
| (7) |
where ΔGο and ΔVο are the free energy difference and volume change of denaturation at standard pressure P0, 1 atm, and a constant experimental temperature, T. Combining Equations 6 and 7, we arrive at the following model for our data:
| (8) |
where <λ>N, <λ>D, ΔGο, and ΔVο are free parameters.
Cavity Volume Calculation and Visualization
Cavity calculations were performed on the atomic structures of L99A (PDB accession code 1L90), L99G/E108V (1QUH), A98L (1QS5), V149G (1G0P), and WT* (1L63). An additional atomic structure of WT* was provided by Marcus Collins (private communication). Cavities were identified using the program MSMS (34) with a 1.2 Å probe. The identified molecular surfaces were viewed using the program PyMol (Delano Scientific LLC, Palo Alto, CA, USA) with a script provided by Warren DeLano (private communication).
Results
Thermodynamic Analysis of Fluorescence Measurements
The fluorescence of tryptophan is sensitive to the polarity of its local environment and can be used to monitor protein conformation changes. Both the solvent and protein contribute to environmental polarity, such that protein denaturation is generally accompanied by a red-shift in tryptophan fluorescence (35). Fluorescence measurements were made on the T4 lysozyme mutants at pH 3.0 and 7.0. Thermodynamic analysis was performed on the pressure-induced change in the center of spectral mass (defined in Experimental Procedures).
All samples exhibited two-state behavior (native versus denatured) under pressure, consistent with observations made in other denaturation studies (36, 37). The peak intensities of the mutants were normalized to that of WT*, which did not denature under our conditions, to correct for pressure-induced changes in fluorescence quenching and solvent transmission. An isosbestic point was evident in the pressure-corrected spectra of each denaturation series (Figure 6), indicative of two-state denaturation. Singular value decomposition (SVD) analysis determined that the data could be adequately described as linear combinations of two independent SVD states. For each data set, denaturation curves reconstructed from the two significant SVD states fit the experimental spectra with a goodness of fit parameter, R2 ≥ 0.9986. The reversibility calculated from the center of spectral mass upon decompression to ambient pressure was 80-93%. The low end of this range applied to samples maintained at high pressure for several hours. The effects of non-reversibility were apparent only at low pressure, and therefore, low-pressure data were collected first in order of increasing pressure. Thermodynamic fits (Equation 8) performed on the centers of spectral mass yielded goodness of fit parameters, R2 ≥ 0.9990.
Figure 6.
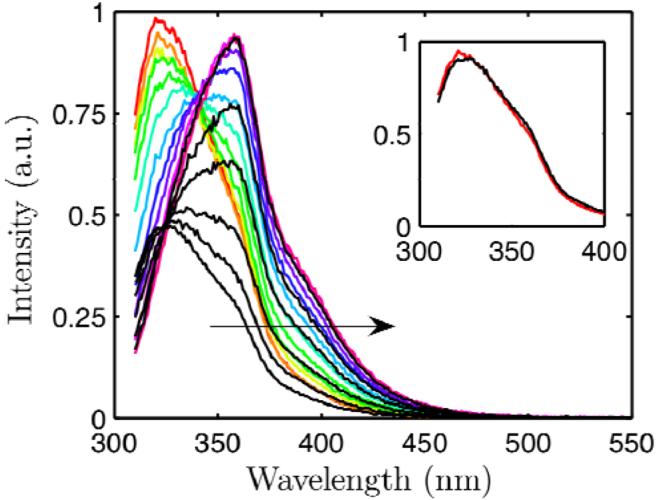
Tryptophan fluorescence spectra of L99A (color) and Se-Met L99A (black) in 50 mM glycine 20 mM NaCl pH 3.0 16 °C with increasing pressure (direction indicated by arrow). The emission intensities were normalized at high pressure (200 MPa) to emphasize the pressure-induced intensity increase of Se-Met L99A compared with L99A. Inset: Spectra of L99A (red) and Se-Met L99A (black) at low pressure (25 MPa) scaled by a constant factor.
Fluorescence measurements were made on L99G, L99G/E108V, A98L, V149G, and WT* at pH 3.0. The thermal denaturation of T4 lysozyme has been extensively characterized at pH 3.0 (15, 16, 18, 38), and therefore this pH was chosen as a major test condition for this work. The low pH also enabled pressure denaturation of all the mutants to complete below 400 MPa, the pressure limit of our apparatus. At pH 7, these mutants are more stable, and not all of them denature at 400 MPa. The temperature was set to 16 °C, where the folded fractions of the least stable mutants, L99G/E108V and V149G were nearly 1 at ambient pressure. The centers of spectral mass of all mutants showed sigmoidal dependence on pressure, indicative of conformational changes (Figure 2), while that of WT* did not change up to 300 MPa (not shown). The centers of spectral mass of the native and denatured states were similar for L99A and L99G/E108V at each solvent condition. This observation is consistent with the structural similarity of L99A and L99G/E108V compared with the other two mutants. The steepness of the denaturation curves is a function of the magnitude of the volume change of denaturation, ΔVο, while the pressure at which the transition begins is indicative of the stability at ambient pressure, ΔGο. The results from the thermodynamic fits are presented in Table 1. At pH 3.0, A98L and V149G exhibited similar volume changes although V149G was less stable, denaturing at a lower pressure. Compared to A98L and V149G, L99A and L99G/E108V showed large volume changes. The magnitude of ΔVο was also dependent on the ionic strength of the solvent. An increase from 20 to 100 mM NaCl resulted in a reduction by approximately 25 Å3 in the magnitudes of ΔVο for all mutants. At both NaCl concentrations, the volume change of L99G/E108V was roughly 50 Å3 greater than that of L99A.
Figure 2.
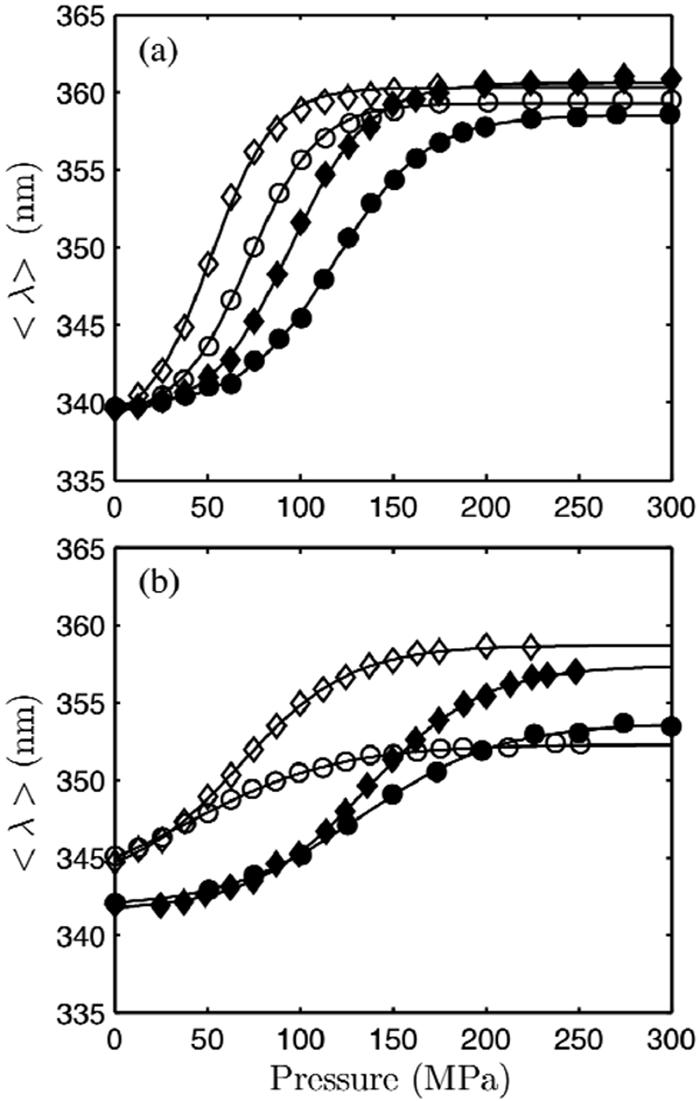
Pressure denaturation of T4 lysozyme mutants in pH 3.0 50 mM glycine 20 mM NaCl (diamond) and 100 mM NaCl (circle) buffers monitored at 16 °C by tryptophan fluorescence spectroscopy. (a) L99G/E108V (open) and L99A (closed). (b) V149G (open) and A98L (closed).
Table 1.
Thermodynamic Quantities Calculated from a Two-State Denaturation Model
| Sample | Condition | <λ>N (nm)a | <λ>D (nm)a | ΔG° (kcal/mol) | ΔV° (Å3) | Pm (MPa)b |
|---|---|---|---|---|---|---|
| pH 3.0, 16 °C | ||||||
| L99G/E108V | 20 mM NaCl | 338.4 ± 1.0 | 360.3 ± 0.3 | 1.75 ± 0.24 | -235.8 ± 25.0 | 51.7 |
| 100 mM NaCl | 339.0 ± 0.5 | 359.3 ± 0.2 | 2.21 ± 0.17 | -212.8 ± 13.8 | 72.3 | |
| L99A | 20 mM NaCl | 339.2 ± 0.4 | 360.6 ± 0.2 | 2.54 ± 0.15 | -187.3 ± 10.0 | 94.5 |
| 100 mM NaCl | 339.7 ± 0.2 | 358.5 ± 0.2 | 2.75 ± 0.14 | -161.4 ± 7.5 | 118.7 | |
| A98L | 20 mM NaCl | 341.6 ± 0.2 | 357.4 ± 0.3 | 2.49 ± 0.13 | -127.2 ± 6.8 | 136.0 |
| 100 mM NaCl | 341.8 ± 0.4 | 353.7 ± 0.2 | 2.07 ± 0.22 | -107.7 ± 10.0 | 133.7 | |
| V149G | 20 mM NaCl | 343.3 ± 0.4 | 358.7 ± 0.2 | 1.30 ± 0.10 | -134.1 ± 7.3 | 67.4 |
| 100 mM NaCl | 342.4. ± 1.0 | 352.3 ± 0.2 | 0.58 ± 0.17 | -99.2 ± 10.0 | 40.8 | |
| pH 7.0, 24 °C | ||||||
| L99G/E108V | 20 mM NaCl | 340.3 ± 0.2 | 355.6 ± 0.3 | 2.56 ± 0.17 | -102.8 ± 7.0 | 172.8 |
| L99A | 20 mM NaCl | 340.9 ± 0.2 | 356.1 ± 0.5 | 3.19 ± 0.26 | -93.1 ± 8.2 | 238.1 |
Results of fitting Equation 8 (Experimental Procedures) to data.
Subscripts “N” and “D” refer to the native and denatured states, respectively.
Pm is the midpoint pressure of denaturation.
For direct comparison with the room-temperature high-pressure crystal structures of L99A and WT* acquired by Collins et al. (1), fluorescence measurements were also made at pH 7.0, 24 °C. L99G/E108V was also studied because of its structural similarity to L99A. We observed sigmoidal transitions for L99A and L99G/E108V as a function of pressure, while no transition was detected for WT* up to 350 MPa (Figure 3). The denaturation curves of the two mutants are similarly shaped with a gradual transition except that of L99G/E108V is shifted by about 65 MPa to a lower pressure with respect to that of L99A. Compared to the values at pH 3.0, the volume changes of denaturation at pH 7.0 were similar and small in magnitude (approximately -100 Å3). The results of thermodynamic fits are given in Table 1.
Figure 3.
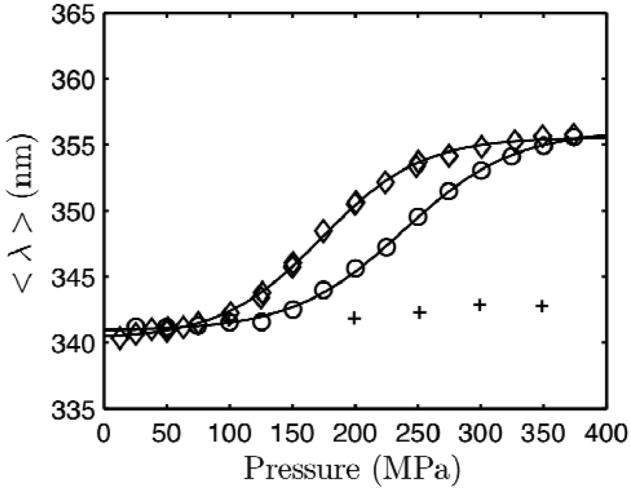
Pressure denaturation of L99G/E108V (diamond), L99A (circle), and WT* (plus) T4 lysozyme in pH 7.0 50 mM Tris HCl 20 mM NaCl buffer monitored at 24 °C by tryptophan fluorescence spectroscopy.
Structural Information from SAXS Measurements
Small-angle X-ray scattering is sensitive to electron density distributions with inhomogeneities on the length scales of 10 to 100 Å and thereby provides structural information for proteins in solution. At pH 3.0 where the SAXS measurements were made, T4 lysozyme is highly positively charged. To minimize the effect of inter-particle interference on the intensity profile, a specific NaCl concentration was required at a given protein concentration. These interaction-free conditions were first identified at ambient pressure (25). SAXS profiles were measured at pressures ranging from 28 to 300 MPa. Due to the exposure limit set by radiation damage, the data presented here were taken on two aliquots of the same sample. The small angle X-ray scattering of L99A T4 lysozyme exhibited pressure-reversibility. SAXS was not attempted at pH 7.0 both to limit the scope of the required beam time and because not all the mutants denature within the accessible pressure range at this pH.
Guinier analysis was performed on the low q region of the SAXS profiles. The results of Guinier fits to the data are shown in Figure 4. We previously reported a radius of gyration of 16.5 +/- 0.3 Å for L99A at ambient pressure (25), which is in agreement with that calculated from the crystal structure of a folded monomer, 16.4 Å. At 28 MPa, the radius of gyration determined over q = 0.024 - 0.076 Å-1 was 17.1 +/- 0.1 Å, indicating that L99A is mostly folded at this pressure. As the applied pressure was increased, the radius of gyration increased to 31.6 +/- 1.7 Å (determined over q = 0.024 - 0.038 Å-1) at 300 MPa. As the radius of gyration (Rg) as determined by Guinier analysis is only accurate for homogeneous samples, we do not place strong meaning on the specific value of Rg at intermediate pressures where L99A was a mixture of folded and denatured forms. Pair distance distribution functions were computed using GNOM (28) from data taken at 28 and 300 MPa with a maximum size, Dmax, of 54 and 134 Å, respectively (Figure 5 (a)). Radii of gyration determined from the pair distance distribution functions were 17.0 and 34.5 Å, in good agreement to the Guinier fits. The Rg of the pressure-denatured state was quite small in comparison to 40.7 Å, the predicted radius of gyration of a fully unfolded polypeptide of the same length (39). WT* did not show change up to 300 MPa (25). The linearity of the Guinier plots at low q indicated that aggregates were not detected. The 28 and 300 MPa scattering profiles are shown in the inset of Figure 5 (a) as Kratky plots, i.e. Iq2 vs. q. Krakty plots emphasize the I(q) decay in the intermediate q region, which is sensitive to the shape of the protein (40). The 28 MPa profile shows a peak around 0.1 Å-1, indicating that L99A is compact and globular at this pressure. At 300 MPa, this peak is greatly reduced and no an additional peak at lower q is evident, indicating that L99A is extended and largely monomeric at high pressure.
Figure 4.
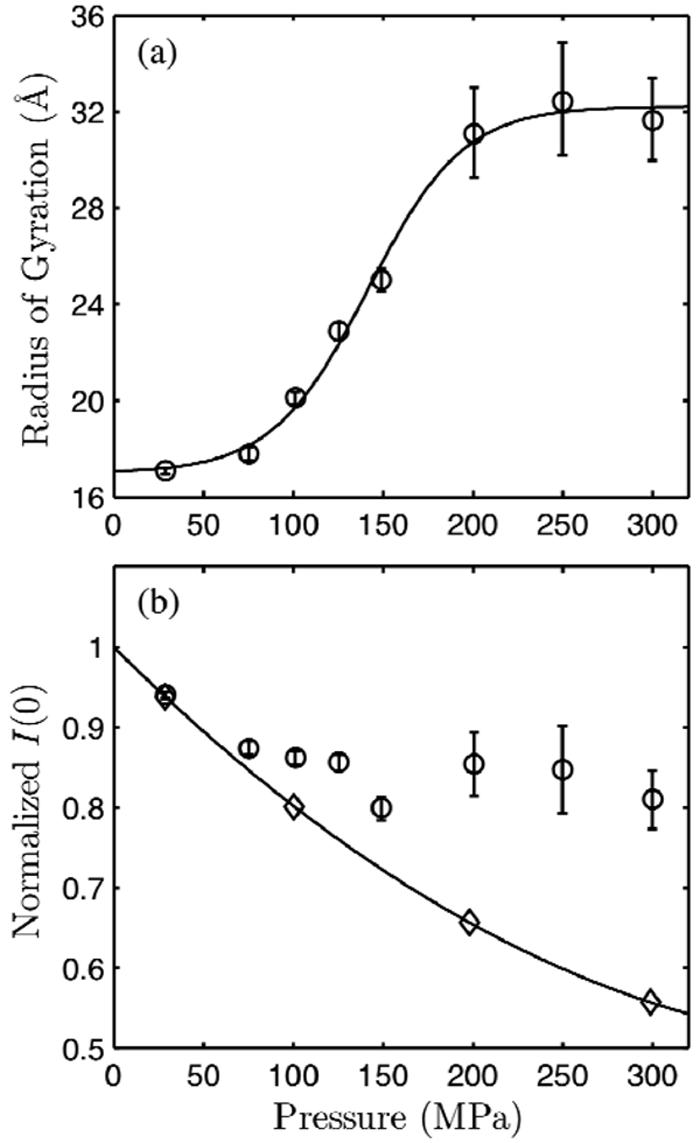
(a) Radius of gyration, Rg, as a function of pressure for 10 g/l L99A in 50 mM glycine 100 mM NaCl pH 3.0 buffer at room temperature. Rg was determined by a Guinier fit to the qRg < 1.3 region of scattering profiles taken at each pressure. The error bars are larger at high pressure because only one exposure was taken at each pressure. At lower pressure, the sample was less susceptible to radiation damage-induced aggregation and multiple exposures were taken at each pressure, which enabled averaging of images. A two-state thermodynamic fit is shown (solid line) to guide the eye. (b) The zero-angle scattering intensity of L99A (circle) and the WT* (diamond) at pH 3.0. WT* does not denature below 300 MPa.
Figure 5.
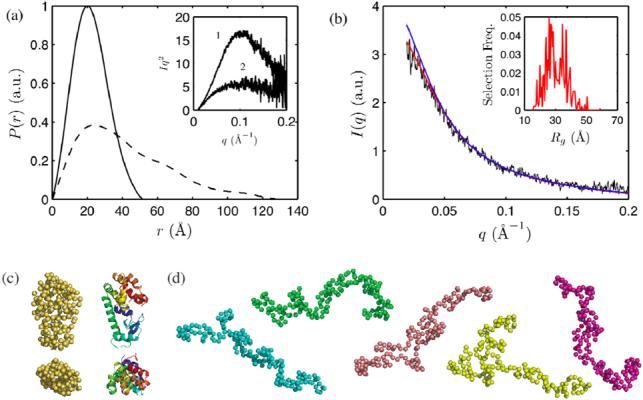
(a) Pair distance distribution functions of native (28 MPa, solid line) and denatured (300 MPa, dotted line) L99A T4 lysozyme at pH 3.0 obtained with GNOM (28). Inset: Kratky representation of the 28 MPa (1) and 300 MPa data (2). (b) The scattering profile of L99A at 300 MPa (black) was examined with the Ensemble Optimization Method (30). An ensemble of unfolded conformers with residual structure (red scattering profile) better described the experimental data compared to an ensemble of random coil conformers (blue scattering profile). The distribution of Rg in the first ensemble is shown in the inset. (c) Side and top views of the crystal structure of L99A (right) and a representative low-resolution structure at 28 MPa obtained with GASBOR (29) (left) show good agreement. (d) GASBOR models that fit well to 300 MPa data were extended.
The zero-angle scattering intensity, I(0), determined by Guinier analysis is shown in Figure 4 (b). I(0) is a function of the electron density contrast between the hydrated protein and solvent, the excluded volume of the protein, molecular weight, and inter-protein interactions. We have previously shown that the electron density contrast of WT* decreases with pressure because of the greater compressibility of the solvent compared to the protein (25). Relative to WT*, L99A shows an increase in I(0). As L99A is highly charged at pH 3.0 and sensitive to the ionic strength, we attribute the increase in I(0) to changes in hydration or protein interactions that accompany denaturation (41-43). Although I(0) is also a function of the protein volume, the volume changes due to denaturation are on the order of 1% of the volume of the protein and therefore do not significantly affect I(0).
For further structural characterization, low-resolution models of L99A were produced using the ab initio reconstruction program GASBOR (29) and the denatured ensemble was modeled with the EOM package (30). Five GASBOR reconstructions were performed on the small X-ray scattering data taken at 28 and 300 MPa on the assumption that L99A is a monomer at both pressures. The results of this reconstruction are shown in Figure 5 (c) - (d). The low-pressure reconstructions resembled the crystal structure of L99A in exterior shape and size. The high-pressure reconstructions do not represent actual structures found in solution, as the denatured state is an ensemble without a unique conformation, but rather, they are models that closely match the experimental scattering profile. While the low-pressure structures are compact, the high-pressure reconstructions are noticeably more extended. The small angle X-ray scattering profile taken at 300 MPa was also examined with EOM. EOM generates a large pool of randomly generated conformations available for a polypeptide of a certain sequence (44) from which an optimization algorithm is used to select the subset that best fits the experimental scattering profile. Two starting pools were generated. The first contained 10,000 random coil conformations with an average Rg of 38.0 Å, and the second contained 10,000 unfolded conformations with residual structure with an average Rg of 29.7 Å. The ensemble selected from the pool with residual structure had an average Rg of 31.2 Å, in agreement with Guinier analysis, and better described our experimental data than the selected random coil ensemble, which had an average Rg of 36.1 Å (Figure 5 (b)).
Structural Information from Fluorescence Quenching Measurements
Additional information on global conformational changes was derived from fluorescence quenching measurements. T4 lysozyme has five methionine residues in close proximity to the three tryptophan residues present in the C-terminal lobe. The sulfur atom in methionine quenches tryptophan emission and provides a probe of the methionine-tryptophan separation. Denaturation of T4 lysozyme is usually accompanied by an increase in emission intensity (45), consistent with an increased methionine-tryptophan distance. In the seleno-methionine variant of L99A (Se-Met L99A), the methionines are replaced with seleno-methionines. As selenium and sulfur have differing quenching strengths (46), a comparison of L99A and Se-Met L99A yields additional information on the spatial compactness of the unfolded state of L99A.
Tryptophan fluorescence measurements were made on Se-Met L99A in pH 3.0, 50 mM glycine, 20 mM NaCl and pH 7.0, 50 mM tris, 20 mM NaCl buffers and compared to those of L99A under the same conditions. The intensity increase accompanying denaturation was more pronounced for Se-Met L99A than for L99A, consistent with the stronger quenching ability of selenium (Figure 6). Figure 7 (a) shows that at each pressure, the centers of spectral mass were similar for L99A and Se-Met L99A in the same solvent, indicating that these two mutants are structurally similar at any given pressure and that the introduction of seleno-methionines does not significantly distort the shape of the fluorescence spectra or change the denaturation behavior.
Figure 7.
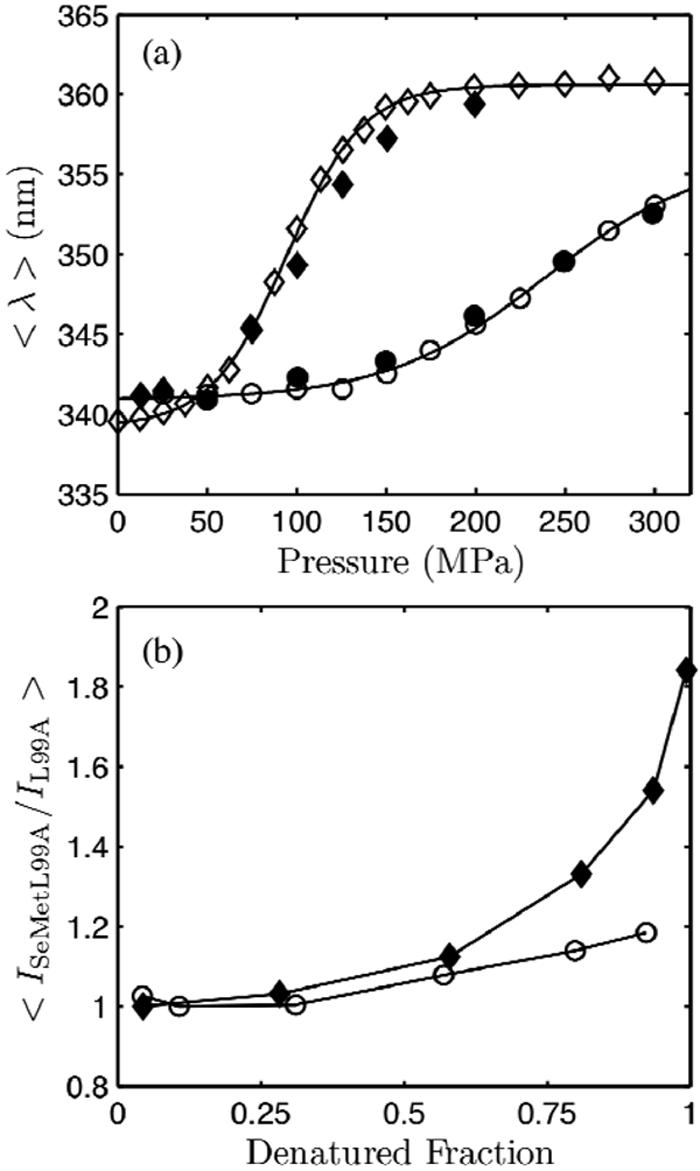
Pressure denaturation of L99A and Se-Met L99A in 50 mM glycine 20 mM NaCl pH 3.0 16 °C and 50 mM Tris HCl 20 mM NaCl pH 7.0 24 °C. (a) Centers of spectral mass of L99A (open) and Se-Met L99A (closed) at pH 3.0 (diamond) and pH 7.0 (circle). L99A and Se-Met L99A show similar denaturation curves, indicative of structural similarity at each pressure. (b) Normalized intensity ratios of Se-Met L99A and L99A emission at pH 3.0 (closed diamond) and pH 7.0 (open circle). At pH 7.0, the ratio shows little change with pressure, while at pH 3.0, there is a large increase. This indicates a greater separation between tryptophan and seleno-methionine residues in the denatured state at pH 3.0 compared with pH 7.0, suggesting that the pH 3.0 denatured state is more unfolded than at pH 7.0.
Pressure-induced changes to the fluorescence yield of tryptophan or transmission through the solvent were factored out in the ratio of the emission intensities of L99A and Se-Met-L99A, IL99A and ISe-MetL99A, at the same pressure. The ratios were calculated as follows at each pressure
| (9) |
The sum was taken over points in the wavelength range 310-400 nm where the fluorescence signal is strong. N is the number of ratios in the sum (91, in this case). Figure 7 (b) shows a comparison of these ratios at pH 3.0, 50 mM glycine, 20 mM NaCl and pH 7.0, 50 mM Tris, 20 mM NaCl as a function of denatured fraction. They are normalized at 0.1 MPa. We see that as denaturation progresses, the ratio is greater at pH 3.0 than at pH 7.0. This result suggests that the denatured state at pH 7.0 is more compact than at pH 3.0.
Cavity Volume Calculations and Internal Waters
The volumes contained by the external surface and the volumes of internal cavities present in the crystal structures of the T4 lysozyme mutants were calculated to determine if a correlation exists between the volume changes of denaturation extracted from thermodynamic analysis and the volume of buried cavities. In order to identify and measure buried cavities, crystal structures (PDB accession codes 1L90 for L99A, 1QUH for L99G/E108V, 1QS5 for A98L, 1G0P for V149G, and 1L63 for WT*) were analyzed with a 1.2 Å probe using MSMS (34). MSMS implements a rolling probe to determine reduced surfaces, from which the solvent excluded surface can be computed. Two sets of molecular surfaces were produced for each structure. In the first set of calculations, all crystallographically determined solvent particles were manually removed from the PDB files prior to analysis to eliminate false surface cavities caused by surface water, and to enable detection of hydrated internal cavities. The external surface and six to nine cavities were identified for each mutant (Fig. 1). In the second set, internal water molecules were retained to determine changes in cavity size due to hydration. Four water molecules, Sol-171, 179, 175, and 208, are considered conserved internal solvent molecules in T4 lysozyme (47). Using a 1.2 Å probe in MSMS (34) to define the molecular surfaces, Sol-213 was also observed within the external surface of all mutants. Sol-175 was found either in a solvent-exposed pocket on the surface or within the external surface. As Sol-175 was considered to fully occupy its cavity, its exact location did not contribute to the total cavity volume. Both L99G/E108V and V149G structures contain two additional poorly ordered water molecules in their respective mutation-enlarged cavities. The L99G/E108V structure contains Sol-401 and 402 in the hydrophobic cavity near the L99G mutation (Figure 1 (c), cavity 6). The V149G mutation enlarges the polar cavity binding Sol-208 (Figure 1 (e), cavity 3) and introduces Sol-323 (alternative site 423) and 324. Molecular surfaces that intersected or were located outside the external surface were not considered internal cavities. For comparison, a second crystal structure of WT* (provided by Marcus Collins, private communication) was also analyzed. The variability of individual cavity volumes in the two WT* structures was within 5.4 Å3 (3% of the total cavity volume of WT*). Table 2 summarizes the results.
Table 2.
Cavity Volumes of T4 lysozyme Mutants
| Cav.# | WT* (1L63) | WT* (MC)a | L99A (1L90) | L99G/E108V (1QUH) | A98L (1QS5) | V149G (1G0P) | Comments |
|---|---|---|---|---|---|---|---|
| 0 | 21044.0 | 21113.6 | 21103.3 | 20866.9 | 20919.7 | 21312.7 | Protein Surface |
| 1 | 50.3 (0) | 44.9 (0) | 51.9 (0) | 47.5 (0) | 46.8 (0) | 49.2 (0) | Sol-171, 179 |
| 2 | 4.6 | - | - | - | 10.4 | - | |
| 3 | 25.3 (0) | 22.2 (0) | 27.6 (0) | 23.6 (0) | 37.8 (0) | 106.3 (0)b | Sol-208, Trp138 |
| 4 | 25.8 (0) | 29.3 (4.2) | 26.7 (0) | - | - | 27.5 (0) | Sol-175 |
| 5 | 7.1 | 11.9 | 9.2 | - | 7.7 | 9.6 | |
| 6 | 39.2 | 39.1 | 161.1 | 222.5 (87.2)c | 53.7 | 40.7 | |
| 7 | 24.6 | 23.9 | 28.9 | 24.3 | 24.3 | 25.0 | Trp126 |
| 8 | - | - | - | 5.0 (0) | 7.1 (0) | - | Sol-213 |
| 9 | - | - | - | 5.5 | 8.5 | - | |
| 10 | - | - | - | - | 12.1 | - | |
Volumes in Å3 of molecular surfaces found in crystallographic structures (Experimental Procedures) of T4 lysozyme mutants with internal solvent molecules removed. Volumes of solvent-containing cavity determined with internal solvent molecules kept in the structures are shown inside the parentheses. The molecular surfaces were found using a 1.2 Å probe in MSMS (34). Molecular surface 0 is the outer surface of the proteins, while 1 - 10 were cavities isolated from the external solvent. Surfaces detected by MSMS but found to intersect with the external surface of the protein are not shown.
WT* structure was provided by Marcus Collins (private communication).
Cavity 3 in V149G contained Sol 323 (alternative site 423) and 324 according to the crystal structure.
Similarly, cavity 6 in L99G/E108V contains Sol 401 and 402.
Discussion
Structural Characterization of the Native and Pressure-Denatured States in Solution
The low-resolution solution structures of native L99A T4 lysozyme at pH 3.0 obtained from SAXS showed good agreement with the crystal structure solved at neutral pH. This supports the reasonable assumption that the shapes and sizes of the internal cavities in the crystal structure are representative of the solution structure. For the pressure-denatured state of L99A at pH 3.0, SAXS results suggest that the average overall conformation is extended while local regions may have residual structure. The radius of gyration of the pressure-denatured state was smaller than that predicted for the chemically-denatured state (39), consistent with the view that the widely accepted model of protein denaturation, i.e. the transfer of core residues to water, does not appropriately describe pressure denaturation. Fluorescence quenching measurements suggest that the pressure-denatured state of L99A at pH 7.0 is even more compact than at pH 3.0.
Unfolding at pH 3.0
The volume change accompanying denaturation is contributed by volume changes of the protein atoms, the solvent-excluded cavities, and the hydration of solvent-exposed residues. As the atoms are the least compressible component, it is thought that the major contributions to the volume change of denaturation are the elimination of solvent-excluded cavities and the hydration of solvent-exposed residues (32, 48). Around charged residues, electrostriction of water molecules is known to occur (32). However, the volume properties associated with the hydration of hydrophobic residues are still under investigation, particularly with respect to pressure (12,32, 49, 50). It is therefore difficult to probe the contributions of the hydration changes. By comparing mutants with various volumes of internal cavities, however, we can investigate how these volumes correlate with the volume change of denaturation.
To quantify cavity volumes, structural changes caused by a mutation must be identified, and the solvent occupancy of each cavity must be quantified. In a manner similar to the estimation of a mutation-induced change in stability (ΔΔG) by solvent transfer free energies, the change in cavity volume as a result of a single buried mutation can - as a first approximation - be estimated as the difference between the van der Waals volumes of the exchanged side chains. This analysis has been performed on staphylococcal nuclease and ribonuclease A mutants to predict the mutation-induced change in cavity volumes (8, 51). Particularly in the case of large-to-small amino acid substitutions in staphylococcal nuclease, the cavity volumes predicted by this method successfully correlated with the ΔVο of pressure denaturation, lending support to the hypothesis that internal hydration of cavities is implicated in pressure denaturation. However, in both studies, atomic resolution structures were not available for most of the mutants to support the assumption that the structural changes were localized at the mutation sites. The same side-chain volume analysis, when applied to T4 lysozyme, fails to predict mutation-induced changes in cavity volumes that correlate with ΔVο. Using the side chain volumes of Leu, Ala, Val, and Gly reported by Richards, et al. (52) and the experimental ΔVο of L99A at pH 3.0, 50 mM glycine, 20 mM NaCl as a reference, ΔVο was predicted for L99G/E108V, A98L, and V149G (8). Note that the mutation, E108V, was not included in this analysis because the 108th residue is on the surface of the protein. We therefore do not expect differences in the hydration of Glu (E) and Val (V), to contribute differently to the volume change of denaturation. The predicted results in Table 3 show in particular, the failure to account for the observed behavior of A98L and V149G. The ΔVο of V149G was predicted to be as large in magnitude as L99A, while that of A98L was predicted to be very small in magnitude, roughly half of the experimentally observed value.
Table 3.
Denaturation volume changes predicted by side-chain volumes
| ΔV°predict (Å3) | δΔV (Å3) | |
|---|---|---|
| L99G/E108V | 206.2 ± 10.0 | 29.6 ± 26.9 |
| A98L | 73.1 ± 10.0 | 54.1 ± 12.1 |
| V149G | 187.1 ± 10.0 | -53.0 ± 12.4 |
The experimental denaturation volume change, ΔV°, of L99A at pH 3.0 20 mM NaCl was used as a reference. δΔV = ΔV°experiment - ΔV°predict. Volume changes of other mutants were predicted with van der Waals volumes of side-chains (52) of exchanged amino acids.
The failure of this method to estimate mutation-induced changes in cavity volumes is not surprising. Previous crystallographic studies of T4 lysozyme have also shown that structural changes due to a mutation cannot be easily predicted. At some buried sites, a large-to-small amino acid substitution in T4 lysozyme created or enlarged a cavity, while at other sites, a similar mutation caused a rearrangement of the protein that filled the cavity (53). Moreover, for mutations that permitted the creation or enlargement of cavities, the new cavity volume could not be simply predicted by a side-chain analysis using the WT* structure and the van der Waals volumes of the exchanged amino acids. In the case of L99A, the change in cavity size is slightly smaller than expected. An extreme case was L99G T4 lysozyme. Unlike L99A, the L99G mutation exposes the large cavity to the solvent. A second mutation, E108V, far from the cavity and on the surface of the protein was required to close the opening of the molecule's surface created by the L99G mutation (16). The structural changes caused by a small-to-large amino acid substitution such as A98L are less predictable (18).
Crystal structures of T4 lysozyme were thus necessary to quantify cavity volumes. The internal cavities of the mutants identified with a 1.2 Å probe are shown in Figure 1 (b)-(f), and the corresponding volumes are presented in Table 2. An inspection of the crystal structures explains the large discrepancies between the experimental ΔVο and that predicted by side-chain volume analysis for A98L and V149G (Table 3). The A98L mutation does not fill the cavity at the mutation site, but instead, causes the strain-induced formation of several small cavities throughout the protein (Fig. 1 (d)). Therefore the total cavity volume is greater than that predicted by side-chain volume analysis. In the case of V149G, the mutation not only enlarges a cavity (Fig. 1 (e)) but this cavity accommodates two additional solvent-binding sites. Thus, the effective volume of this cavity is likely smaller than that predicted by side-chain volume analysis.
The volume of a single water molecule in bulk is approximately 30 Å3, which is roughly the size of a small cavity in T4 lysozyme. Therefore, the solvent occupancy of each cavity can have large effects on its effective volume. Cavity 1 (Fig. 1, Table 2) is a conserved solvent-binding site in T4 lysozyme (47) and contains Sol 171 and 179. Similarly, cavity 3 binds another conserved internal water molecule, Sol 208. Cavities 4 and 8 contain Sol 175 and 213, respectively. Crystal structures enable the identification of internal water molecules that otherwise may not be identified. However, the average occupancy of internal water is not easily determined by conventional model-based crystallographic refinement methods (54). Sol 171, 175, 179, and 208 have been observed in a large number of mutant structures solved over many independent experiments, and several of these water molecules show evidence of hydrogen bonding stabilization (47). Sol 213 was also observed in all of the mutants studied in this work. We therefore made the reasonable assumption that these water molecules have full occupancy in all of the studied mutants.
The occupancies of poorly ordered water molecules introduced by V149G and L99G/E108V were more difficult to assign. In the crystal structures, the enlarged cavity at the V149G mutation site (Fig. 1 (e), cavity 3) contains two additional water molecules (Sol 323 and 324) in addition to Sol 208, while that of L99G/E108V (Fig. 1 (c), cavity 6) contains Sol 401 and 402. We first assume that both of these cavities are fully occupied. Under this assumption, however, no correlation is apparent in the relationship between ΔVο at pH 3.0 with the calculated total cavity volumes (Fig. 8 (a)). As discussed earlier, the ΔVο of V149G predicted by side-chain volume analysis was much larger than the experimentally determined value because solvent binding of the mutation-enlarged cavity was not accounted for. As this mutation site is the location of the only buried polar network in T4 lysozyme (17), we believe that it is reasonable that Sol-323 and 324 occupy this cavity at least partially under the solvent conditions used in this study. This may explain why the tryptophan fluorescence spectrum of V149G in its folded state was the most red-shifted of all the mutants (Figure 2). It is possible that the tryptophan residue lining this polar cavity (Trp 138) reports the increased hydration of V149G.
Figure 8.
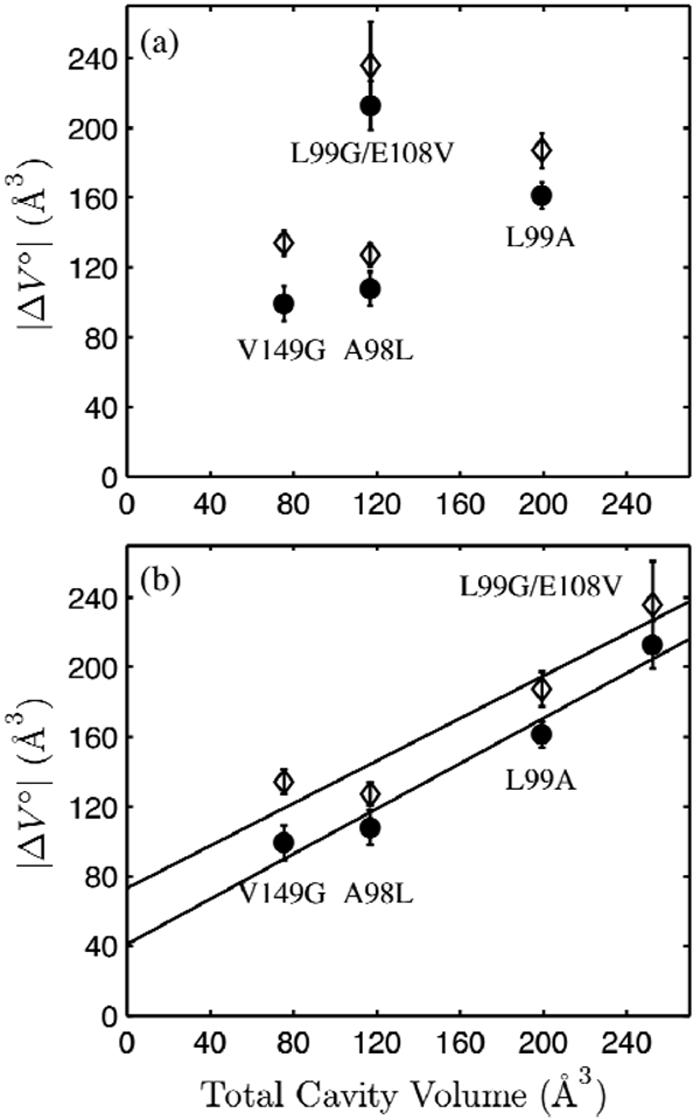
The volume changes of denaturation (see Table 1) of L99G/E108V, L99A, V149G, and A98L T4 lysozyme in pH 3.0 buffer, 16 °C at 20 mM NaCl (open diamond) and 100 mM NaCl (closed circle) compared to the total cavity volumes calculated from crystal structures using two methods. (a) No correlation is apparent when full occupancy of all crystallographically determined internal solvent-biding sites was assumed in the calculation of cavity volumes. (b) The large cavity of L99G/E108V was assumed to be empty, while full occupancy was assumed for all other internal solvent molecules. The volume changes of denaturation correlate with the total cavity volumes.
In contrast, we speculate that the enlarged hydrophobic cavity in L99G/E108V is empty at pH 3.0. The enlarged cavity of L99A is believed to be empty (1, 15), and the crystal structures of L99G/E108V and L99A are very similar except for the differing volumes of the enlarged cavities (16). At pH 3.0, the magnitude of the volume change upon pressure denaturation was roughly 50 Å3 greater for L99G/E108V compared to L99A. This value is similar to the difference in volumes of the enlarged cavities (Figure 1 (b)-(c), cavity 6) in their empty states (61.4 Å3), which are the dominant contributors to the total cavity volumes of the two mutants. This suggests that the occupancies of the two cavities are similar at pH 3.0. The total cavity volumes were recalculated with the new assumption that the enlarged cavity in L99G/E108V is empty and that all other crystallographically determined solvent-binding sites are fully occupied. A correlation between denaturation volume changes and total cavity volume is now observed (Fig. 8 (b)). The proportionality of the two quantities suggests that all the cavities are hydrated under pressure denaturation at this pH. The slope of the linear fits was dependent on the probe size used to calculate the cavity volumes. Using a 1.4 Å probe gave a smaller slope as fewer small cavities could be detected, reducing the estimated total cavity volume of small cavity mutants by a greater fraction than that of the large cavity mutants.
The magnitude of the volume change of denaturation was also dependent on the ionic strength, reflected by the ~ 40 Å3 change in vertical positions of linear fits in Figure 8 (b). We consider two possible mechanisms of hydration changes around solvent-exposed residues to explain this ionic strength dependence. At pH 3.0, T4 lysozyme is highly positively charged. Most of the charged residues are on the surface of T4 lysozyme, and no change in the hydration of these residues is expected from denaturation. However, there are several buried and semi-buried salt bridges in T4 lysozyme. Crystal structures of T4 lysozyme solved at various ionic strengths demonstrate that a change in ionic strength does not affect buried salt bridges in the native state (55). These salt bridges are likely exposed in the pressure-denatured state, and the presence of counter ions around the dissociated salt pairs would reduce the effect of electrostriction on the denaturation volume change. The presence of counter ions may also affect the compactness of the denatured state, and thus change the number of solvent-exposed residues. Refolding of an acid-denatured protein has been observed at high anion concentrations (56). This is thought to be due to reduced charge repulsions in the protein. A similar behavior can be expected to occur to T4 lysozyme at pH 3.0. This may be the basis for the apparent blue-shift in the tryptophan fluorescence of the denatured state with increased NaCl concentration. Regardless of mechanism, our data indicate that under the conditions investigated at low pH, all of the internal cavities are hydrated while the ionic strength of the solvent affects the volume contribution of hydration around solvent-exposed residues.
Cavity Filling at pH 7.0
Fluorescence measurements were made at pH 7.0, 24 °C as a direct comparison to the high-pressure, room-temperature crystal structures of L99A and WT* T4 lysozyme (1). According to the high-pressure crystal structures acquired by Collins et al., cooperative water filling of the large hydrophobic cavity in L99A (Fig. 1 (b), cavity 6) is favorable under pressure even without unfolding (1). We emphasize that in the crystal state, large conformational changes such as unfolding do not occur. Only the large cavity of L99A showed increased internal electron density with pressure (PDB structures 2B6T, 2B6W, 2B6X, 2B6Y, 2B6Z, 2B70, 2B72, 2B73, 2B74, 2B75). No other pressure-induced changes in internal hydration were observed in the crystal structures. WT*, which has a smaller corresponding cavity (Fig. 1 (a), cavity 6), showed no water filling up to 200 MPa (PDB structures 2OE4, 2OE7 are at 100 MPa; 2OE9, 2OEA at 200 MPa). Molecular dynamics simulations performed on the high-pressure structures of L99A suggested cooperative filling of four water molecules in the large cavity (1).
In solution, L99A underwent a pressure-induced change in fluorescence corresponding to a denaturation volume change of ΔVο = - 93.4 ± 8.2 Å3, nearly a factor of two smaller than at pH 3.0, with a pressure midpoint of roughly 240 MPa. Structural information obtained from comparative fluorescence quenching of L99A and Se-Met L99A indicated that the pressure-denatured state of L99A at pH 7.0 is even more compact than that at pH 3.0. While our methods do not provide direct evidence of preferential water filling of the large cavity in L99A in solution at this pH, we explored this is a possible mechanism to explain the small volume change and compactness of the denatured state. Assuming that the volume change of transferring each water molecule from the bulk to the large cavity is 30 Å3, this mechanism would suggest that in solution, the large cavity is filled by three water molecules (93.4 Å3 ~ 3 × 30 Å3). This is in reasonable agreement to the predictions made by the MD simulations (1). For comparison with the change in water occupancy of the large cavity determined experimentally by integrated electron densities in the crystal structures (1), the centers of spectral mass of L99A were converted to change in water occupancy under the assumption that under pressure, the large cavity is filled by three water molecules (Fig. 9). A two-state thermodynamic fit to the converted fluorescence data with a fixed volume change of three bulk water molecules shows good agreement with the crystallographic results. This suggests that preferential water filling of the large cavity is a plausible mechanism to explain the small volume change and size of the denatured state at this pH.
Figure 9.
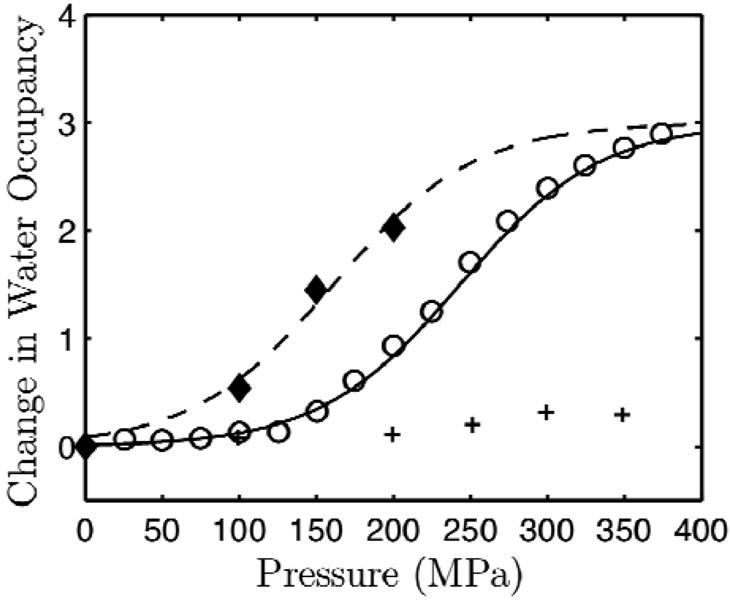
Comparison of change in water occupancy of L99A at neutral pH determined by X-ray crystallography (diamond, reprinted with permission from Collins, et al. (1)) and fluorescence spectroscopy (circle). A two-state (0 or 3 water molecules) thermodynamic fit to the data is shown on each data set with a fixed volume change of 90 Å3 (volume of three water molecules in bulk). The fit to the crystallography data is shifted to lower pressure by 82 MPa relative to the fluorescence data. This stability difference may be due to subtle differences between structure and dynamics of the T4 lysozyme molecule in solution and in the crystal. The change in water occupancy of WT* determined by fluorescence is shown for reference (cross).
As a comparison to L99A, fluorescence measurements were also made on the structurally similar mutant, L99G/E108V. Although the large cavity of L99G/E108V is roughly 61 Å3 greater in volume than the corresponding cavity in L99A, the denaturation volume changes were the same to within error (approximately 10 Å3). As mentioned earlier, the crystal structure of L99G/E108V shows two poorly ordered water molecules in the large cavity (Sol 401 and 402). We interpret this to mean that at neutral pH, where the crystal structure was solved, the large cavity of L99G/E108V is partially filled with a maximum occupancy of two water molecules. By taking two water molecules into consideration, the difference in volume changes of cavity-filling for L99A and L99G/E108V can be as small as 1 Å3 (61 Å3 ~ 2 × 30 Å3).
One of the paradoxes of pressure denaturation has been the small magnitudes of observed volume changes. It has been thought that a positive volume contribution must exist, possibly due to the hydration of hydrophobic residues (32). While this is plausible, it cannot be assumed that all the existing cavities and packing defects of a protein are hydrated or eliminated with the application of pressure. The high-pressure crystallography study of L99A showed that water penetration of the large cavity and not other, smaller cavities in the native structure of L99A was possible in part due to the cooperativity (1). Only the hydrophobic cavity large enough to accommodate multiple waters interacting with hydrogen bonds was filled in the folded structure.
Conclusions
Our results imply the existence of multiple pressure-denatured states of the T4 lysozyme family with differing levels of internal hydration and unfolding that were dependent on the solvent conditions and particularly pH. The results were thus consistent with water penetration of the protein rather than transfer of hydrophobic residues from the core of the protein to the water as the mechanism of pressure denaturation. At pH 3.0, the pressure-denatured state was extended but more compact than the predicted size of a chemically denatured protein of the same length (39). We were able to relate the volume change of denaturation to cavity volume and hydration changes by comparing multiple mutants of T4 lysozyme with known crystal structures and varying the ionic strength of the solvent. The magnitudes of the volume changes of denaturation for L99A, L99G/E108V, A98L, and V149G T4 lysozyme positively correlated with the total cavity volume. These results suggest that at pH 3.0, all the cavities were hydrated in the pressure-denatured state. Increasing the ionic strength reduced the magnitude of the volume change likely as a result of counter ion interactions with salt pairs exposed in the denatured state. At pH 7.0, the denaturation thermodynamics of L99A in solution was in good agreement with the preferential cavity filling observed by high-pressure crystallography and MD simulations (1). The pressure-denatured state at pH 7.0 was more compact in size than at pH 3.0. These results suggest that at pH 7.0, not all the cavities are filled. We speculate several mechanisms for the pH-dependence on the hydration of the denatured state. pH may affect the polarity of internal cavities. It may also affect the dynamics of the protein and therefore affect the water-penetration pathways. Finally, it is also possible that the charge state of the protein affects the compactness of the denatured state, which in turn may lead to preferential cavity filling as is the case for folded structures (1).
We showed that the problem of pressure denaturation not only requires understanding of the change in water occupancy as a function of pressure, but also in the volume properties of the protein and interacting waters. The availability of crystal structures was necessary for the quantification of cavity volumes and solvent occupancy of the T4 lysozyme mutants investigated in our work. However, we recognize that a crystal structure represents a static picture of the average conformation of a protein under a specific condition. As a protein is a fluctuating system, the cavities and internal solvent molecules are not static. The calculation of a cavity volume from an atomic structure also depends on the probe size and the van der Waals radii of the amino acids used for identifying molecular surfaces. For the T4 lysozyme mutants used in this study, a 1.2 Å probe was preferred over a 1.4 Å probe. Small cavities that were found in all mutants by a 1.2 Å probe were not consistently identified with a 1.4 Å probe. The magnitudes of the experimentally observed volume changes of denaturation were roughly the size of several water molecules. The interpretation of volume changes from native structures, therefore, requires certainty in the occupancy of internal water molecules in the native state and its dependence on the solvent condition.
In this work, we investigated the contribution of cavity volumes and electrostriction to the volume change of pressure denaturation. The volume properties of hydration waters, particularly around hydrophobic residues as a function of pressure, remain to be established. Finally, it was assumed in this study that the volume change accompanying denaturation was pressure-independent because the magnitude and sign of isothermal compressibility change with denaturation, i.e. the pressure dependence of the denaturation volume change, are still under debate. Direct measurements of the isothermal compressibility change associated with pressure denaturation have not yet been made (57). With the exception of the study by Seemann, et al. (33), the isothermal compressibility change has only been determined from fitting a thermodynamic model with a second-order expansion of ΔG to denaturation data (57). It is noted that the sign of the isothermal compressibility is defined inconsistently in the literature, causing confusion on the sign of the second-order term of ΔG (32, 48, 57, 58, 59, 60). A second-order fit also increases the number of free fitting parameters such that over-fitting of data is also a concern. Further investigations of the pressure-dependence of the volume change accompanying denaturation are suggested.
The magnitude of the volume change accompanying pressure denaturation of a protein is generally less than 1% of the protein volume and on the order of several bulk water molecules in volume. Our results support the growing view that the pressure-denatured state of a protein is one in which the penetration by a few water molecules is favored and emphasizes the importance of the role of water in protein folding and biological processes.
Supplementary Material
Acknowledgments
The authors thank Marcus Collins (Univ. of Washington), Mark Tate (Cornell University), Lois Pollack (Cornell University), and Jessica Lamb (Cornell University) for helpful discussions. For their help with data acquisition and experiments, the authors are grateful to Fred Heberle, Raphael Kapfer, Chae Un Kim, Lucas Koerner, Darren Southworth, and Gil Toombes as well as CHESS staff members, Arthur Woll, Peter Busch, and Richard Gillilan. The authors thank Warren DeLano (Delano Scientific LLC) for providing a script for viewing molecular surfaces in PyMol, and Lucas Koerner (Cornell University) for critical reading of this manuscript.
Supported by grants from the Cornell Center for Materials Research (NSF DMR-0079992), NIH GM074899 grant to the Hauptmann-Woodward Institute, DOE BER DEFG-02-97ER62443, and CHESS, an NSF-DMR/NIH-NIGMS supported National Facility under NSF award DMR-0225180.
Abbreviation List
- SAXS
small-angle X-ray scattering
- SVD
singular value decomposition
- EOM
the SAXS program Ensemble Optimization Method
- WT*
a cysteine-free pseudo-wild-type mutant of T4 lysozyme
- L99A
a leucine-99 to alanine mutant of WT*
- L99G/E108V
a mutant of WT* with the mutations, leucine-99 to glycine and glutamic acid-108 to valine
- A98L
a alanine-98 to leucine mutant of WT*
- V149G
a valine-149 to glycine mutant of WT*
- Se-Met L99A
L99A with methionines replaced by selenomethionines
References
- 1.Collins MD, Hummer G, Quillin ML, Matthews BW, Gruner SM. Cooperative water filling of a nonpolar protein cavity observed by high-pressure crystallography and simulation. Proc. Nat. Acad. Sci U.S.A. 2005;102:16668–16671. doi: 10.1073/pnas.0508224102. [published erratum appears in (2006) Proc. Nat. Acad. Sci U.S.A. 103, 4793] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dill KA, Shortle D. Denatured states of proteins. Annu. Rev. Biochem. 1991;60:795–825. doi: 10.1146/annurev.bi.60.070191.004051. [DOI] [PubMed] [Google Scholar]
- 3.Kauzmann W. Denaturation of proteins and enzymes. In: McElroy WD, Glass B, editors. The mechanism of enzyme action. Johns Hopkins Press; Baltimore, MD: 1954. pp. 70–110. [Google Scholar]
- 4.Kauzmann W. Some factors in the interpretation of protein denaturation. Adv. Protein Chem. 1959;14:1–63. doi: 10.1016/s0065-3233(08)60608-7. [DOI] [PubMed] [Google Scholar]
- 5.Dill KA. Dominant forces in protein folding. Biochemistry. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 6.Murphy KP, Privalov PL, Gill SJ. Common features of protein unfolding and dissolution of hydrophobic compounds. Science. 1990;247:559–561. doi: 10.1126/science.2300815. [DOI] [PubMed] [Google Scholar]
- 7.Zipp A, Kauzmann W. Pressure denaturation of metmyoglobin. Biochemistry. 1973;12:4217–4228. doi: 10.1021/bi00745a028. [DOI] [PubMed] [Google Scholar]
- 8.Frye KJ, Royer CA. Probing the contribution of internal cavities to the volume change of protein unfolding under pressure. Protein Sci. 1998;7:2217–2222. doi: 10.1002/pro.5560071020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Panick G, Malessa R, Winter R, Rapp G, Frye KJ, Royer CA. Structural characterization of the pressure-denatured state and unfolding/refolding kinetics of staphylococcal nuclease by synchrotron small-angle X-ray scattering and Fourier-transform infrared spectroscopy. J. Mol. Biol. 1998;275:389–402. doi: 10.1006/jmbi.1997.1454. [DOI] [PubMed] [Google Scholar]
- 10.Hummer G, Garde S, García AE, Paulaitis ME, Pratt LR. The pressure dependence of hydrophobic interactions is consistent with the observed pressure denaturation of proteins. Proc. Nat. Acad. Sci U.S.A. 1998;95:1552–1555. doi: 10.1073/pnas.95.4.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paliwal A, Asthagiri D, Bossev DP, Paulaitis ME. Pressure denaturation of staphylococcal nuclease studied by neutron small-angle scattering and molecular simulation. Biophys. J. 2004;87:3479–3492. doi: 10.1529/biophysj.104.050526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Day R, García AE. Water penetration in the low and high pressure native states of ubiquitin. Proteins Struct. Funct. Genet. 2008;70:1175–1184. doi: 10.1002/prot.21562. [DOI] [PubMed] [Google Scholar]
- 13.Matsumura M, Matthews BW. Control of enzyme activity by an engineered disulfide bond. Science. 1989;243:792–794. doi: 10.1126/science.2916125. [DOI] [PubMed] [Google Scholar]
- 14.Matthews BW. Structural and genetic analysis of protein stability. Annu. Rev. Biochem. 1993;62:139–160. doi: 10.1146/annurev.bi.62.070193.001035. [DOI] [PubMed] [Google Scholar]
- 15.Eriksson AE, Baase WA, Zhang X-J, Heinz DW, Blaber M, Baldwin EP, Matthews BW. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science. 1992;255:178–183. doi: 10.1126/science.1553543. [DOI] [PubMed] [Google Scholar]
- 16.Wray JW, Baase WA, Lindstrom JD, Weaver LH, Poteete AR, Matthews BW. Structural analysis of a non-contiguous second-site revertant in T4 lysozyme shows that increasing the rigidity of a protein can enhance its stability. J. Mol. Biol. 1999;292:1111–1120. doi: 10.1006/jmbi.1999.3102. [DOI] [PubMed] [Google Scholar]
- 17.Xu J, Baase WA, Quillin ML, Baldwin EP, Matthews BW. Structural and thermodynamic analysis of the binding of solvent at internal sites in T4 lysozyme. Protein Sci. 2001;10:1067–1078. doi: 10.1110/ps.02101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu R, Baase WA, Matthews BW. The introduction of strain and its effects on the structure and stability of T4 lysozyme. J. Mol. Biol. 2000;295:127–145. doi: 10.1006/jmbi.1999.3300. [DOI] [PubMed] [Google Scholar]
- 19.Poteete AR, Sun D-P, Nicholson H, Matthews BW. Second-site revertants of an inactive T4 lysozyme mutant restore activity by restructuring the active site cleft. Biochemistry. 1991;30:1425–1432. doi: 10.1021/bi00219a037. [DOI] [PubMed] [Google Scholar]
- 20.Vetter IR, Baase WA, Heinz DW, Xiong J-P, Snow S, Matthews BW. Protein structural plasticity exemplified by insertion and deletion mutants in T4 lysozyme. Protein Sci. 1996;5:2399–2415. doi: 10.1002/pro.5560051203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 22.Muchmore DC, McIntosh LP, Russell CB, Anderson DE, Dahlquist FW. Expression and nitrogen-15 labeling of proteins for proton and nitrogen-15 nuclear magnetic resonance. Methods Enzymol. 1989;177:44–73. doi: 10.1016/0076-6879(89)77005-1. [DOI] [PubMed] [Google Scholar]
- 23.Neuman RC, Jr., Kauzmann W, Zipp A. Pressure dependence of weak acid ionization in aqueous buffers. J. Phys. Chem. 1973;77:2687–2691. [Google Scholar]
- 24.Kauzmann W, Bodanszky A, Rasper J. Volume changes in protein reactions. II. Comparison of ionization reactions in proteins and small molecules. J. Am. Chem. Soc. 1962;84:1777–1788. [Google Scholar]
- 25.Ando N, Chenevier P, Novak M, Tate MW, Gruner SM. High hydrostatic pressure small-angle X-ray scattering cell for protein solution studies featuring diamond windows and disposable sample cells. J. Appl. Crystallogr. 2008;41:167–175. [Google Scholar]
- 26.Svergun DI, Koch MHJ. Small-angle scattering studies of biological macromolecules in solution. Rep. Prog. Phys. 2003;66:1735–1782. [Google Scholar]
- 27.Glatter O. Data Treatment. In: Glatter O, Kratky O, editors. Small Angle X-ray Scattering. 1st ed. Academic Press; London: 1982. pp. 119–165. [Google Scholar]
- 28.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003;36:1277–1282. [Google Scholar]
- 29.Svergun DI, Koch MHJ. Advances in structure analysis using small-angle scattering in solution. Curr. Opin. Struct. Biol. 2002;12:654–660. doi: 10.1016/s0959-440x(02)00363-9. [DOI] [PubMed] [Google Scholar]
- 30.Bernadó P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 31.Silva JL, Miles EW, Weber G. Pressure dissociation and conformational drift of the beta dimer of tryptophan synthase. Biochemistry. 1986;25:5780–5786. doi: 10.1021/bi00367a065. [DOI] [PubMed] [Google Scholar]
- 32.Royer CA. Revisiting volume changes in pressure-induced protein unfolding. Biochim. Biophys. Acta. 2002;1595:201–209. doi: 10.1016/s0167-4838(01)00344-2. [DOI] [PubMed] [Google Scholar]
- 33.Seemann H, Winter R, Royer CA. Volume, expansivity and isothermal compressibility changes associated with temperature and pressure unfolding of Staphylococcal nuclease. J. Mol. Biol. 2001;307:1091–1102. doi: 10.1006/jmbi.2001.4517. [DOI] [PubMed] [Google Scholar]
- 34.Sanner MF, Olson AJ, Spehner J-C. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 1996;38:305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 35.Vivian JT, Callis PR. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys. J. 2001;80:2093–2109. doi: 10.1016/S0006-3495(01)76183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peng Q, Li H. Atomic force microscopy reveals parallel mechanical unfolding pathways of T4 lysozyme: evidence for a kinetic partitioning mechanism. Proc. Natl. Acad. Sci. U.S.A. 2008;105:1885–1890. doi: 10.1073/pnas.0706775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elwell ML, Schellman JA. Stability of phage T4 lysozymes. I. Native properties and thermal stability of wild type and two mutant lysozymes. Biochim. Biophys. Acta. 1977;494:367–383. doi: 10.1016/0005-2795(77)90166-0. [DOI] [PubMed] [Google Scholar]
- 38.Becktel WJ, Baase WA. Thermal denaturation of bacteriophage T4 lysozyme at neutral pH. Biopolymers. 1987;26:619–623. doi: 10.1002/bip.360260505. [DOI] [PubMed] [Google Scholar]
- 39.Kohn JE, Millett IS, Jacob J, Zagrovic B, Dillon TM, Cingel N, Dothager RS, Seifert S, Thiyagarajan P, Sosnick TR, Hasan MZ, Pande VS, Ruczinski I, Doniach S, Plaxco KW. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12491–12496. doi: 10.1073/pnas.0403643101. [published erratum appears in (2004) Proc. Natl. Acad. Sci. U.S.A. 102, 14475-a] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pollack L, Tate MW, Finnefrock AC, Kalidas C, Trotter S, Darnton NC, Lurio L, Austin RH, Batt CA, Gruner SM, Mochrie SGJ. Time resolved collapse of a folding protein observed with small angle x-ray scattering. Phys. Rev. Lett. 2001;86:4962–4965. doi: 10.1103/PhysRevLett.86.4962. [DOI] [PubMed] [Google Scholar]
- 41.Arai M, Ito K, Inobe T, Nakao M, Maki K, Kamagata K, Kihara H, Amemiya Y, Kuwajima K. Fast compaction of alpha-lactalbumin during folding studied by stopped-flow X-ray scattering. J. Mol. Biol. 2002;321:121–132. doi: 10.1016/s0022-2836(02)00566-1. [DOI] [PubMed] [Google Scholar]
- 42.Chen L, Wildegger G, Kiefhaber T, Hodgson KO, Doniach S. Kinetics of lysozyme refolding: structural characterization of a non-specifically collapsed state using time-resolved X-ray scattering. J. Mol. Biol. 1998;276:225–237. doi: 10.1006/jmbi.1997.1514. [DOI] [PubMed] [Google Scholar]
- 43.Tardieu A, Le Verge A, Malfois M, Bonneté F, Finet S, Riès-Kautt M, Belloni L. Proteins in solution: from x-ray scattering intensities to interaction potentials. J. Cryst. Growth. 1999;196:193–203. [Google Scholar]
- 44.Bernadó P, Blanchard L, Timmins P, Marion D, Ruigrok RWH, Blackledge M. A structural model for unfolded proteins from residual dipolar couplings and small-angle x-ray scattering. Proc. Natl. Acad. Sci. U. S. A. 2005;102:17002–17007. doi: 10.1073/pnas.0506202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elwell M, Schellman J. Phage T4 lysozyme: Physical properties and reversible unfolding. Biochim. Biophys. Acta. 1975;386:309–323. doi: 10.1016/0005-2795(75)90273-1. [DOI] [PubMed] [Google Scholar]
- 46.Yuan T, Weljie A, Vogel HJ. Tryptophan fluorescence quenching by methionine and selenomethionine residues of calmodulin: Orientation of peptide and protein binding. Biochemistry. 1998;37:3187–3195. doi: 10.1021/bi9716579. [DOI] [PubMed] [Google Scholar]
- 47.Zhang XJ, Matthews BW. Conservation of solvent-binding sites in 10 crystal forms of T4 lysozyme. Protein Sci. 1994;3:1031–1039. doi: 10.1002/pro.5560030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heremans K, Smeller L. Protein structure and dynamics at high pressure. Biochim. Biophys. Acta. 1998;1386:353–370. doi: 10.1016/s0167-4838(98)00102-2. [DOI] [PubMed] [Google Scholar]
- 49.Kitchen DB, Reed LH, Levy RM. Molecular dynamics simulation of solvated protein at high pressure. Biochemistry. 1992;31:10083–10093. doi: 10.1021/bi00156a031. [DOI] [PubMed] [Google Scholar]
- 50.Harpaz Y, Gerstein M, Chothia C. Volume changes on protein folding. Structure. 1994;2:641–649. doi: 10.1016/s0969-2126(00)00065-4. [DOI] [PubMed] [Google Scholar]
- 51.Torrent J, Connelly JP, Coll MG, Ribó M, Lange R, Vilanova M. Pressure versus heat-induced unfolding of ribonuclease A: the case of hydrophobic interactions within a chain-folding initiation site. Biochemistry. 2000;38:15952–15961. doi: 10.1021/bi991460b. [DOI] [PubMed] [Google Scholar]
- 52.Richards FM. The interpretation of protein structures: total volume, group volume distributions and packing density. J. Mol. Biol. 1974;82:1–14. doi: 10.1016/0022-2836(74)90570-1. [DOI] [PubMed] [Google Scholar]
- 53.Eriksson AE, Baase WA, Matthews BW. Similar hydrophobic replacements of Leu99 and Phe153 within the core of T4 lysozyme have different structural and thermodynamic consequences. J. Mol. Biol. 1993;229:747–769. doi: 10.1006/jmbi.1993.1077. [DOI] [PubMed] [Google Scholar]
- 54.Hodel A, Kim S-H, Brunger AT. Model bias in macromolecular crystal structures. Acta Crystallogr. 1992;A48:851–858. [Google Scholar]
- 55.Bell JA, Wilson KP, Zhang X-J, Faber HR, Nicholson H, Matthews BW. Comparison of the crystal structure of bacteriophage T4 lysozyme at low, medium, and high ionic strengths. Proteins Struct., Funct., and Genet. 1991;10:10–21. doi: 10.1002/prot.340100103. [DOI] [PubMed] [Google Scholar]
- 56.Goto Y, Calciano LJ, Fink AL. Acid-induced folding of proteins. Proc. Natl. Acad. Sci. USA. 1990;87:573–577. doi: 10.1073/pnas.87.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taulier N, Chalikian TV. Compressibility of protein transitions. Biochim. Biophys. Acta. 2002;1595:48–70. doi: 10.1016/s0167-4838(01)00334-x. [DOI] [PubMed] [Google Scholar]
- 58.Prehoda KE, Mooberry ES, Markley JL. Pressure denaturation of proteins: evaluation of compressibility effects. Biochemistry. 1998;37:5785–5790. doi: 10.1021/bi980384u. [DOI] [PubMed] [Google Scholar]
- 59.Smeller L. Pressure-temperature phase diagrams of biomolecules. Biochim. Biophys. Acta. 2002;1595:11–29. doi: 10.1016/s0167-4838(01)00332-6. [DOI] [PubMed] [Google Scholar]
- 60.Meersman F, Smeller L, Heremans K. Protein stability and dynamics in the pressure-temperature plane. Biochim. Biophys. Acta. 2006;1764:346–354. doi: 10.1016/j.bbapap.2005.11.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.