

Abstract
BACKGROUND: Posttranslational protein modifications have been implicated in the development of autoimmunity. Protein L-isoaspartate (D-aspartate) O-methyltransferase (PIMT) repairs modified proteins and is encoded by PCMT1, located in a region linked to type 1 diabetes (T1D), namely IDDM5. AIM: To evaluate the association between genetic variations in the PCMT1 gene and T1D. METHODS: Firstly, PCMT1 was sequenced in 26 patients with T1D (linked to IDDM5) and 10 control subjects. The variations found in PCMT1 were then tested (alone and interacting with a functional polymorphism in SUMO4 and with HLA) for association with T1D in 253 families (using transmission disequilibrium test). In a third step, the association of the functional variation in PCMT1 (rs4816) with T1D was analyzed in 778 T1D patients and 749 controls (using chi-square test). In vitro promoter activity was assessed by transfecting INS-1E cells with PCMT1 promoter constructs and a reporter gene, with or without cytokine stimulation. RESULTS: Four polymorphisms in complete linkage disequilibrium were identified in PCMT1 (5' to the gene (rs11155676), exon 5 (rs4816) and exon 8 (rs7818 and rs4552)). In the whole cohort of 253 families, the allele associated with increased PIMT enzyme activity (rs4816, allele A) was less frequently transmitted to the affected than to the non-affected offspring (46% vs. 53%, p = 0.099). This finding was even more evident in the subset of families where the proband had high-risk SUMO4 (p = 0.069) or low-risk HLA (p = 0.086). Surprisingly, in the case-control study with 778 cases and 749 controls, an inverse trend was found (40.36% of patients and 36.98% of controls had the allele, p = 0.055). PCMT1 promoter activity increased with cytokine stimulation, but no differences were detected between the constructs adjacent to rs11155676. CONCLUSION: PCMT1 was virtually associated with T1D in groups defined by other risk genes (SUMO4 and HLA). A general association in a not further defined sample of T1D patients was not evident. Verification in a larger population is needed.
Keywords: type 1 diabetes, INS-1E cells, promoter activity, protein isomerisation, polymorphism, SUMO4
Introduction
Posttranslational protein modifications may create new antigenic epitopes and elicit an autoimmune response. In fact, many of these modifications have been shown to play a role in both animal models and human autoimmune disease [1-5]. We proposed that cytokines, produced by macrophages and dendritic cells in the islet of Langerhans, might cause posttranslational protein modifications triggering multiple antigen-specific B- and T-lymphocyte responses in the initiation phase of the pathogenesis of type 1 diabetes (T1D) [6]. In addition, we recently published data supporting a role for the repair enzyme protein L-isoaspartate (D-aspartate) O-methyltransferase (PIMT) in the pathogenesis of T1D. It is highly expressed in human beta-cells, and an inducer of this enzyme delays diabetes onset and reduces the severity of the disease in diabetes-prone BB rats [7]. Pcmt knock-out mice display an autoimmune phenotype, with increased T cell proliferative responses to mitogen and receptor-mediated stimulation. Furthermore, anti-DNA auto-antibodies develop in wild-type mice transferred with bone marrow from animals lacking Pcmt [8].
In humans, the gene encoding PIMT (PCMT1) is located on chromosome 6q24-25, in a genomic region which has been previously linked to T1D (IDDM5) [9].
The aim of this study was to further elucidate the role of PIMT in T1D through assessing the results from PCMT1 genetic studies. These included screening the gene for variations in a selected group of individuals, assessing their potential functional implications and evaluating the association between these genetic variations in the PCMT1 gene (on their own or through their interaction with other genes) and T1D.
Methods
Subjects
For mutational scanning of the PCMT1 gene, a group of 26 patients with T1D, in whom the disease was linked to the region of interest (IDDM5), and 10 non-affected subjects were selected from a cohort of 147 Danish multiplex families with T1D [10]. In order to genotype the variants found in this initial screening, DNA from 253 families with type 1 diabetes (1097 individuals) was used; 155 sib-pair families and 98 simplex families, including the 147 families mentioned above [11]. The same individuals were genotyped for a functional variant in SUMO4 (rs237025) and for HLA. The former is located less than 1 cM from PCMT1, and has been proposed to be associated with T1D [12, 13], whereas HLA is the major genetic risk determinant for T1D [14].
All probands were less than 30 years of age at the time of diagnosis. All participants gave written informed consent for the performance of the study, which had been approved by the appropriate local Ethics Committee.
Association of PCMT1 with T1D was also assessed in 778 T1D patients and 749 controls obtained from a large Danish population-based case-control study [15].
PCMT1 sequencing and genotyping
For the family study, DNA was extracted from peripheral blood leukocytes using a standard salting-out method. PCMT1 sequencing primers were based on the reference sequence available from the National Center for Biotechnology Information (NCBI; Bethesda, MD, http://www.ncbi.nlm.nih.gov) and the University of California Santa Cruz Genome (http://www.genome.ucsc.edu) databases. Primers were designed to give PCR products covering the gene’s 8 exons (1800 base pairs) and 800 base pairs upstream of exon 1. Each PCR product (210-550 nucleotides) was amplified and sequenced on automated sequencing equipment (ABI 3100, Applied Biosystems, Foster City, CA, USA) using Big Dye terminator 3 chemistry (Applied Biosystems). Data were analyzed using SeqScape version 2.0, Applied Biosystems.
The variants identified in PCMT1 during initial sequencing were subsequently genotyped in the cohort of 253 Danish families, by PCR-based restriction fragment length polymorphism, using the enzymes Pvu II, BseM II, Hga I and Afl II for the polymorphisms in the 5’ region, exon 5 and exon 8, respectively.
In the case-control study, genotyping of rs4816 was performed on a Sequenome platform (RSKC2 core facility) on whole genome amplified DNA [16]. The following primers were used: first primer: ACG TTG GAT GTG GAT CGT CCT TCC TGA CAT, second primer: ACG TTG GAT GGT ACT GGA AAA GTC ATA GG, mass extension primer: CAC CGT AAA GAG CTA GTA GAT GAC TCA. PCR programmes followed instructions provided by Sequenom (Sequenom, San Diego, CA, USA).
For the family study, DNA was extracted from peripheral blood leukocytes using a standard salting-out method. PCMT1 sequencing primers were based on the reference sequence available from the National Center for Biotechnology Information (NCBI; Bethesda, MD, http://www.ncbi.nlm.nih.gov) and the University of California Santa Cruz Genome (http://www.genome.ucsc.edu) databases. Primers were designed to give PCR products covering the gene’s 8 exons (1800 base pairs) and 800 base pairs upstream of exon 1. Each PCR product (210-550 nucleotides) was amplified and sequenced on automated sequencing equipment (ABI 3100, Applied Biosystems, Foster City, CA, USA) using Big Dye terminator 3 chemistry (Applied Biosystems). Data were analyzed using SeqScape version 2.0, Applied Biosystems.
The variants identified in PCMT1 during initial sequencing were subsequently genotyped in the cohort of 253 Danish families, by PCR-based restriction fragment length polymorphism, using the enzymes Pvu II, BseM II, Hga I and Afl II for the polymorphisms in the 5’ region, exon 5 and exon 8, respectively.
In the case-control study, genotyping of rs4816 was performed on a Sequenome platform (RSKC2 core facility) on whole genome amplified DNA [16]. The following primers were used: first primer: ACG TTG GAT GTG GAT CGT CCT TCC TGA CAT, second primer: ACG TTG GAT GGT ACT GGA AAA GTC ATA GG, mass extension primer: CAC CGT AAA GAG CTA GTA GAT GAC TCA. PCR programmes followed instructions provided by Sequenom (Sequenom, San Diego, CA, USA).
SUMO4 and HLA genotyping
The functional variant in the SUMO4 gene (rs237025, 55Met/Val) previously shown to be associated with T1D [12] was genotyped using Tetra-primer ARMS-PCR [17]. Briefly, two primer pairs were used to amplify the two alleles of a given SNP in a single PCR reaction, yielding three different PCR products: a non-specific product, determined by the "outer primers" and two allele-specific products, determined by each specific "inner" primer and its "outer" counterpart. HLA-DRB1 genotype was determined by allele-specific PCR for the DR3 and DR4 alleles [10].
Genetic analysis
A combined Transmission Disequilibrium Test (TDT) sib-TDT was performed [18] to determine the association of the variants found in PCMT1 and T1D, as well as for the functional variant in SUMO4. Furthermore, data on PCMT1 was stratified according to the proband's SUMO4 genotype, as well as risk HLA genotype. DR3/DR4 was defined as high-risk HLA [14], whereas all other genotypes were classified as non-high risk HLA. Parent homozygocity TDT [19] was also performed to assess the interaction between SUMO4 and PCMT1. For this purpose, parents with SUMO4 genotype 55Val/Val, previously defined as high-risk for the development of T1D [12], were selected. In addition, the number of high-risk alleles was compared in affected and non-affected individuals.
In the case-control study, association of rs4816 to T1D was tested with allelic and genotypic tests, using a chi-squared test with 1 and 2 degrees of freedom, respectively.
Functional studies
Information available from both the National Center for Biotechnology Information (NCBI; Bethesda, MD, http://www.ncbi.nlm.nih.gov) and the University of California Santa Cruz Genome (http://www.genome.ucsc.edu) genomic databases and previously published data were reviewed to assess the potential functional consequences of the variations found in PCMT1.
In silico analysis of the 5' region in search of potential binding sites for transcription factors was performed using several open access, web-based programmes: Alibaba 2.1 (www.alibaba2.com), Genomatix MatInspector (www.genomatix.de/cgi-bin/ matinspector) and Genomatix PromoterInspector (www.genomatix.de/cgi-bin/promoterinspector).
In vitro promoter activity was assessed using 8 promoter constructs cloned upstream of the firefly luciferase (luc) reporter gene, comprising 1817-201 nucleotides upstream of the starting codon of PCMT1 in exon 1 (Figure 1). All constructs were verified by sequencing and restriction enzyme cleavage. INS-1e cells (passage numbers 65-89) were co-transfected with each reporter construct and renilla-luciferase as an internal control (pRL-TK vector, Promega, Madison, WI, USA, catalogue #E2241). Transfections were performed using a cationic lysosome-based method (SuperFect, Qiagen, Ballerup, Denmark), in the absence or presence of IL-1 (150pg/ml), IFNγ (5ng/ml) or both. Luciferase activity was measured using an Orion micro-plate luminometer (Berthold Technologies, Bad Wildbad, Germany). The ratio of firefly/renilla luciferase activity was calculated for each construct and treatment. Five experiments were performed, with each condition in duplicate. Each treatment and promoter construct was compared with the other ones using a two-sided ANOVA test and Tukey's post-hoc correction.
Figure 1.
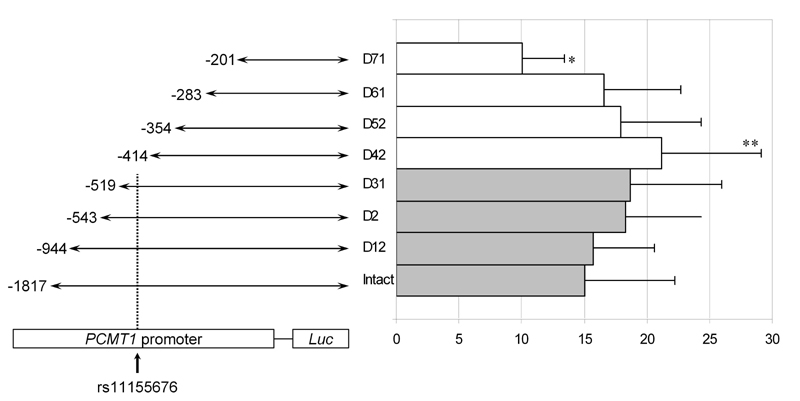
Promoter activity (firefly/renilla luciferase) in INS1e cells transfected with 8 different promoter constructs. * p < 0.05 (vs. rest of constructs except Intact and D12). ** p < 0.05 (vs. intact). The diagram to the left shows the length (out of scale) of the 8 promoter constructs used and the position of the polymorphism described.
Results
Genetic analysis
Four variants were identified through initial sequencing of the PCMT1 gene in the screening samples: one in position 509 upstream of the starting codon (-509 C>G, rs 11155676), one in exon 5 (358 G>A, rs4816) and two in exon 8, within the 3’UTR (845 A>G and 1072 A>T, rs7818 and rs4552, respectively). These four variants were in complete linkage disequilibrium, yielding 2 haplotypes only (Figure 2). After confirming the latter in 96 additional subjects, only one of the variants was genotyped in the rest of the family cohort (genotyping success 99.1%). In the 436 parents genotyped, 35.2% were homozygous for the wildtype haplotype, 15.5% were homozygous for the variant haplotype and 47.7% were heterozygous (expected distribution according to Hardy Weinberg Equilibrium (HWE): 36%, 16% and 48%, respectively, p = 0.98). The variant found in exon 5 (rs4816) leads to an aminoacid substitution (119 Val>Ile) associated with higher enzymatic repair activity [20]. The variant found in the region 5' of the gene changed a putative transcription factor binding site (from AP2 to MDGP).
Figure 2.
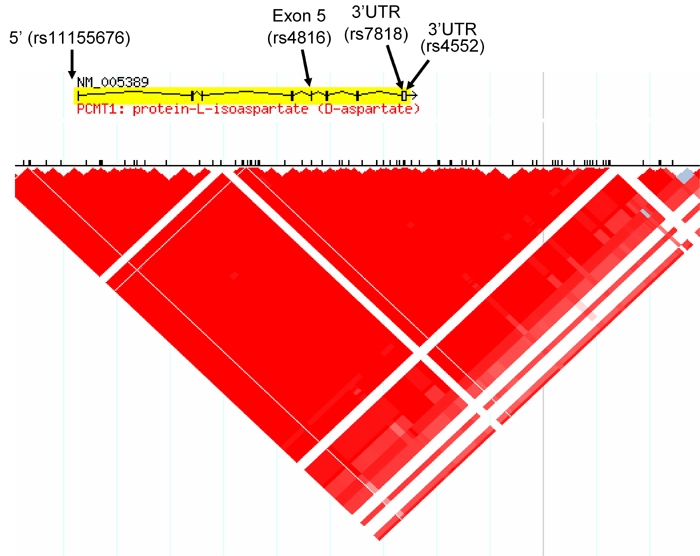
HapMap diagram showing the complete linkage disequilibrium block the PCMT1 gene is contained in. The arrows indicate the localization of the four polymorphisms found.
The genotyping success for the functional SUMO4 polymorphism was 97.3%. Of the 428 parents genotyped, 29.8% displayed genotype AA, 47.7% AG and 19.8% GG. (HWE: 30.4%, 49.5% and 20.1%, respectively, p = 0.93).
The variant PCMT1 haplotype was transmitted to 46% of the affected siblings in the 253 Danish families with T1D (chi-square 1.78, p = 0.18), and to 53% of the non-affected ones (p = 0.099 for transmission to affected vs. non-affected). The functional variant in SUMO4 was transmitted to 50.9% of the affected offspring (chi-square 0.088, p = 0.767) and to 50.5% of the non-affected offspring. When stratified according to the reported SUMO4 risk genotypes, there was a trend towards a decreased transmission of the variant PCMT1 haplotype to the affected offspring, when compared to the non-affected offspring in the families where the proband had 163 GG or AG genotype (high-risk, dominant model) (p = 0.069) (Table 1). Parent homozygocity TDT and sib-TDT did not add significant information (data not shown). The total number of high-risk PCMT1 and SUMO4 alleles was similar in affected and unaffected subjects. Regarding HLA stratification, there was a trend towards an uneven transmission of the PCMT1 variant in the non-high risk group only (Table 1).
Table 1. Transmission of the PCMT1 variant haplotype to affected and non-affected offspring, stratified according to SUMO4 and HLA risk genotypes.
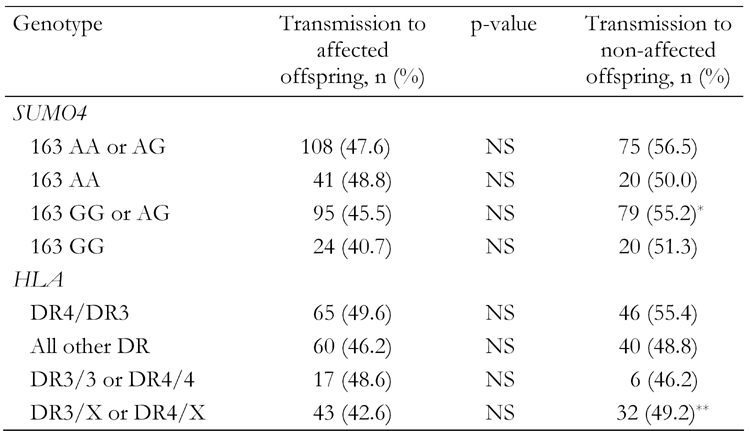
* p = 0.069. ** p = 0.086 (vs. transmission to affected offspring). NS: not signifi-cant.
In the case-control study, rs4816 was also in Hardy-Weinberg equilibrium (using the exact test, p > 0.001). The minor allele (A), associated with higher PIMT activity [20], was present in 40.36% of the affected and in 36.98% of the unaffected subjects (OR 1.15, p = 0.055). For genotype analysis, the dominant model showed a significantly higher frequency of carriers of the A allele among subjects with T1D (490/778 vs. 431/749, p = 0.03).
Promoter activity
Exposure to cytokines significantly increased promoter activity for all the constructs tested (firefly/renilla luciferase ratio 11.6 ± 7.0 for unstimulated vs. 18.6 ± 5.6 for IL1 vs. 16.4 ± 5.3 for IFNγ and 20.1 ± 6.8 for the cytokine mix, p ≤ 0.003 for all cytokines vs. no treatment). The shorter construct showed significantly lower promoter activity (10.0 ± 3.4, p ≤ 0.05) than the rest (except for the 2 longest, see Figure 1). However, there was no difference between the constructs adjacent to the previously described variant, namely constructs D31 and 42, see Figure 1 (18.7 ± 7.3 vs. 21.2 ± 8.0, p = 0.92).
Discussion
The genetic studies performed in the Danish type 1 diabetes family cohort, showed suggestive, but not conclusive, trends of PCMT1 haplotypes' involvement in modulating diabetes susceptibility in global and stratified analysis. The low-risk PCMT1 haplotype is associated with higher enzymatic activity and tends to be preferentially transmitted to the unaffected offspring, especially in certain risk groups defined by other genes (HLA and SUMO4). However, the non-stratified results of the case-control study pointed in the opposite direction, which weakens our initial conclusion.
The class II MHC plays a principal role in antigen presentation to CD4 cells. Inherited differences in HLA molecules between individuals can determine to which antigens and, in particular, which epitopes an individual can respond. Thus, given a certain HLA genotype, the role of other players in the development of the disease may differ. Indeed, specific HLA genotypes are the most critical determinants of genetic risk to develop T1D; it is estimated that they account for 40-50% of the genetic risk [14]. Posttranslational modifications can also change a protein's binding affinity for class II MHC molecules [1]. Furthermore, protein deamidation/isomerisation can modify cleavage sites for the enzyme asparaginase endopeptidase, potentially having important consequences for protein processing and antigen presentation by class II MHC molecules [3, 21]. In the present study, PCMT1 appeared to play a more important role in subjects with any HLA genotype except DR3/DR4, i.e. those with lower-risk DR genotypes. On the other hand, SUMO4, located within IDDM5, less than 1 cM away from PCMT1, encodes a protein which binds to IκBα inhibiting NFκB transcriptional activity, and may play a central role in apoptosis. Thus, the genetic interaction between PCMT1 and both HLA and SUMO4 is biologically plausible. Although the functional variant of SUMO4 has previously been shown to be associated with T1D [12, 13], neither the present, nor other larger, studies have been able to replicate these findings in Caucasian populations [22]. Nevertheless, as with other minor genes involved in the pathogenesis of T1D, its effect might not be significant on its own, but only through interaction with other genes, as suggested by a recent Swedish study. This work showed a positive association between the functional SUMO4 variant and T1D in high-risk HLA DR3/DR4 subjects [23].
Our finding that PCMT1 promoter activity is enhanced in INS-1E cells, in response to cytokines, in an in vitro model of the disease, argues in favor of a role for PIMT in the pathogenesis of T1D. However, although in silico analysis of the polymorphism described in the PCMT1 promoter region (-509 C>G) revealed a change in a putative transcription factor binding site, no difference was seen in promoter activity between the constructs adjacent to this position. This suggested that eliminating this binding does not have a strong effect on the expression of PCMT1. However the effect of creating a new binding site was not addressed by the reporter gene studies.
Some of the proteins recently described as substrates for PIMT in the brain [24, 25] change their expression in pancreas over time in diabetes-prone, but not in diabetes-resistant BB rats [26]. Furthermore, in a cellular model of beta-cell maturation, proteomic studies suggested that posttranslational protein modifications may reflect the acquired sensitivity of the beta cell to cytokines [27]. In the same model, the exposure to cytokines led to a 1.5-fold decrease in Pcmt1 expression in mature, but not in immature pre-beta-cells [28], suggesting that PIMT response may be part of the specific phenotype making the beta-cell prone to immune-mediated destruction. However, the present study does not support such a decrease, indeed in vitro PCMT1 promoter activity increased rather than decreased in response to cytokines. Interestingly, PCMT1 contains several putative targets for miRNAs (miR), which may contribute to the transcriptional regulation of this gene. Of the two polymorphisms identified in exon 8, rs7818 is included within known miRNA targets (for has-miR-361, has-miR-627 and has-miR 589, miRanda miRNA targets, www.t1dbase.org, last accessed on the 23rd July 2008). This suggests that the functional consequences of the 2 different haplotypes described may not only depend on the polymorphism found in exon 5, but also on the regulation of translation mediated by miRNAs.
In conclusion, the present study adds to existing data suggesting a role of PCMT1 in the pathogenesis of T1D. To date, the following observations support such a role: PIMT is highly expressed in human pancreas, the induction of PCMT1 delays and reduces diabetes severity in diabetes-prone BB rats, changes in PIMT substrate expression precede the development of diabetes in these animals, PCMT1 promoter activity is enhanced by in vitro exposure of an insulin-producing cell-line to cytokines and, finally, the low-risk PCMT1 haplotype tends to be preferentially transmitted to non-affected offspring, especially in certain risk groups.
The lack of statistical significance might be due to the size of the population analyzed, which is insufficient to detect a small increase in risk. In addition, the case-control study showed an opposite trend. Power calculations revealed that the present case-control sample had a power of 43% to detect the differences found, with an alpha of 0.05, and that a total of 1898 cases would be needed to reach a power of 80%. Due to the reduced statistical power in the present study, definite conclusions cannot be made. Although a larger sample is needed to confirm the detected association between PCMT1 and type 1 diabetes, the findings of our present study, together with our previous findings [7], are the first to suggest a possible role of this gene in the development of type 1 diabetes.
Acknowledgments
We are very grateful to Bodil Bosmann Jørgensen for her excellent technical assistance. The present study was partially funded by a grant from Sehested Hansen's Fund.
References
- 1.Doyle HA, Mamula MJ. Posttranslational modifications of self-antigens. Ann N Y Acad Sci. 2005;1050:1–9. doi: 10.1196/annals.1313.001. [DOI] [PubMed] [Google Scholar]
- 2.Young AL, Carter WG, Doyle HA, Mamula MJ, Aswad DW. Structural integrity of histone H2B in vivo requires the activity of protein L-isoaspartate O-methyltransferase, a putative protein repair enzyme. J Biol Chem. 2001;276(40):37161–37165. doi: 10.1074/jbc.M106682200. [DOI] [PubMed] [Google Scholar]
- 3.Cloos PA, Christgau S. Posttranslational modifications of proteins: implications for aging, antigen recognition, and autoimmunity. Biogerontology. 2004;5(3):139–158. doi: 10.1023/B:BGEN.0000031152.31352.8b. [DOI] [PubMed] [Google Scholar]
- 4.Mamula MJ, Gee RJ, Elliott JI, Sette A, Southwood S, Jones PJ, Blier PR. Isoaspartyl post-translational modification triggers autoimmune responses to self-proteins. J Biol Chem. 1999;274(32):22321–22327. doi: 10.1074/jbc.274.32.22321. [DOI] [PubMed] [Google Scholar]
- 5.Yang ML, Doyle HA, Gee RJ, Lowenson JD, Clarke S, Lawson BR, Aswad DW, Mamula MJ. Intracellular protein modification associated with altered T cell functions in autoimmunity. J Immunol. 2006;177(7):4541–4549. doi: 10.4049/jimmunol.177.7.4541. [DOI] [PubMed] [Google Scholar]
- 6.Nerup J, Mandrup-Poulsen T, Helqvist S, Andersen HU, Pociot F, Reimers JI, Cuartero BG, Karlsen AE, Bjerre U, Lorenzen T. On the pathogenesis of IDDM. Diabetologia. 1994;37(Suppl 2):S82–S89. doi: 10.1007/BF00400830. [DOI] [PubMed] [Google Scholar]
- 7.Wagner AM, Cloos P, Bergholdt R, Boissy P, Andersen TL, Henriksen DB, Christiansen C, Christgau S, Pociot F, Nerup J. Post-translational protein modifications in type 1 diabetes: a role for the repair enzyme protein-L-isoaspartate (D-aspartate) O-methyltransferase? Diabetologia. 2007;50(3):676–681. doi: 10.1007/s00125-006-0556-1. [DOI] [PubMed] [Google Scholar]
- 8.Doyle HA, Gee RL, Mamula MJ. A failure to repair self-proteins leads to T cell hyperproliferation and autoantibody production. J Immunol. 2003;171(6):2840–2847. doi: 10.4049/jimmunol.171.6.2840. [DOI] [PubMed] [Google Scholar]
- 9.Cox NJ, Wapelhorst B, Morrison VA, Johnson L, Pinchuk L, Spielman RS, Todd JA, Concannon P. Seven regions of the genome show evidence of linkage to type 1 diabetes in a consensus analysis of 767 multiplex families. Am J Hum Genet. 2001;69(4):820–830. doi: 10.1086/323501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nerup J, Pociot F. A genomewide scan for type 1-diabetes susceptibility in Scandinavian families: identification of new loci with evidence of interactions. Am J Hum Genet. 2001;69(6):1301–1313. doi: 10.1086/324341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bergholdt R, Taxvig C, Eising S, Nerup J, Pociot F. CBLB variants in type 1 diabetes and their genetic interaction with CTLA4. J Leukoc Biol. 2005;77(4):579–585. doi: 10.1189/jlb.0904524. [DOI] [PubMed] [Google Scholar]
- 12.Guo D, Li M, Zhang Y, Yang P, Eckenrode S, Hopkins D, Zheng W, Purohit S, Podolsky RH, Muir A et al. A functional variant of SUMO4, a new I kappa B alpha modifier, is associated with type 1 diabetes. Nat Genet. 2004;36(8):837–841. doi: 10.1038/ng1391. [DOI] [PubMed] [Google Scholar]
- 13.Owerbach D, Pina L, Gabbay KH. A 212-kb region on chromosome 6q25 containing the TAB2 gene is associated with susceptibility to type 1 diabetes. Diabetes. 2004;53(7):1890–1893. doi: 10.2337/diabetes.53.7.1890. [DOI] [PubMed] [Google Scholar]
- 14.Pociot F, McDermott MF. Genetics of type 1 diabetes mellitus. Genes Immun. 2002;3(5):235–249. doi: 10.1038/sj.gene.6363875. [DOI] [PubMed] [Google Scholar]
- 15.Eising S, Svensson J, Skogstrand K, Nilsson A, Lynch K, Andersen PS, Lernmark A, Hougaard DM, Pociot F, Norgaard-Pedersen B et al. Type 1 diabetes risk analysis on dried blood spot samples from population-based newborns: design and feasibility of an unselected case-control study. Paediatr Perinat Epidemiol. 2007;21(6):507–517. doi: 10.1111/j.1365-3016.2007.00846.x. [DOI] [PubMed] [Google Scholar]
- 16.Brorsson C, Bergholdt R, Sjogren M, Eising S, Sorensen KM, Hougaard DM, Orho-Melander M, Groop L, Pociot F. A non-synonymous variant in SLC30A8 is not associated with type 1 diabetes in the Danish population. Mol Genet Metab. 2008;94(3):386–388. doi: 10.1016/j.ymgme.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 17.Ye S, Dhillon S, Ke X, Collins AR, Day IN. An efficient procedure for genotyping single nucleotide polymorphisms. Nucleic Acids Res. 2001;29(17):E88. doi: 10.1093/nar/29.17.e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spielman RS, Ewens WJ. A sibship test for linkage in the presence of association: the sib transmission/disequilibrium test. Am J Hum Genet. 1998;62(2):450–458. doi: 10.1086/301714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lie BA, Todd JA, Pociot F, Nerup J, Akselsen HE, Joner G, Dahl-Jorgensen K, Ronningen KS, Thorsby E, Undlien DE. The predisposition to type 1 diabetes linked to the human leukocyte antigen complex includes at least one non-class II gene. Am J Hum Genet. 1999;64(3):793–800. doi: 10.1086/302283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeVry CG, Clarke S. Polymorphic forms of the protein L-isoaspartate (D-aspartate) O-methyltransferase involved in the repair of age-damaged proteins. J Hum Genet. 1999;44(5):275–288. doi: 10.1007/s100380050161. [DOI] [PubMed] [Google Scholar]
- 21.Moss CX, Matthews SP, Lamont DJ, Watts C. Asparagine deamidation perturbs antigen presentation on class II major histocompatibility complex molecules. J Biol Chem. 2005;280(18):18498–18503. doi: 10.1074/jbc.M501241200. [DOI] [PubMed] [Google Scholar]
- 22.Smyth DJ, Howson JM, Lowe CE, Walker NM, Lam AC, Nutland S, Hutchings J, Tuomilehto-Wolf E, Tuomilehto J, Guja C et al. Assessing the validity of the association between the SUMO4 M55V variant and risk of type 1 diabetes. Nat Genet. 2005;37(2):110–111. doi: 10.1038/ng0205-110. [DOI] [PubMed] [Google Scholar]
- 23.Sedimbi SK, Luo XR, Sanjeevi CB, Lernmark A, Landin-Olsson M, Arnqvist H, Bjorck E, Nystrom L, Ohlson LO, Schersten B et al. SUMO4 M55V polymorphism affects susceptibility to type I diabetes in HLA DR3- and DR4-positive Swedish patients. Genes Immun. 2007;8(6):518–521. doi: 10.1038/sj.gene.6364406. [DOI] [PubMed] [Google Scholar]
- 24.Vigneswara V, Lowenson JD, Powell CD, Thakur M, Bailey K, Clarke S, Ray DE, Carter WG. Proteomic identification of novel substrates of a protein isoaspartyl methyltransferase repair enzyme. J Biol Chem. 2006;281(43):32619–32629. doi: 10.1074/jbc.M605421200. [DOI] [PubMed] [Google Scholar]
- 25.Zhu JX, Doyle HA, Mamula MJ, Aswad DW. Protein repair in the brain, proteomic analysis of endogenous substrates for protein L-isoaspartyl methyltransferase in mouse brain. J Biol Chem. 2006;281(44):33802–33813. doi: 10.1074/jbc.M606958200. [DOI] [PubMed] [Google Scholar]
- 26.Sparre T, Christensen UB, Gotfredsen CF, Larsen PM, Fey SJ, Hjerno K, Roepstorff P, Pociot F, Karlsen AE, Nerup J. Changes in expression of IL-1 beta influenced proteins in transplanted islets during development of diabetes in diabetes-prone BB rats. Diabetologia. 2004;47(5):892–908. doi: 10.1007/s00125-004-1382-y. [DOI] [PubMed] [Google Scholar]
- 27.Nielsen K, Sparre TM, Larsen R, Nielsen M, Fey SJ, Larsen PM, Roepstorff P, Nerup P, Karlsen AE. Protein expression changes in a cell system of beta-cell maturation reflect an acquired sensitivity to IL-1beta. Diabetologia. 2004;47:62–74. doi: 10.1007/s00125-003-1277-3. [DOI] [PubMed] [Google Scholar]
- 28.Nielsen K, Kruhoffer M, Orntoft T, Sparre T, Wang H, Wollheim C, Jorgensen MC, Nerup J, Karlsen AE. Gene expression profiles during beta cell maturation and after IL-1beta exposure reveal important roles of Pdx-1 and Nkx6.1 for IL-1beta sensitivity. Diabetologia. 2004;47(12):2185–2199. doi: 10.1007/s00125-004-1578-1. [DOI] [PubMed] [Google Scholar]