
Table 1. Data-collection and structure-refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | ||
| Space group | P322 | P1 |
| Unit-cell parameters (Å, °) | a = 66.03, b = 66.03, c = 46.72, α = β = 90, γ = 120 | a = 30.17, b = 41.32, c = 41.29, α = 80.05, β = 68.63, γ = 68.52 |
| Molecules per ASU | 1 | 2 |
| Resolution range (Å) | 20–2.20 | 20–1.75 (1.81–1.75) |
| Completeness (%) | 97.9 (94.9) | 91.5 (89.3) |
| I/σ(I) | 10.4 (1.6) | 9.3 (3.3) |
| Rmerge† (%) | 4.65 (28.11) | 4.92 (14.81) |
| Structure refinement | ||
| Resolution (Å) | 20–1.75 (1.81–1.75) | |
| Rcryst/Rfree‡ (%) | 20.6 (33.0)/23.9 (34.0) | |
| No. of protein atoms | 1241 | |
| No. of waters | 203 | |
| R.m.s.d. from ideal values | ||
| Bond lengths (Å) | 0.015 | |
| Bond angles (°) | 1.545 | |
| Average B factor (Å2) | 27.3 | |
| Ramachandran plot (%) | ||
| Most favoured regions (%) | 93.6 | |
| Allowed regions (%) | 6.4 |
†
R
merge = 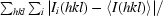
 , where I
i(hkl) is the intensity of the measured reflection and 〈I(hkl)〉 is the mean intensity of all symmetry-related reflections.
, where I
i(hkl) is the intensity of the measured reflection and 〈I(hkl)〉 is the mean intensity of all symmetry-related reflections.
‡
R
cryst = 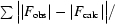
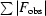 , where F
obs and F
calc are observed and calculated structure factors. R
free =
, where F
obs and F
calc are observed and calculated structure factors. R
free = 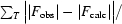
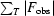 , where T is a test data set of about 10% of the total reflections that were randomly chosen and set aside prior to refinement.
, where T is a test data set of about 10% of the total reflections that were randomly chosen and set aside prior to refinement.