

Recombinant ATP-dependent DNA ligase from the archaeon Thermococcus sp. 1519 was overexpressed, purified and used to produce crystals that diffracted to 3.09 Å resolution.
Keywords: DNA ligases, archaea
Abstract
DNA ligases catalyze the sealing of 5′-phosphate and 3′-hydroxyl termini at single-strand breaks in double-stranded DNA and their function is essential to maintain the integrity of the genome in DNA metabolism. An ATP-dependent DNA ligase from the archaeon Thermococcus sp. 1519 was overexpressed, purified and crystallized. Crystals were obtained using the hanging-drop vapour-diffusion method employing 35%(v/v) Tacsimate pH 7.0 as a precipitant and diffracted X-rays to 3.09 Å resolution. They belonged to space group P41212, with unit-cell parameters a = b = 79.7, c = 182.6 Å.
1. Introduction
DNA ligases catalyze the sealing of 5′-phosphate and 3′-hydroxyl termini at single-strand breaks in double-stranded DNA or at two fragments containing either complementary single-strand or blunt ends. These roles are essential in DNA replication, recombination and repair (Lehman, 1974 ▶). DNA ligases are universally present in eukarya, bacteria and archaea and may be classified into two families according to their high-energy cofactor requirements for either ATP or NAD+: ATP-dependent DNA ligases (EC 6.5.1.1) and NAD+-dependent DNA ligases (EC 6.5.1.2). ATP-dependent ligases are present in eukaryotic viruses, bacteriophages, eukarya, archaea and bacteria, whereas NAD+-dependent ligases have exclusively been found in bacteria. ATP-dependent and NAD+-dependent DNA ligases catalyze reactions via three sequential nucleotidyl-transfer reactions: covalent addition of AMP from ATP or NAD+ (step 1), AMP transfer from the enzyme to the 5′-phosphoryl group of the nick on the DNA (step 2) and phosphodiester-bond formation with AMP release (step 3) (Pascal, 2008 ▶; Pascal et al., 2006 ▶).
Little is known about DNA ligases from archaea, which represent the third domain of life. However, thermostable DNA ligases from several hyperthermophilic archaea have been isolated and functionally characterized (see, for example, Sriskanda et al., 2000 ▶; Lai et al., 2002 ▶; Keppetipola & Shuman, 2005 ▶). All of them utilized ATP as a cofactor and it was presumed that archaeal ligases would exclusively utilize ATP as their nucleotide substrate. However, this notion was challenged by studies of DNA ligases from the hyperthermophilic archaeons Thermococcus kodakarensis KOD1 and T. fumicolans, which used either ATP or NAD+ as a cofactor (Nakatani et al., 2000 ▶; Rolland et al., 2004 ▶).
Archaeal DNA ligases are composed of three structural domains: a DNA-binding domain (DBD), a nucleotidyltransferase domain (NTase) and an oligonucleotide-binding domain (OB-domain). The NTase and OB-domain form the catalytic core. The OB-domain extends from the C-terminus of the NTase and assists in the formation of the ligase–AMP intermediate (Pascal, 2008 ▶; Pascal et al., 2004 ▶). The DBD, which is an N-terminal extension of the NTase, forces the binding of the substrate by supporting the distorted shape of the DNA. The three domains establish a ligase protein clamp that fully encircles the DNA substrate. The DBD acts cooperatively with the other domains and supports the active conformation of the catalytic core by physical interaction with the NTase and OB-domain (Pascal, 2008 ▶).
Crystal structures of archaeal DNA ligases from the euryarchaeon Pyrococcus furiosus (PfuLig) at 1.8 Å resolution (PDB code 2cfm; Nishida et al., 2006 ▶) and from the crenarchaeon Sulfolobus solfataricus (SsLig) at 2.1 Å resolution (PDB code 2hiv; Pascal et al., 2006 ▶) have been reported. PfuLig consists of 561 amino acids packed into three domains: DBD (residues 1–220), NTase (residues 221–423 residues) and OB-domain (residues 424–561) (Nishida et al., 2006 ▶). SsLig consists of 601 residues and displays a sequence identity with PfuLig of 38% (Pascal et al., 2006 ▶). In the absence of nicked DNA, SsLig has an open extended conformation (Nishida et al., 2006 ▶). The structure of substrate-free PfuLig revealed that the catalytic core domains are connected by a C-terminal extension helix and adopt a closed conformation corresponding to the OB-domain orientation observed upon formation of the ligase–AMP complex (Pascal et al., 2006 ▶). The mutual orientations of DBD and NTase in the structures of PfuLig and SsLig are almost identical.
Here, we report the overexpression, purification, crystallization and preliminary crystallographic analysis of a DNA ligase from the hyperthermophilic euryarchaeon Thermococcus sp. 1519, in which the OB-domain is supposed to adopt a new orientation. The Thermococcus sp. 1519 DNA ligase (LigTh1519), which has previously been purified and characterized (Smagin et al., 2008 ▶), is a monomeric enzyme of 559 residues with a molecular mass of 64.7 kDa. It shares a high level of amino-acid sequence identity with the DNA ligases from T. kodakaraensis (91%) and T. fumicolans (89%), suggesting that it has similar cofactor requirements and multi-domain architecture to these enzymes. The sequence identity of LigTh1519 with PfuLig and SsLig is 80% and 38%, respectively. LigTh1519 can use ATP as a cofactor and displays temperature and pH optima for activity at 338 K and 9.0, respectively (Smagin et al., 2008 ▶).
2. Expression and purification
The LigTh1519 gene (GenBank FJ713107) was cloned from Thermococcus sp. 1519 genomic DNA and inserted into the expression vector pQE30 (Qiagen; Smagin et al., 2008 ▶). The encoded recombinant protein contained an N-terminal MRGSHHHHHHGS tag followed by the amino-acid sequence of the native LigTh1519. The recombinant vector, pQELigTh1519, was introduced into Escherichia coli DLT1270/pRARE-2 strain (Smagin et al., 2008 ▶). The transformants were grown at 310 K in Luria–Bertani medium containing 100 µg ml−1 ampicillin and 20 µg ml−1 chloramphenicol to an optical density of 0.5 at 600 nm. Isopropyl β-d-1-thiogalactopyranoside was added to a final concentration of 1 mM for induction of gene expression and cultivation was continued for an additional 15 h.
The harvested cells were incubated in buffer A [50 mM Tris–HCl pH 8.0, 250 mM NaCl, 200 µg ml−1 lysozyme, 1 mM phenylmethanesulfonyl fluoride (PMSF), 5%(v/v) glycerol] for 30 min and then disrupted by sonication (4 × 30 s on ice). The cell debris was removed by centrifugation at 12 000g for 20 min. The supernatant was subjected to heat-treatment at 338 K for 15 min followed by centrifugation (12 000g for 20 min) to remove precipitate. The supernatant was incubated in the presence of 0.15%(v/v) polyethyleneimine for 15 min at 277 K and the nucleic acids were removed by centrifugation (12 000g for 20 min). The resulting nucleotide-free soluble fraction was applied onto an affinity column (Ni-Sepharose FF, 5 ml; Amersham Pharmacia Biotech) equilibrated with buffer B (50 mM Tris–HCl pH 8.0, 500 mM NaCl, 1 mM PMSF, 20 mM imidazole). After washing with buffer B, the bound protein was eluted with a linear imidazole gradient from 100 to 500 mM in buffer B. Fractions containing LigTh1519, which eluted at 300 mM imidazole, were dialyzed overnight against buffer C [50 mM Tris–HCl pH 8.0, 50 mM NaCl, 0.5 mM dithiothreitol (DTT), 5 mM EDTA, 5%(v/v) glycerol] and then applied onto an anion-exchange column equilibrated with buffer B (Mono Beads Q, 10 ml; Amersham Pharmacia Biotech). Elution was with a linear NaCl gradient from 0 to 500 mM in buffer B. Fractions containing LigTh1519 were concentrated by ultrafiltration and subjected to gel filtration (Superdex 200 10/300 column, Amersham Pharmacia Biotech) employing buffer D (50 mM Tris–HCl pH 8.0, 100 mM NaCl, 0.5 mM DTT). The purified recombinant LigTh1519 was used for crystallization. The homogeneity of the purified LigTh1519 was judged using 12%(w/v) SDS–PAGE (Fig. 1 ▶). The protein concentration was determined by the Bradford method using bovine serum albumin as a standard.
Figure 1.
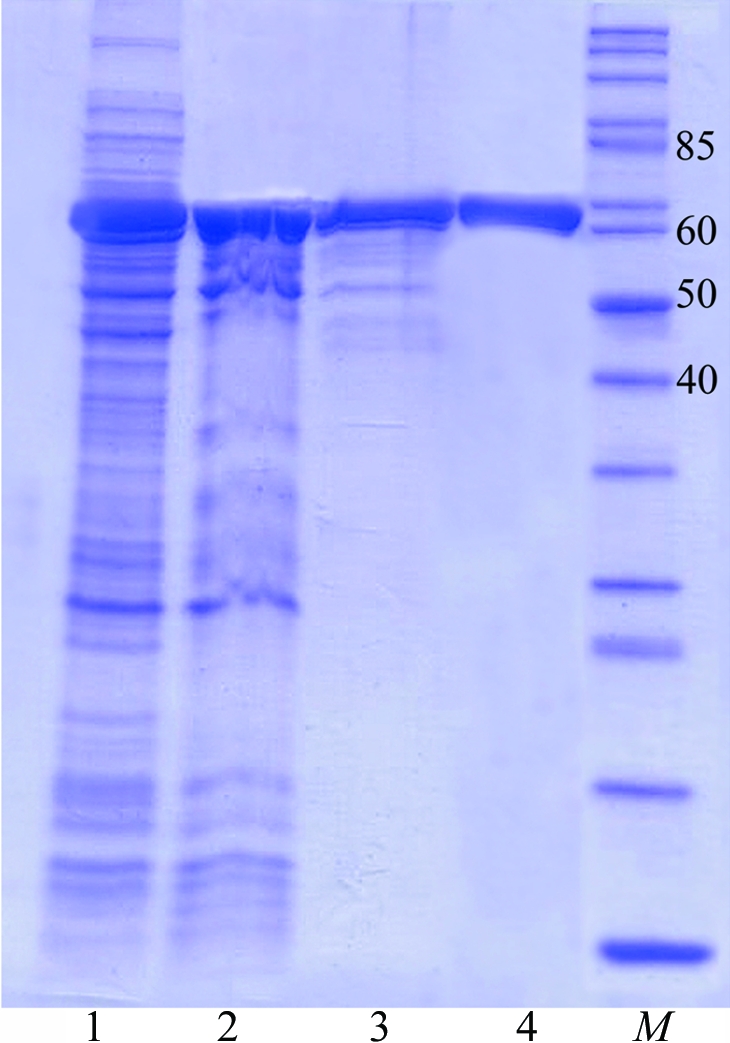
SDS–PAGE analysis of LigTh1519 during purification. Proteins were analysed on 12%(w/v) SDS–PAGE and were stained with Coomassie Blue. Lane 1, supernatant after removal of cell debris. Lane 2, supernatant after heat-treatment at 338 K. Lane 3, LigTh1519 after Ni-Sepharose FF column chromatography. Lane 4, purified LigTh1519 after gel filtration. Lane M, molecular-weight markers (kDa).
3. Crystallization
For crystallization, freshly prepared recombinant LigTh1519 (12.6 mg ml−1) in 50 mM Tris–HCl pH 8.0 containing 100 mM NaCl and 0.5 mM DTT was used. Initial screening was performed at 291 K using the hanging-drop vapour-diffusion method and employing Crystal Screen and Index Screen crystallization solutions (Hampton Research). Crystals of LigTh1519 were obtained by mixing 1 µl protein solution with 1 µl reservoir solution [35%(v/v) Tacsimate pH 7.0; Crystal Screen condition No. 28]. Single crystals with a truncated square bipyramidal form with dimensions of about 200–300 µm appeared after 3–4 months (Fig. 2 ▶).
Figure 2.

Crystals of recombinant LigTh1519.
4. X-ray analysis
Preliminary X-ray diffraction measurements were made at the Kurchatov Institute synchrotron (Moscow, Russia). A complete data set was collected to 3.09 Å resolution (Fig. 3 ▶) on beamline X13 of the Deutsches Electronen Synchrotron (EMBL/DESY, Hamburg, Germany) using a MAR CCD 165 detector (MAR Research, Germany). The crystals were directly flash-cooled in a stream of cold nitrogen gas at 100 K using an Oxford Cryosystems cooling device (Oxford Cryosystems Ltd, England). There was no need for prior crystal transfer to a cryoprotectant solution. The crystals belonged to space group P41212, with unit-cell parameters a = b = 79.7, c = 182.6 Å. For data collection, the automated collection of data (DNA) software package (http://www.dna.ac.uk) was used.
Figure 3.

A typical 0.45° oscillation image obtained during data collection from LigTh1519 crystals. The edge of the oscillation image corresponds to 3.09 Å resolution.
X-ray diffraction images were indexed, integrated and subsequently scaled using the program XDS (Kabsch, 1993 ▶). The CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶) was used for data reduction. Crystal and data-collection statistics are summarized in Table 1 ▶.
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| Space group | P41212 |
| Radiation source | X13, EMBL/DESY, Hamburg, Germany |
| Unit-cell parameters (Å) | a = b = 79.7, c = 182.6 |
| Temperature (K) | 100 |
| Wavelength (Å) | 0.803 |
| Oscillation range (°) | 0.45 |
| Mosaicity (°) | 0.41 |
| No. of frames | 167 |
| Resolution limit (Å) | 19.89–3.08 (3.10–3.08) |
| Total reflections | 64365 (472) |
| Molecules per ASU | 1 |
| Solvent content (%) | 45 |
| Matthews coefficient (Å3 Da−1) | 2.24 |
| Independent reflections | 11403 (191) |
| Average redundancy | 5.6 (2.5) |
| Average I/σ(I) | 24.4 (3.1) |
| Completeness (%) | 98 (94.0) |
| Rmerge† | 0.05 (0.35) |
R
merge = 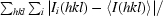
 , where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the average intensity of reflection hkl.
, where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the average intensity of reflection hkl.
The structure of LigTh1519 was solved by the molecular-replacement method (MOLREP; Vagin & Teplyakov, 1997 ▶), using the two domains (DBD and NTase) of PfuLig and SsLig as models. For the model based on the structure of PfuLig, the correct solution with an R factor of 0.578 and a correlation coefficient of 0.362 at a resolution of 4 Å corresponds to the first peak of the rotation function and the first peak of the translation function. Several refinement cycles of crystallographic refinement at 3.09 Å resolution using the program REFMAC (Vagin et al., 2004 ▶) decreased the R factor to 0.40. The solution based on the SsLig model gives similar results (R factor = 0.42 at 3.09 Å resolution). However, the structure of LigTh1519 could not be solved using the complete three-domain structures of these DNA ligases as models. This could be explained by a different orientation of the OB-domain in the PfuLig or SsLig full-length molecular models and in LigTh1519. In the crystal structure of SsLig the two catalytic core domains are in an open conformation (Pascal et al., 2006 ▶). In the structure of PfuLig the NTase and OB-domain adopt a closed conformation (Pascal, 2008 ▶; Nishida et al., 2006 ▶). It is therefore suggested that the OB-domain in LigTh1519 adopts a different or an intermediate orientation between the open and closed conformations. The low resolution of the LigTh1519 crystals does not permit reliable definition of the orientation of the OB-domain. We are also attempting to crystallize recombinant LigTh1519 with ATP and Mg2+. Refinement of the structure is currently in progress and will be published elsewhere.
Acknowledgments
This work was supported by the Federal Agency for Science and Innovations of Russia (contract 02.512.12.2001). The study of NVR was supported by the grant from the President of the Russian Federation (project No. MD-971.2007.4).
References
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Kabsch, W. (1993). J. Appl. Cryst.26, 795–800.
- Keppetipola, N. & Shuman, S. (2005). J. Bacteriol.187, 6902–6908. [DOI] [PMC free article] [PubMed]
- Lai, X., Shao, H., Hao, F. & Huang, L. (2002). Extremophiles, 6, 469–477. [DOI] [PubMed]
- Lehman, I. R. (1974). Science, 186, 790–797. [DOI] [PubMed]
- Nakatani, M., Ezaki, S., Atomi, H. & Imanaka, T. (2000). J. Bacteriol.182, 6424–6433. [DOI] [PMC free article] [PubMed]
- Nishida, H., Kiyonari, S., Ishino, Y. & Morikawa, K. (2006). J. Mol. Biol.360, 956–967. [DOI] [PubMed]
- Pascal, J. M. (2008). Curr. Opin. Struct. Biol.18, 96–105. [DOI] [PubMed]
- Pascal, J. M., O’Brien, P. J., Tomkinson, A. E. & Ellenberger, T. (2004). Nature (London), 432, 473–478. [DOI] [PubMed]
- Pascal, J. M., Tsodikov, O. V., Hura, G. L., Song, W., Cotner, E. A., Classen, S., Tomkinson, A. E., Tainer, J. A. & Ellenberger, T. (2006). Mol. Cell, 24, 279–291. [DOI] [PubMed]
- Rolland, J., Gueguen, Y., Persillon, C., Masson, J. & Dietrich, J. (2004). FEMS Microbiol. Lett.236, 267–273. [DOI] [PubMed]
- Smagin, V. A., Mardanov, A. V., Bonch-Osmolovkaya, E. A. & Ravin, N. V. (2008). Prikl. Biokhim. Mikrobiol.44, 523–528. [PubMed]
- Sriskanda, V., Kelman, Z., Hurwitz, J. & Shuman, S. (2000). Nucleic Acids Res.28, 2221–2228. [DOI] [PMC free article] [PubMed]
- Vagin, A. A., Steiner, R. A., Lebedev, A. A., Potterton, L., McNicholas, S., Long, F. & Murshudov, G. N. (2004). Acta Cryst. D60, 2184–2195. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.