

Abstract
Cyclin dependent kinase 1 (Cdk1)/cyclin B1 complex is the driving force for mitotic entry, and its activation is tightly regulated by the G2/M checkpoint. We originally reported that a novel protein C53 (also known as Cdk5rap3 and LZAP) potentiates DNA damage-induced cell death by modulating the G2/M checkpoint (1). More recently, Wang et al (2007) found that C53/LZAP may function as a tumor suppressor via inhibiting NF-κB signaling (2). We report here identification of C53 protein as a novel regulator of Cdk1 activation. We found that knockdown of C53 protein causes delayed Cdk1 activation and mitotic entry. During DNA damage response, activation of checkpoint kinase 1 and 2 (Chk1 and Chk2) is partially inhibited by C53 overexrepsssion. Intriguingly, we found that C53 interacts with checkpoint kinase 1 (Chk1) and antagonizes its function. Moreover, a portion of C53 protein is localized at the centrosome, and centrosome-targeting C53 potently promotes local Cdk1 activation. Taken together, our results strongly suggest that C53 is a novel negative regulator of checkpoint response. By counteracting Chk1, C53 promotes Cdk1 activation and mitotic entry in both unperturbed cell cycle progression and DNA damage response.
Keywords: C53, Cdk1, Checkpoint kinases
Cells utilize the checkpoint mechanisms to co-ordinate cell cycle progression and cellular response to cell cycle irregularities such as replication stress and DNA damage, thereby maintaining accurate transmission of genetic material during cell division. One of the key checkpoints is the G2/M checkpoint that regulates activation of Cdk1 (cyclin dependent kinase 1) and mitotic entry. In response to DNA damage, cells elicit the checkpoint response to either halt cell cycle progression for DNA repair or induce cell death (3). Activation of ATM/ATR (ataxia telangiectesia mutated/ATM and Rad3 related), two members of the PI(3)K (phosphatidyl-inositol-3-OH kinase)-like kinases (PIKKs), is the first step for DNA damage signal transduction (4,5). Locally active ATM/ATR further phosphorylate and activate checkpoint kinases 1 and 2 (Chk1 and Chk2) that rapidly spread the signal to downstream target proteins, such as p53 and Cdc25 phosphatases (6-9). Phosphorylation and inactivation of Cdc25s by Chk1/Chk2 lead to accumulation of Thr15-phosphorylated inactive Cdk1 and the G2/M arrest (6). In an unperturbed cell cycle, the components of the checkpoints are also essential for surveillance of cell cycle progression. For example, both ATR and Chk1 are essential for animal development and proper cell cycle progression (8,10-11). Inhibition of Chk1-mediated signaling may lead to increased DNA replication and DNA breakage, aberrant mitotic entry and apoptosis (8,12).
Interestingly, a number of recent studies have demonstrated an important role of the centrosome in regulation of the G2/M transition (13). The centrosome is the major microtubule-organizing center (MTOC) that contributes to the regulation of cell shape, polarity, adhesion and motility (14). Recent studies have demonstrated that the centrosome also serves as a solid-phase platform for a multitude of signaling networks, especially cell cycle control and checkpoint signaling (15). The Cdk1/cyclin B1 complex accumulates at the centrosome during the interphase, and its initial activation occurs at the centrosome in the late prophase (16). Both positive and negative pathways that regulate Cdk1 activation integrate at the centrosome (13). Mitotic kinases such as Polo-like kinase 1 (Plk1) and aurora-A kinase are localized at the centrosome to promote Cdk1 activation at the onset of mitosis (16-18). Negative regulators of mitotic entry such as Chk1 and Chk2 are also reported to localize at the centrosomes (19-20). In the interphase, centrosome-associated Chk1 shields centrosomal Cdk1 from unscheduled activation by Cdc25B, thereby preventing premature mitotic entry (19).
C53 protein (also known as Cdk5rap3 and LZAP) was originally isolated as a binding protein of Cdk5 activator p35, but its function remains largely elusive (21,22). It is highly conserved during evolution, and its orthologues are found in vertebrate, invertebrate and plants but not in yeast and bacteria. Human C53 consists of 506 amino acid residues without well-defined domains except a small region of leucine zipper. Our previous work demonstrated that C53 acts as an important regulator of genotoxin-induced apoptosis (1). We found that C53 overexpression overrides the G2/M DNA damage checkpoint to promote Cdk1 activation, thereby sensitizing cancer cells to various DNA damage agents (1). In this study, we further characterized its important role in normal cell cycle progression and DNA damage response. Intriguingly, we found that C53 interacts and antagonizes checkpoint kinases in both unperturbed cell cycle progression and DNA damage response. Our finding provides a molecular mechanism for C53’s chemo-sensitizing activity, and sheds light on a novel regulatory mechanism for checkpoint kinases and cell cycle control.
Experimental Procedures
Tissue culture cells and reagents
U-2 OS osteosarcoma cell (from ATCC) were grown in MaCoy’s 5A medium supplemented with 10% fetal bovine serum (FBS), while HeLa, MCF7, T47D and HCT116 and HCT116 (p53-/-) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and antibiotics. Genotoxic reagents and chemicals were purchased from Sigma.
Construction of C53 expression and shRNA vectors
Human full-length C53 cDNA and the cDNAs for its truncated mutants (C53N and C53C1-3) were PCR-amplified and subcloned in-frame into pCMV-5a (Sigma), and the resulting constructs were used for expression of C-terminal Flag-tagged C53 in mammalian cells. Primers for full length C53 were 5′ GCGTCGACATGGAGGACCATCAGCAC3′ (forward) and 5′ (GCGGTACCCAGAGAGGTTCCCAG3′ (reverse). Primers for C53C1, C2 and C3 were 5′GCGTCGACATGTGGGGCGGACTTTGGGGTAG3′ (C1, forward), 5′GCGTCGACATGTCTGGCATCTCTGCCGAGG3′ (C2, forward), and 5′ GCGTCGACATGGATGCTGTTGCTTTGCAG AT3′ (C3, forward), respectively. Primer for C53N was 5′CCGGATCCTTCAATCGCATCTTCTGCCAC3′ (reverse). Human cDNAs for Chk1 and Chk2 were amplified from their full-length MGC cDNA clones (ATCC), and subcloned in-frame into pcDNA-Myc (Clontech). Centrosome-targeting C53 (C53-GFP-PACT) was constructed by two-step PCR. Specifically, C53-GFP cDNA was amplified using specific primers (primer 1: 5′ GCGTCGACATGGAGGACCATCAGCAC3′ and primer 2: 5′ GGCAATGATGGCTTCAATGTTGGCCTTGTACAGCTCGTCCATGCCGAG3′) and pEGFP-N3-C53 as the template, while PACT cDNA was amplified using primers (primer 3: 5′ CTCGGCATGGACGAGCTGTACAAGGCCAACATTGAAGCCATCATTGCC3′ and primer 4: 5′ GGATCGATTTATGCACCTTGATTCAGTCCAAAGCC3′) and GFP-Chk1-PACT (a gift from Dr. J. Bartek) as the template. Since primers 2 and 3 were complementary, the second PCR was performed using primer 1 and 4 as primers and purified C53-GFP and PACT cDNAs as templates. The recombinant C53-GFP-PACT cDNA was digested with Sal I and Cla I, and subsequently cloned into pLPCX vector (Clontech). All cDNA clones were verified by DNA sequencing.
C53 shRNA vectors were described in Jiang et al. (2005) (1). Retroviruses expressing control shRNA or C53 shRNA were prepared using 293GP packaging cell line and VSVG envelope protein according to the manufacturer’s instruction (BD Clontech).
Cell transfection, cell synchronization and C53 knockdown by shRNA and siRNA
HeLa cells were synchronized by double thymidine block. Briefly, HeLa cells were plated at 40% confluency and arrested with 2 □M thymidine. After 19-hr incubation, cells were washed 4 times with fresh medium and transfected with shRNA vectors (C53 and control) using Lipofectamine 2000 (Invitrogen). After incubation with DNA-lipid mixture for 3 hours, cells were washed twice and incubated in fresh medium for additional 5 hrs. Subsequently, cells were cultured in medium containing 2 □M thymidine and 2 μg/ml puromycin for the second arrest and drug selection. After 16-hr incubation, cells were released into the cell cycle by incubation in fresh medium. Cells were collected or fixed at indicated time points and subjected to specific analyses. C53 depletion was also performed using C53 siRNAs that were purchased from Ambion. Negative control siRNA Allstar was obtained from Qiagen. 10 nM of siRNA was used in reverse transfection of HeLa cells.
For overexpression of C53 and its derivatives, cells were transfected with the expression vectors using either Lipofectamine or Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
Antibodies, co-immunoprecipitation, immunoblotting and immunofluorescence staining
Immunoblotting was performed as described (23). For co-immunoprecipitation assays, cells (4×106) were harvested and washed in cold PBS, and then resuspended in 0.5 ml of lysis buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 0.5% NP-40, 50 mM NaF and a cocktail of protease inhibitors) for 15 min on ice. After centrifugation, the supernatant was incubated with the primary antibody for 3 hours at 4°C, and 10-20 μl of ultralink protein A/G beads (Pierce) for additional 1 hour. For immunoprecipitation using anti-Flag M2 antibody, the M2-conjugated agarose beads (Sigma) were used. After three washes in the lysis buffer, the immunoprecipitate was eluted with 0.1 M Glycine (pH 2.5), and subsequently subjected to SDS-PAGE and immunoblotting with specific antibodies. For immunofluorescence staining, cells were fixed in 100% methanol (-20°C) for 5 min, followed by three washes of phosphate buffered saline (PBS). After incubated with blocking buffer (10% goat serum, 2 % BSA in 1xPBS) for 1 hour at room temperature (RT), cells were stained with specific primary antibodies overnight at 4°C. After three washes (5 min each) in PBS, cells were incubated with specific secondary antibodies for 30 min at RT, and subsequently washed in PBS three times (5 min each). Cells were mounted in anti-fade reagent Fluosaver (Calbiochem) and analyzed by fluorescence microscopy. Confocal images were acquired using Zeiss 510 META confocal microscope, while epifluorescence images were obtained using Leica DMR-HC inverted microscope with Openlab software.
The following antibodies at indicated dilutions were used for immunoblotting: C53 polyclonal rat antibody (affinity purified, 1:1,000 dilution), β-actin (Sigma, clone AC-15, 1:10,000), Flag (Sigma, M2, 1:1,000), Cdk1 and p-Y15-Cdk1 (Cell Signaling, 1:1,000), Cdc25C (Calbiochem, DCS193, 1: 500) and p-S216-Cdc25C (Cell Signaling, 1:500), Chk1 (Sigma, DCS-310, 1: 3,000) and p-S345-Chk1 (Cell Signaling, 1:1,000), Chk2 (LabVision, DCS-293, 1:1,000) and p-T68-Chk2 (Cell Signaling, 1:1,000), p-S139-H2AX (BioLegend, 1:2,000), p-S10-Histone H3 (Upstate, 1:3,000), Myc (Santa Cruz, 9E10, 1:1,000), α-tubulin (Sigma, clone DM 1A, 1:10,000). The following antibodies at indicated dilutions were used for immunofluorescence staining: C53 (1:200), γ-tubulin (Sigma, GTU-88, 1:1,000), p-S10-Histone H3 (Upstate, 1:500), p-Y15-Cdk1 (Cell Signaling, 1:800), Chk1 (Sigma, DCS-310, 1:600). All affinity-purified and species-specific fluorophore-conjugated secondary antibodies were obtained from Jackson ImmunoResearch, and used at dilutions between 1:400 to 1:800.
BrdU labeling and mitotic index
BrdU labeling was used to evaluate DNA synthesis. After released from the second thymidine arrest at indicated time points, cells grown in 12-well plate were pulse labeled with BrdU (50 μM) for 30 min. After three washes of PBS, cells were fixed with 1 ml of Carnoy’s fixative (3 parts methanol:1 part glacial acetic acid) at -20°C for 20 min, and followed by three washes of PBS. Subsequently, DNA was denatured by incubation of 2M HCl at 37°C for 60 min, followed by three washes in borate buffer (0.1 M borate buffer, pH 8.5). After incubation with the blocking buffer, cells were stained with anti-BrdU antibody (BD Biosciences, 1: 100) overnight at 4°C. After washes in PBS, cells were incubated with Texas Red-conjugated anti-mouse goat IgG for 30 min at RT. After washes, cells were mounted and BrdU positive cells were manually scored under immunofluorescence microscope.
Mitotic events were scored by time-lapse videomicroscopy and DNA staining. Cells were synchronized as described above. Real-time images were captured every 10 min with Openlab software. Mitotic events of control and C53-depleted cells were scored by their morphological change (from flat to round-up). For each experiment, at least 800 cells of control or C53-depleted cells were videotaped, tracked and analyzed. Alternatively, nocodazole (100 ng/ml) was added into the medium after release, cells were collected, fixed and stained with DNA dye (Hoechst 33258). Mitotic cells were scored by nuclear morphology and DNA condensation. Alternatively, the mitotic index was evaluated by phospho-H3 staining and flow cytometry.
In vitro kinase assay
In vitro Cdk1 kinase assay was performed as described in Jiang et al (2005) (1) with minor modification. Cdk1 was immunoprecipitated with Cdk1 antibody (Cell Signaling).
Results
1. C53 knockdown delays mitotic entry and Cdk1 activation
Our previous study suggests that C53 plays an important role in regulation of the G2/M checkpoint in DNA damage response. In this study we attempted to investigate its role in the G2/M transition during unperturbed cell cycle progression. Control and C53-knockdown HeLa cells were synchronized at the G1/S boundary by double thymidine block, and then released into mitosis. To avoid potential carry-over effects of C53 depletion-induced cell cycle defects in the previous cycle on the following mitotic entry during the next cycle, we transfected shRNAs into HeLa cells during the interval between two thymidine blocks, so that we were able to evaluate direct impact of C53 knockdown on mitotic entry (Figure 1A) (17). Expression of endogenous C53 was usually knocked down to more than 50% after 24 hours by both C53 shRNA and siRNAs (Figure 1E and data not shown). BrdU was added into the medium at indicated time points to evaluate DNA synthesis. As shown in Figure 1B, incorporation of BrdU into the control and C53-knockdown cells did not differ significantly during cell cycle progression (Figure 1B), indicating that C53 knockdown may not affect the G1/S transition and DNA synthesis. In contrast, accumulation of mitotic cells was significantly delayed in C53-knockdown cells (Figure 1C). To further examine the specific effect of C53 knockdown on mitotic entry, we repeated this experiment using two C53 siRNAs and evaluated the mitotic entry using phospho-H3 staining and flow cytometry. As shown in Figure 1D, C53 knockdown by two C53 siRNAs significantly delayed mitotic entry. The variation on mitotic percentage between Figure 1C and 1D may be due to the different toxicity of transfection methods. Mitotic delay was further confirmed by our observation that there was less phosphorylation of histone H3 in C53-knockdown cells than in the control cells (Figure 1E, panel of p-histone H3). We also examined whether C53 knockdown delays Cdk1 activation. Consistently, C53-knockdown cells contained more inactive Cdk1 than the control cells did (Figure 1E, panels of Cdk1 and p-Y15-Cdk1). Moreover, in vitro kinase assay showed that Cdk1 in C53-knockdown cells was less active (Figure 1F). Together, our data strongly indicate that C53 may serve as a positive regulator for Cdk1 activation at the onset of mitosis.
Fig 1.

C53 knockdown delays mitotic entry of HeLa cells. A. Experimental procedure for double thymidine arrest and C53 knockdown. B. Knockdown of endogenous C53 did not affect DNA synthesis. HeLa cells were transfected with shRNAs and synchronized at the G1/S transition as described in the methods. Cells were pulse labeled with BrdU (50 μM) for 30 min at indicated time points after release from the second thymidine block. BrdU positive cells were detected by immunostaining and scored manually. More than 500 cells were counted in each of three independent experiments. C. shRNA-mediated knockdown of C53 protein delayed mitotic entry. Cell cycle progression of more than 1,000 cells were recorded by time-lapse videomicroscopy. The number of mitotic cells was scored by examination of individual cells. D. siRNA-mediated C53 depletion led to delayed mitotic entry. Two C53 siRNAs (10 nM) were transfected into HeLa cells by HiPerfect transfection agent. The negative control was Allstar siRNA from Qiagen. Nocodazole (100 ng/ml) was added in the medium after the second release. Cells were harvested at indicated time points and subject to phospho-H3 staining and flow cytometry analysis. E. C53 knockdown causes accunluaiton of inactive Cdk1. Cell lysates were collected at indicated time points after release from the second block, and subjected to immunoblotting using indicated antibodies. F. C53 knockdown delayed Cdk1 activation. Cdk1 was precipitated from cell lysates at indicated time, and subject to in vitro kinase assay with histone H1 as a substrate. Relative intensity was measured with Openlab software and marked at the bottom of the gel. All data are represented as mean ± SEM.
2. C53 modulates checkpoint kinase-mediated DNA damage response
To further understand how C53 regulates Cdk1 activation, we took advantage of the experimental system that was used to evaluate the effect of C53 on DNA damage response (1). We previously demonstrated that C53 overexpression in HeLa cells abolished the G2/M checkpoint-mediated Cdk1 inactivation induced by DNA damage agent etoposide (1). Therefore, we speculated that by dissecting the effect of C53 on activation of individual components of DNA damage response pathway, we would identify potential C53 targets. Etoposide treatment induced the control cells to undergo a prominent G2 arrest, as indicated by the increased number of G2/M cells and enlarged cell body and nuclei (Supplemental Figure S1, and data not shown). In agreement with our previous work, C53 overexpression abolished accumulation of G2/M cells induced by etoposide, suggesting that C53 expression abolishes the G2/M arrest induced by etoposide treatment (Supplemental Figure S1).
We next examined how C53 overexpression influenced checkpoint activation at various molecular levels. As shown in Figure 2, treatment of HeLa cells with DNA damage agents such as etoposide (Etop) and doxorubicin (Dox) and DNA synthesis inhibitor hydroxyurea (HU) caused Cdk1 inactivation, as indicated by the increase of inhibitory phosphorylation of Cdk1 (p-Y15-Cdk1) (Figure 2, panels of p-Y15-Cdk1 and Cdk1, lanes 1, 3, 5 and 7). In agreement with our previous report, C53 overexpression attenuated inhibitory phosphorylation of Cdk1 induced by genotoxic stresses (Figure 2, panels of p-Y15-Cdk1 and Cdk1, lanes 3-8). Moreover, C53 overexpression blocked inhibitory Ser216 phosphorylation of Cdc25C, the activator of Cdk1 (Figure 2, panels of p-S216-Cdc25C and Cdc25C). As Cdc25C (Ser216) is a bona fide substrate of Chk1 and Chk2 in DNA damage response (6), we further examined the effect of C53 on activation of Chk1 and Chk2. Intriguingly, C53 overexpression reduced phosphorylation of Chk1 at Ser345 (Figure 2, panels of p-S345-Chk1 and Chk1) as well as Chk2 at Thr68 (Figure 2, panels of p-T68-Chk2 and Chk2), indicating that C53 overexpression may attenuate activation of checkpoint kinases in DNA damage response. We also consistently observed that C53 overexpression slightly increased protein level of total Chk1 in the presence or absence of DNA damage (Figure 2, panel of Chk1). Active ATM/ATR are normally responsible for phosphorylation and activation of Chk1 and Chk2. To examine whether C53 overexpression interferes with ATM/ATR activation and activity, we used phosphorylation of histone H2X (Ser139) as the indicator for ATM/ATR activity (24). Etoposide treatment induced a remarkable increase on phosphorylation of H2X, while the effect of doxorubicin and hydroxyurea was moderate (Figure 2, panel of p-Ser139-H2X, lanes 1, 3, 5 and 7). C53 overexpression slightly increased H2X phosphorylation in the absence of genotoxic stress (Figure 2, panel of p-S139-H2X, lanes 1 and 2), probably due to a small percentage of cell death induced by C53 overexpression. Importantly, C53 overexpression did not significantly affect elevated H2.X phosphorylation induced by the genotoxic stresses (Figure 2, compare lanes 4. 6 and 8 with lanes 3, 5. and 7). These results suggest that C53 may act at the level of Chk1/Chk2 in the hierarchy of DNA damage response events.
Fig 2.
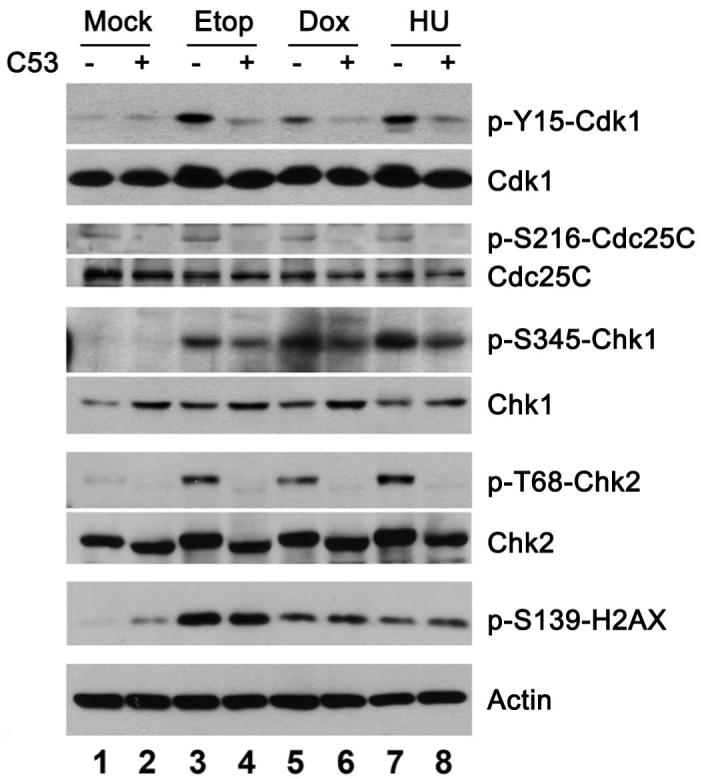
C53 modulates checkpoint kinase-mediated DNA damage response. Ectopic expression of C53 suppressed the DNA damage checkpoint response. HeLa cells were transfected with the C53 or control vectors. At 24 hours after transfection, the cells were treated with etoposide (Etop, 20 μM, 18 hours), doxorubicin (Dox, 1 μM, 5 hours), hydroxyurea (HU, 4 mM, 18 hours). Cell lysates were subjected to SDS-PAGE and immunoblotting of indicated antibodies.
3. C53 interacts with checkpoint kinase 1 and antagonizes its activity
Our finding that C53 negatively influences checkpoint kinase activation may reflect the possible functional interaction between C53 and checkpoint kinases. We first examined whether C53 interacted directly with Chk1 and Chk2. As shown in Figure 3A, overexpressed Myc-Chk1 and Myc-Chk2 were present in the immunoprecipitate of C53-Flag fusion protein. Additionally, endogenous Chk1 was co-immunoprecipitated with endogenous C53, but not with control rat IgG (Figure 3B). A doublet of Chk1 may represent different forms of phosphorylated Chk1 as previously reported (25,26). We were unable to detect Chk2 in the same C53 immunoprecipitate (data not shown), probably due to the weak interaction between endogenous C53 and Chk2.
Fig 3.

C53 interacts with Chk1 and antagonizes its activity. A. C53 co-immunoprecipitated with Chk1 and Chk2. Both C53-Flag and Myc-Chk1/Chk2 were overexpressed in HeLa cells. C53-Flag fusion protein was pulled down by Flag (M2) antibody-conjugated agarose beads, and Chk1/Chk2 were detected by immunoblotting using Myc antibody. B. Co-immunoprecipitation of endogenous C53 and Chk1. Endogenous C53 was pulled down with C53 antibody, and Chk1 was detected by immunoblotting using Chk1 antibody. IgG HC indicated IgG heavy chain. C. Mapping of C53’s Chk1-interacting domains. Myc-Chk1 and C53-Flag and its derivatives were overexpressed in HeLa cells. C53-Flag protein was pulled down by M2-conjugated agarose beads. The presence of Chk1 was detected by Myc immunoblotting. D. Full-length and C-terminal fragment of C53 protein antagonized Chk1-mediated Cdk1 inactivation. HeLa cells were transiently transfected with Myc-Chk1 and C53 constructs. Cells were collected at 24 hours after transfection. Total cell lysates were subjected to immunoblotting using indicated antibodies. E. Chk1 inhibitor UCN-01 prevented delayed Cdk1 activation induced by C53 knockdown. HeLa cells were synchronized by double thymidine block. UCN-01 (300 nM) was added into the medium at 5 hours after release from the second block. Cells were collected at 10 hours after release, and the total cell lysates were subjected to SDS-PAGE and immunoblotting using indicated antibodies. F. C53 constructs were used in this study.
Given the fact that Chk1 is essential for unperturbed cell cycle progression (8,10), it is likely that C53 exerts its function through modulating Chk1 activity in an unperturbed cell cycle. To test this hypothesis, we first determined the domain(s) of C53 responsible for C53-Chk1 interaction. C53 is cleaved by caspase-3 at three sites (D268, D286 and D311, our unpublished results and Figure 3F). As shown in Figure 3C, both full-length and the carboxy-terminal fragments (C53C1-3) of C53 protein interacted with Myc-Chk1, while the control protein Bcl-2 and the amino-terminal fragment of C53 (C53N, residues 1-256) did not (Figure 3C). Among C-terminal cleavage fragments, C53-C1 appeared to bear the highest affinity to Myc-Chk1, indicating that the region between residues #268-286 may be important for C53-Chk1 interaction. We also tested the Chk1-C53 interaction in vitro using purified C53 proteins. In agreement with our Co-IP results, GST-C53-C1 pulled down most 35S-labeled Chk1 (Supplemental figure S2), indicative of its highest affinity to Chk1. We next tested whether C53 can directly inhibit Chk1 kinase activity. Purified GST-C53 or His-C53 protein did not inhibit GST-Chk1 kinase activity in vitro (data not shown). In HeLa cells, ectopic expression of Myc-Chk1 increased inhibitory phosphorylation of Cdk1 (Figure 3D, panels of Cdk1 and p-Y15-Cdk1, lanes 1 and 2), a result which was in agreement with previous studies (19). Intriguingly, only C53 and C53C1, but not C53N, attenuated Chk1-induced inactivation of Cdk1 (Figure 3D, panel of p-Y15-Cdk1, lanes 3-5). Of note, there was more total active Chk1 (indicated by Ser345 phosphorylation) in C53 or C53C1-expressing cells than C53N and control cells (data not shown). Moreover, overexpression of C53 or its derivatives alone did not alter phosphorylation of Cdk1 (Supplemental figure S3). This result suggests that C53 may directly inhibit Chk1 kinase activity in vivo through an activity localized at its C-terminal fragment.
If C53 indeed negatively regulates Chk1 activity, we speculated that Chk1 inhibition would reverse delayed Cdk1 activation induced by C53 knockdown. Chk1 inhibitor UCN-01 (7-hydroxystaurosporine) (300 nM) was added at 5 hours after the cells were released from the second thymidine block. As shown in Figure 3E, the addition of UCN-01 abolished delayed Cdk1 activation caused by C53 knockdown (Figure 3E, compare the lanes 4 and 8 in panels of p-Y15-Cdk1 and p-Histone H3). This result indicates that Chk1 activity is indeed responsible for delayed Cdk1 activation in C53 depleted cells at the onset of mitosis, further supporting the notion that C53 is an antagonist of Chk1.
4. A portion of C53 is localized at the centrosome and regulates local activation of Cdk1 at the centrosome
To further elucidate the mechanism of C53-mediated regulation of checkpoint kinases, we raised and affinity purified C53 rat polyclonal antibodies, and examined the subcellular localization of C53. As shown in Figure 4A, our purified polyclonal C53 antibody recognized a single protein with molecular weight (MW) 66 kilodalton, which corresponds to the MW of endogenous C53 protein. C53 is largely localized in the cytoplasm around the nucleus of U-2 OS cells (Figure 4B). A portion of C53 is also localized at the centrosomes, as indicated by its co-localization with centrosomal marker γ-tubulin (Figure 4B). To confirm antibody specificity, we depleted either C53 antibody with immobilized GST-C53 fusion protein, or endogenous C53 with C53 shRNA. Both depletions resulted in negative staining of C53 in the cytoplasm and at the centrosome, suggesting that the antibody staining is C53-specific (Figure 4B). Similar subcellular localization of C53 has also been observed in other cell types including HeLa cells (Figure 4C), MCF7, DU145 and T47 cells (data not shown). Interestingly, more C53 appeared to be recruited to the centrosome in the prophase (Figure 4C).
Fig 4.

A portion of C53 is localized at the centrosome. A. Immunoblotting of cell lysates with or without C53 knockdown using purified C53 rat polyclonal antibody. B. Immunostaining of endogenous C53. U-2 OS cells were stained with C53 polyclonal rat antibody and γ-tubulin monoclonal antibody (GTU-88, Sigma). For depletion, the primary antibodies were incubated with immobilized GST or GST-C53 fusion proteins. For shRNA knockdown, U-2 OS cells were transfected with C53 shRNA. After 4-day puromycin selection, the cells were subjected to immunostaining. C. C53 staining in HeLa cells. To better visualize the centrosomal staining of C53, HeLa cells were pre-extracted with 0.5% Triton in PBS for 5 min at RT. The prophase is indicated by centrosomal separation. All centrosomes were marked by arrowheads.
5. C53 regulates local activation of Cdk1 at the centrosome
In an unperturbed cell cycle, the Cdk1/cyclin B1 complex is recruited to the centrosome in the late G2 phase, and initially activated by centrosome-associated Cdc25B at the centrosome in the prophase (16-18). Centrosome-associated Chk1 prevents its premature activation in the interphase (19). Given our finding that a portion of C53 is localized at the centrosomes and C53 affects overall activation of Cdk1, we postulated that C53 may also regulate initial activation of Cdk1 at the centrosome. C53 and Chk1 were co-localized at the centrosome (Supplemental Figure S4). We reasoned if C53 functions as a negative regulator of Chk1, C53 depletion would lead to elevated Chk1 activity, which in turn would cause accumulation of inactive Cdk1 at the centrosome during the G2/M transition. As shown in Supplemental Figure S5, a fraction of inactive Cdk1 (p-Y15-Cdk1) was located at the interphase centrosomes of U-2 OS cells. In asynchronous U-2 OS cells transfected with control shRNA, more than 90% of the centrosomes displayed C53 staining. In contrast, only 28.9% (±5.3%) of the centrosomes were stained by C53 antibody in C53 knockdown cells. Interestingly, C53 knockdown led to higher percentage of cells in which the centrosomes were decorated with inactive Cdk1 (p-Y15-Cdk1) (58.5% ± 2.1% of C53-knockdown cells vs. 27.0% ± 3.0% of control cells. p value =0.001), indicating that C53 may influence local activation of Cdk1 (Figure 5A). The level of total inactive Cdk1 was also higher in C53-knockdown cells as examined by immunoblotting (Figure 5B).
Figure 5.

C53 regulates local activation of Cdk1 at the centrosomes. A. More centrosomes were decorated by inactive Cdk1 in C53-depleted cells. U-2 OS cells were infected with retroviruses expressing either control shRNA or C53 shRNA. After 4-day puromycin selection, U-2 OS cells were immunostained with p-Y15-Cdk1 and tubulin antibodies. The numbers of cells containing inactive Cdk1-decorated centrosomes were scored manually. More than 200 cells were examined in each of three independent experiments. Data are represented as mean ± SEM. B. Total p-Y15-Cdk1 was evaluated by immunoblotting. U-2 OS cells were infected with retroviruses expressing control shRNA or C53 shRNA. After 4-day drug selection (puromycin 2 μg/ml), cells were collected and subjected to immunoblotting using indicated antibodies. C. Centrosome-targeting C53 reduced the number of inactive Cdk1-decorated centrosomes in U-2 OS cells. C53-GFP-PACT or GFP-PACT constructs were transfected into U-2 OS cells. After 24 hours, cells were fixed and immunostained with p-Y15-Cdk1 antibody. p-Y15-Cdk1-decorated centrosomes were scored as described above. More than 200 cells were examined in each of three independent experiments. Data are represented as mean + SEM. D. Overexpression of centrosome-targeting C53 promoted overall Cdk1 activation. U-2 OS cells (6-well plate) were transfected with C53-GFP-PACT (200 ng) or GFP-PACT construct (200 ng). Cells were collected after 24 hours, and subjected to immunoblotting using indicated antibodies.
If C53 functions as an antagonist of Chk1, it is conceivable that centrosome-targeting C53 would counteract centrosomal Chk1 to promote local activation of Cdk1 at the centrosome. We fused the PACT (pericentrin-AKAP450 centrosomal targeting) domain of AKAP450 to the C-terminus of C53-GFP protein to generate C53-GFP-PACT, a fusion protein which mainly targeted the centrosomes (Supplemental Figure S6). As shown in Figure 5C, introduction of C53-GFP-PACT into U-2 OS cells remarkably reduced the number of the centrosomes decorated by p-Y15-Cdk1 (9.3% ± 0.8% of C53-GFP-PACT expressing cells vs. 30.1% ± 1.9% of GFP-PACT expressing cells, p value=0.0006), suggesting that forced expression of centrosomal C53 is able to antagonize Chk1 and promote Cdk1 activation at the centrosome. Additionally, centrosome-targeting C53-GFP-PACT was also able to decrease the total pool of p-Y15-Cdk1, as examined by immunoblotting of total cell lysate (Figure 5D). This result supports the notion that centrosome-associated regulators such as Chk1 are capable of regulating Cdk1 activation both locally and globally (19).
Discussion
C53 protein (also known as Cdk5rap3 and LZAP) was originally isolated as a Cdk5 activator p35 binding protein in a yeast two-hybrid screening using p35 as the bait protein (21,22), and subsequently identified as a caspase substrate (our unpublished results). More recently, C53 was identified as a binding partner of CREB (cAMP response element-binding protein)-binding protein (27) and ARF (22). It is a highly conserved protein, yet its function remains largely elusive. We report here that C53 protein functions as a novel regulator of checkpoint kinases in cell cycle control. In this study, we demonstrated that C53 acts to antagonize checkpoint kinases to promote Cdk1 activation. We found that C53 depletion delays Cdk1 activation and mitotic entry in unperturbed cell cycle progression. Moreover, C53 overexpression partially inhibits checkpoint kinase activation in DNA damage response, thereby overriding the G2/M checkpoint-mediated Cdk1 inactivation. Intriguingly, we found that C53 interacts with Chk1 and counteract its activity to promote Cdk1 activation both locally and globally. Taken together, our results strongly suggest that C53 acts as a promoter for Cdk1 activation through antagonizing checkpoint kinases.
Mammalian Chk1 and Chk2 are structurally unrelated yet functionally overlapping serine/threonine kinases that play critical roles in relaying checkpoint signaling in response to various genotoxic insults (28). Chk1 is usually activated by ATR-mediated phosphorylation of Ser317 and Ser345 in response to UV and replication stress (8,29), while Chk2 can be activated mainly by ATM-mediated phosphorylation of its residue Thr68 in response to double-strand DNA breaks (DSBs) (7). In the absence of DNA damage, Chk2 appears to be largely inactive. In contrast, Chk1 is active even in unperturbed cell cycle. It is essential for embryonic development and normal cell cycle progression (8,10). Chk1 deficient embryos display many condensed and fragmented nuclei that apoptose through a p53-independent pathway (8,10). Inhibition of Chk1 activity by its pharmacological inhibitors causes premature Cdk1 activation and mitotic catastrophe (30). Many proteins have been identified to play important roles in activation of checkpoint kinases, yet inactivation of the checkpoint response remains poorly understood. Several phosphatases have been shown to inactivate checkpoint kinases. Phosphatases PPM1D and PP2 are capable of dephosphorylating Chk1 and Chk2 and inactivating checkpoint response (31-35).
Interestingly, our findings suggest that C53 may be a novel negative regulator of checkpoint kinases. Our results implicate that checkpoint kinase Chk1 may be a target of C53 action. Firstly, we found that C53 and Chk1 interact with each other and are co-localized at the centrosomes (Figure 4). Secondly, using Cdk1 inactivation as the readout assay for Chk1 intracellular activity, we found that C53 overexpression antagonizes Myc-Chk1-mediated Cdk1 inactivation (Figure 3D), and that the ability of C53 fragments to antagonize Chk1 correlated well with their affinity to Chk1 (Figure 3C and D). Thirdly, Chk1 inhibition by UCN-01 reversed delayed Cdk1 activation induced by C53 knockdown (Figure 3E). Finally, forced expression of centrosome-targeting C53 promotes local Cdk1 activation in U-2 OS cells (Figure 5), indicating that a centrosomal fraction of C53 may counteract centrosome-associated Chk1 during mitotic entry.
Nonetheless, inability of purified C53 to inhibit Chk1 activity in vitro suggests that direct interaction of C53 and Chk1 alone may not be sufficient for C53 antagonizing activity. One possibility is that other protein factors may be needed for C53 function in cells. Another intriguing possibility may come from our observation that a portion of endogenous C53 protein is localized in the centrosome. The centrosome is emerging as an important platform for many signaling pathways (14). Interestingly, both Chk1 and Chk2 were reportedly found at the centrosomes (19,20). Importantly, Kramer et al (2004) demonstrated that centrosome-associated checkpoint kinase Chk1 shields centrosomal Cdk1 from unscheduled activation by Cdc25B, thereby preventing premature mitotic entry (19). Our result demonstrated that centrosome-targeting C53 is very potent to promote Cdk1 activation. Therefore, it is possible that recruitment of C53 to the centrosome may generate high concentration of local C53, thereby effectively inhibiting centrosomal Chk1. Consistent with this idea, we observed that more C53 was localized at the prophase centrosomes than the interphase ones (Figure 4C). Recently Wang et al (2007) reported an intriguing finding that LZAP/C53 may function as a tumor suppressor by direct binding and inhibition of RelA, thereby blocking NF-kB signaling (2). Our study provides an alternative mechanism of C53 function, which may also contribute to its tumor suppressor activity. Whether these two mechanisms function in an independent manner or they influence each other remains to be further investigated. Interestingly, we found that C53 localizes at multiple subcellular compartments including cytosol, nucleus, centrosome, endoplasmic reticulum and microtubules (our unpublished results), indicating that C53 may be multi-functional.
Recent studies strongly suggest that cell cycle checkpoints are essential for maintenance of replication accuracy and genome stability, and defects in the checkpoints are largely responsible for tumorigenesis and genome instability of cancer cells (35). Meanwhile, the DNA damage response network is among the many factors that influence the effectiveness of cancer treatment (36). Many efforts have been made to identify checkpoint kinase inhibitors as chemo- and radio-sensitizing agents in cancer treatment (37). Our identification of C53 as a negative regulator of checkpoint kinases sheds light on a novel regulatory mechanism for checkpoint response, and provides a basis for development of novel chemo-sensitizing agents.
Supplementary Material
Acknowledgements
We thank Dr. J. Bartek for Chk1-GFP-PACT construct. We are grateful for the insightful advice from Drs. Junying Yuan, Qingshen Gao, Jacek Topczewski, Kathy Randell, Marcus Peter and Bernard Mirkin, and the help from Drs. Francis Szele and Ed Kang on time-lapse videomicroscopy and William Goossens on confocal microscopy. The project was supported by NIH grant R21 AG027840 and R01 GM081776 (to H Li), Children’s Memorial Research Center and the Clarke family.
References
- 1.Jiang H, Luo S, Li H. Cdk5 Activator-binding Protein C53 Regulates Apoptosis Induced by Genotoxic Stress via Modulating the G2/M DNA Damage Checkpoint. J Biol Chem. 2005;280:20651–20659. doi: 10.1074/jbc.M413431200. [DOI] [PubMed] [Google Scholar]
- 2.Wang J, An H, Mayo MW, Baldwin AS, Yarbrough WG. LZAP, a putative tumor suppressor, selectively inhibits NF-kappaB. Cancer Cell. 2007;12:239–251. doi: 10.1016/j.ccr.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 4.Cimprich KA, Shin TB, Keith CT, Schreiber SL. cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc Natl Acad Sci U S A. 1996;93:2850–2855. doi: 10.1073/pnas.93.7.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shiloh Y. Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu Rev Genet. 1997;31:635–662. doi: 10.1146/annurev.genet.31.1.635. [DOI] [PubMed] [Google Scholar]
- 6.Furnari B, Rhind N, Russell P. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science. 1997;277:1495–1497. doi: 10.1126/science.277.5331.1495. [DOI] [PubMed] [Google Scholar]
- 7.Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 8.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 9.Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- 10.Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, Ikeda K, Nakayama K, Nakanishi M, Nakayama K. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes Dev. 2000;14:1439–1447. [PMC free article] [PubMed] [Google Scholar]
- 11.Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- 12.Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, Bartek J. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol. 2005;25:3553–3562. doi: 10.1128/MCB.25.9.3553-3562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kramer A, Lukas J, Bartek J. Checking out the centrosome. Cell Cycle. 2004;3:1390–1393. doi: 10.4161/cc.3.11.1252. [DOI] [PubMed] [Google Scholar]
- 14.Doxsey S, McCollum D, Theurkauf W. Centrosomes in Cellular Regulation. Annu Rev Cell Dev Biol. 2005;21:411–434. doi: 10.1146/annurev.cellbio.21.122303.120418. [DOI] [PubMed] [Google Scholar]
- 15.Doxsey S, Zimmerman W, Mikule K. Centrosome control of the cell cycle. Trends Cell Biol. 2005;15:303–311. doi: 10.1016/j.tcb.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 16.Jackman M, Lindon C, Nigg EA, Pines J. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat Cell Biol. 2003;5:143–148. doi: 10.1038/ncb918. [DOI] [PubMed] [Google Scholar]
- 17.Hirota T, Kunitoku N, Sasayama T, Marumoto T, Zhang D, Nitta M, Hatakeyama K, Saya H. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114:585–598. doi: 10.1016/s0092-8674(03)00642-1. [DOI] [PubMed] [Google Scholar]
- 18.Dutertre S, Cazales M, Quaranta M, Froment C, Trabut V, Dozier C, Mirey G, Bouche JP, Theis-Febvre N, Schmitt E, Monsarrat B, Prigent C, Ducommun B. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J Cell Sci. 2004;117:2523–2531. doi: 10.1242/jcs.01108. [DOI] [PubMed] [Google Scholar]
- 19.Kramer A, Mailand N, Lukas C, Syljuasen RG, Wilkinson CJ, Nigg EA, Bartek J, Lukas J. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat Cell Biol. 2004;6:884–891. doi: 10.1038/ncb1165. [DOI] [PubMed] [Google Scholar]
- 20.Tsvetkov L, Xu X, Li J, Stern DF. Polo-like kinase 1 and Chk2 interact and co-localize to centrosomes and the midbody. J Biol Chem. 2003;278:8468–8475. doi: 10.1074/jbc.M211202200. [DOI] [PubMed] [Google Scholar]
- 21.Ching YP, Qi Z, Wang JH. Cloning of three novel neuronal Cdk5 activator binding proteins. Gene. 2000;242:285–294. doi: 10.1016/s0378-1119(99)00499-0. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, He X, Luo Y, Yarbrough WG. A novel ARF-binding protein (LZAP) alters ARF regulation of HDM2. Biochem J. 2006;393:489–501. doi: 10.1042/BJ20050960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Bergeron L, Cryns V, Pasternack MS, Zhu H, Shi L, Greenberg A, Yuan J. Activation of caspase-2 in apoptosis. J Biol Chem. 1997;272:21010–21017. doi: 10.1074/jbc.272.34.21010. [DOI] [PubMed] [Google Scholar]
- 24.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 25.Ng CP, Lee HC, Ho CW, Arooz T, Siu WY, Lau A, Poon RY. Differential mode of regulation of the checkpoint kinases CHK1 and CHK2 by their regulatory domains. J Biol Chem. 2004;279:8808–8819. doi: 10.1074/jbc.M312215200. [DOI] [PubMed] [Google Scholar]
- 26.Shiromizu T, Goto H, Tomono Y, Bartek J, Totsukawa G, Inoko A, Nakanishi M, Matsumura F, Inagaki M. Regulation of mitotic function of Chk1 through phosphorylation at novel sites by cyclin-dependent kinase 1 (Cdk1) Genes Cells. 2006;11:477–485. doi: 10.1111/j.1365-2443.2006.00955.x. [DOI] [PubMed] [Google Scholar]
- 27.Yin X, Warner DR, Roberts EA, Pisano MM, Greene RM. Novel interaction between nuclear co-activator CBP and the CDK5 activator binding protein - C53. Int J Mol Med. 2005;16:251–256. [PubMed] [Google Scholar]
- 28.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 29.Guo Z, Kumagai A, Wang SX, Dunphy WG. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000;14:2745–2756. doi: 10.1101/gad.842500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niida H, Tsuge S, Katsuno Y, Konishi A, Takeda N, Nakanishi M. Depletion of Chk1 leads to premature activation of Cdc2-cyclin B and mitotic catastrophe. J Biol Chem. 2005;280:39246–39252. doi: 10.1074/jbc.M505009200. [DOI] [PubMed] [Google Scholar]
- 31.Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005;19:1162–1174. doi: 10.1101/gad.1291305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujimoto H, Onishi N, Kato N, Takekawa M, Xu XZ, Kosugi A, Kondo T, Imamura M, Oishi I, Yoda A, Minami Y. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006;13:1170–1180. doi: 10.1038/sj.cdd.4401801. [DOI] [PubMed] [Google Scholar]
- 33.Oliva-Trastoy M, Berthonaud V, Chevalier A, Ducrot C, Marsolier-Kergoat MC, Mann C, Leteurtre F. The Wip1 phosphatase (PPM1D) antagonizes activation of the Chk2 tumour suppressor kinase. Oncogene. 2007;26:1449–1458. doi: 10.1038/sj.onc.1209927. [DOI] [PubMed] [Google Scholar]
- 34.Dozier C, Bonyadi M, Baricault L, Tonasso L, Darbon JM. Regulation of Chk2 phosphorylation by interaction with protein phosphatase 2A via its B’ regulatory subunit. Biol Cell. 2004;96:509–517. doi: 10.1016/j.biolcel.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 35.Nojima H. G1 and S-phase checkpoints, chromosome instability, and cancer. Methods Mol Biol. 2004;280:3–49. doi: 10.1385/1-59259-788-2:003. [DOI] [PubMed] [Google Scholar]
- 36.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 37.Zhou BB, Bartek J. Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nat Rev Cancer. 2004;4:216–225. doi: 10.1038/nrc1296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.