

Abstract
Autoimmunity occurs when the immune system recognizes and attacks host tissue. In addition to genetic factors, environmental triggers (in particular viruses, bacteria and other infectious pathogens) are thought to play a major role in the development of autoimmune diseases. In this review, we (i) describe the ways in which an infectious agent can initiate or exacerbate autoimmunity; (ii) discuss the evidence linking certain infectious agents to autoimmune diseases in humans; and (iii) describe the animal models used to study the link between infection and autoimmunity.
Keywords: autoimmune disease, molecular mimicry, virus infection
Introduction
There are more than 80 identified autoimmune diseases [1]. Multiple factors are thought to contribute to the development of immune response to self, including genetics, age and environment. In particular, viruses, bacteria and other infectious pathogens are the major postulated environmental triggers of autoimmunity.
Multiple arms of the immune system may be involved in autoimmune pathology. Antigens are taken up by antigen-presenting cells (APCs) such as dendritic cells (DCs) and processed into peptides which are loaded onto major histocompatibility complex (MHC) molecules for presentation to T cells via clonotypic T cell receptors (TCRs). Cytolytic T cells (Tc, activated by MHC Class I on APC) can directly lyse a target, while T helper cells (Th, activated by MHC class II) release cytokines that can have direct effects or can activate macrophages, monocytes and B cells. B cells themselves have surface receptors that can bind surface antigens. Upon receiving signals from Th cells, the B cell secretes antibodies specific for the antigens. Antibody may bind its specific target alone or may bind to and activate macrophages simultaneously via the Fc receptor.
There are multiple mechanisms by which host infection by a pathogen can lead to autoimmunity (Fig. 1). The pathogen may carry elements that are similar enough in amino acid sequence or structure to self-antigen that the pathogen acts as a self-‘mimic’. Termed ‘molecular mimicry’, T or B cells that are activated in response to the pathogen are also cross-reactive to self and lead to direct damage and further activation of other arms of the immune system. The pathogen may also lead to disease via epitope spreading. In this model the immune response to a persisting pathogen, or direct lysis by the persisting pathogen, causes damage to self-tissue. Antigens released from damaged tissue are taken up by APCs, and this initiates a self-specific immune response. ‘Bystander activation’ describes an indirect or non-specific activation of autoimmune cells caused by the inflammatory environment present during infection. A domino effect can occur, where the non-specific activation of one arm of the immune system leads to the activation of other arms. Lastly, infection may lead autoimmunity through the processing and presentation of ‘cryptic antigens’. In contrast to dominant antigenic determinants, subdominant cryptic antigens are normally invisible to the immune system. The inflammatory environment that arises after infection can induce increased protease production and differential processing of released self-epitopes by APCs.
Fig. 1.
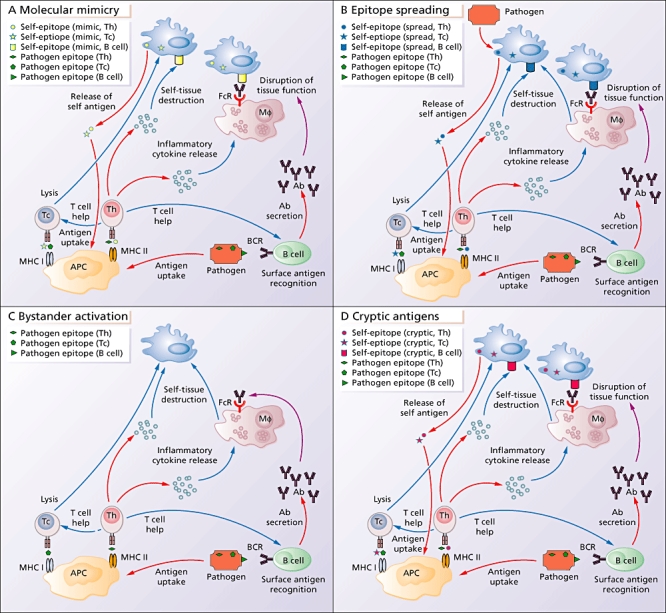
Mechanisms by which pathogens may cause autoimmunity. (a) Molecular mimicry occurs when pathogen-derived epitopes are cross-reactive with self-derived epitopes. Pathogen-derived epitopes are taken up by antigen-presenting cells (APCs) and presented to cytolytic T cells (Tc) via major histocompatibility complex (MHC) class I or to helper T cells (Th) via MHC class II. T cells activated by pathogenic epitopes that are cross-reactive with self-epitopes can then damage self-tissue via lysis (Tc) or release of cytokines (Th). Cytokines released by activated Th cells can activate macrophages (Mφ) or provide help to B cells. Pathogen-derived surface antigens are recognized by a B cell's B cell receptor (BCR), which triggers the secretion of antibodies. These antibodies can cause damage by binding to cross-reactive epitopes on the surface of tissues and disrupting tissue function, or the Fc portion of the antibody can bind simultaneously to the Fc receptor (FcR) on Mφ; this will trigger the Mφ to produce tissue-damaging cytokines. Damaged tissue will release more cross-reactive antigens, which will be taken up by APCs, propagating further damage. (b) In epitope spreading, the immune response to a persisting pathogen, or direct lysis of self-tissue by the persisting pathogen, causes damage to self-tissue. Antigens released from damaged tissue are taken up by APCs, and this initiates an immune response directed towards self-antigens. (c) In bystander activation, the various parts of the immune system respond to the invading pathogens. The inflammatory environment triggered by this response damages self-tissue in an antigen non-specific manner, and in addition triggers non-specific activation of immune cells. (d) In contrast to dominant antigenic determinants, subdominant cryptic antigens are normally invisible to the immune system. The inflammatory environment that arises after infection can induce increased protease production and differential processing of released self-epitopes by APCs.
In this review, we discuss the evidence available for the involvement of specific pathogens in the initiation or exacerbation of representative autoimmune diseases. As will be mentioned, there is evidence for the involvement of different arms of the immune systems by many mechanisms, in both human disease and in animal models.
Coxsackievirus B
Coxsackievirus B (CVB) is the most common cause of infectious myocarditis. Infectious virus and viral RNA can be isolated from patients' hearts [2–4]. CVB3 can cause myocarditis in mice; in most mouse strains, the virus titre peaks at day 4 post-infection and is undetectable after 14 days [5]. The chronic stage of the disease (day 28 onwards) is characterized by mononuclear cell infiltration into the myocardium and the production of antibodies to cardiac myosin which, because of the absence of virus, argues for autoimmunity as the pathophysiological mechanism at this stage of disease. In vitro, cardiac myocytes can be infected and lysed by the virus [6] and CVB infection causes myocardial destruction in SCID mice (which lack T and B cells), showing that the virus can directly infect and lyse cells [7,8]. This damage may lead to autoimmunity via epitope spreading. In mice, virus-specific antibodies arise soon after infection, followed by antibodies to several cardiac proteins such as myosin, tropomyosin and actin [9–11]. T cells also play an important role, as T cells can transfer disease to naive recipients and athymic or T cell-depleted mice exhibit reduced disease following infection [12–15]. Depletion of CD8+ T cells increases myocarditis in infected mice, showing the importance of this subset in mice [16]. Neutralizing anti-mCVB3 monoclonal antibodies (mAb), which could cause cardiac pathology when transferred into mice, were also cross-reactive to cardiac myosin and surface epitopes on cardiac fibroblasts, suggesting mimicry as a possible mechanism [17,18]. Similarly, T cell clones from infected mice proliferate in response to cardiac myosin [19,20]. Some studies have failed to detect cross-reactive T or B cells or mimic sequences within the virus capsid [21]. The same group found that tumour necrosis factor (TNF)-α or interleukin (IL)-1 treatment of genetically resistant mice could render them susceptible to cardiac disease, suggesting that bystander activation may be a mechanism of autoimmunity. CVB3 infection increases ubiquitinization of cellular proteins [9–11], and this increased cellular degradation may also lead to the release of cryptic epitopes. While these studies show that numerous autoimmune mechanisms can lead to cardiomyopathy in infected mice, it remains uncertain if autoimmunity accounts for the pathology seen in humans [22–24].
Streptococcus pyogenes: group A strepcococcus
Infection with S. pyogenes can lead to inflammation of the heart, and the involvement of lymphocytes in cardiac pathology has been suggested for some time [25,26]. Studies have shown that bacterial materials and DNA can persist in host tissue for some years after infection, so it is possible that ongoing immunity against the bacteria may lead to bystander damage to the organ [23]. However, it is accepted most predominantly that the autoimmune reaction is caused by molecular mimicry. Myosin has been identified as the dominant autoantigen in the heart, and myosin-reactive mAb derived from patients with acute rheumatic fever were shown be cross-reactive to both M protein (the major virulence factor of group A streptococci) [27] and the streptococcus carbohydrate epitope N-acetylglucosamine [28]. Similar cross-reactivity was seen with mAb derived from mice immunized with S. pyogenes membranes [29,30]. Cross-reactive mAb has been found to other heart proteins such as tropomyosin and laminin [31,32]. T cell clones from heart lesions of rheumatic heart disease patients, as well as their peripheral blood mononuclear cells (PBMC), can recognize simultaneously streptococcal M protein and heart tissue-derived proteins such as myosin, tropomyosin and laminin [33–36]. BALB/c mice immunized with human cardiac myosin developed T cells cross-reactive with M protein [37], and T cell lines from rats immunized with M protein were also cross-reactive with myosin [38]. These M protein-immunized rats develop cardiac lesions, presenting a good argument that mimicry is a major mechanism of pathology in human rheumatic heart disease. Cardiac lesions can also be induced in rabbits infected with the bacteria [39] and mice immunized with bacterial components [40].
Although somewhat controversial [41,42], infection with S. pyogenes has also been associated with the development of movement and behavioural disorders such as Sydenham chorea, Tourette's syndrome and obsessive–compulsive disorder [43,44]. Patients with these disorders often have antibodies to the basal ganglia in the brain, and molecular mimicry between basal ganglia and S. pyogenes-derived proteins remains the major postulated mechanism of disease induction. Rabbits immunized with streptococcal M protein developed antibodies cross-reactive with several human brain proteins, and synthetic M-derived peptides inhibited brain-cross-reactive antibodies from the serum of a patient with Sydenham chorea [45]. An early paper demonstrated antibody cross-reactivity between S. pyogenes membrane and neuronal cytoplasm in patients with Sydenham chorea [46]. Using serum, cerebrospinal fluid (CSF) and mAb derived from Sydenham chorea patients, dual-specific antibodies were found that react with both the immunodominant carbohydrate epitope on S. pyogenes cell wall (GlcNAc) and with lysoganglioside GM1 on the surface of neurones [47]. The same group demonstrated that GlcNAc-reactive antibodies from the sera of patients with paediatric autoimmune neuropsychiatric disorders associated with streptococci was inhibited by lysoganglioside GM1 [48], and that lysoganglioside GM1-reactive mAb from Sydenham chorea patients could also react with intracellular brain protein beta-tubulin [49]. Animal models are scarce, but Hoffman et al. showed that a subset of Swiss–Jackson Laboratory (SJL)/J mice primed with S. pyogenes homogenate developed movement and behavioural disorders [50]. These mice were found to have antibody deposits in their brains and serum antibody reactive to several regions of the brain.
Trypanosoma cruzi
Chagas disease is caused by infection with the protozoan parasite T. cruzi[51,52]; 10–30% of infected individuals develop the disease, which occurs in two major clinical phases, acute and chronic. The acute phase is characterized by parasitaemia, preferentially in heart muscle cells, and inflammatory infiltration of infected tissue. This is followed by an asymptomatic indeterminant phase, which can last up to 30 years [53]. Patients who progress to the chronic phase of the disease are affected mainly by irreversible cardiomyopathy.
Although it has been suggested that parasite persistence can contribute to chronic Chagas disease cardiomyopathy (CCC), T. cruzi antigens and DNA can also be detected in infected people who remain asymptomatic [54–56]. This suggests that the tissue destruction that characterizes this phase may be largely autoimmune. CCC is characterized histopathologically by mononuclear cell infiltrates, with CD8+ T cells outnumbering CD4+ T cells 2:1. Local production of interferon (IFN)-γ, TNF-α, IL-4 and IL-6 has been reported [57–59]. In addition, real-time polymerase chain reaction (PCR) analysis showed selective up-regulation of IFN-γ-inducible chemokines and chemokine receptors in CCC heart tissue [60]. Collectively, these data suggest that bystander tissue destruction mediated by inflammatory cytokines (especially IFN-γ) may play a role in CCC pathology. PBMC from CCC patients showed cytotoxicity against non-infected cardiac myocytes [61] and cytokine production against cardiac tissue homogenate [62,63], suggesting that the cell-mediated damage can also be tissue-specific. Antibodies to the cardiac protein Galectin-1 were found in both the sera and cardiac tissue of CCC patients; levels correlated with severity of cardiac damage, and interestingly were absent in cardiomyopathies that were not related to T. cruzi infection. There is also evidence for molecular mimicry in CCC. The T. cruzi protein B13 was found to elicit cross-reactive responses to cardiac myosin in from both the humoral [64,65] and CD4+ T cell arms [66,67] of the immune system. Furthermore, cross-reactive antibodies were present in 100% of CCC patients but only 14% of asymptomatic infected individuals [65].
Most of the animal studies of CCC utilize T. cruzi infection of mice as a model. In the C3H/HeJ strain, the heart infiltrate of chronically infected mice is composed predominantly of CD8+ T cells that secrete IFN-γ and TNF-α, which mirrors well the histopathology in humans [68]. In other strains, however, the CD4+ compartment is responsible for the pathology. Chronically infected BALB/c or CBA mice develop CD4+ T cells that proliferate in response to cardiac myosin, but not cardiac actin [69]. Chronically infected BALB/c mice rejected syngeneic newborn hearts unless treated with anti-CD4 (but not anti-CD8) antibody [70]. A CD4+ T cell line derived from chronically infected DBA/2 mice, cross-reactive with both cardiac and T. cruzi-derived proteins, was able to cause intense heart inflammation when transferred into infected or heart-immunized BALB/c nude mice [71]. Girones et al. also published a study indicating that T and B cell mimicry existed between murine and T. cruzi-derived proteins. Here, they showed that T cells from T. cruzi infected mice were reactive to both the SAPA antigen on T. cruzi and the homologous, newly identified Cha autoantigen [72]. Transfer of these T cells into naive mice produced anti-Cha autoantibodies and heart lesions. Several other studies have demonstrated cross-reactive antibodies that recognize cardiac proteins such as myosin and T. cruzi antigens [73–77].
Although the chronic phase usually affects the heart, a subset of patients develop motor dysfunction of the gastrointestinal tract, essentially through the destruction of neurones of the enteric nervous system [78]. It was discovered that antibodies raised in rabbit against a flagellum-associated surface protein on T. cruzi (FL-160) are cross-reactive with a 48-kDa protein found exclusively in nervous tissue [79]. It was then found that antibodies raised against the amino terminus of FL-160 react to a different epitope on mammalian sciatic nerve than antibodies raised against the carboxyl terminus [80]. The medical relevance of this apparent mimicry is uncertain, as the ability of human sera to react to FL-160 did not correlate with clinical disease [81]. Other studies have also shown molecular similarity between T. cruzi antigens and antigens from mammalian nervous tissue [82,83].
Borrelia burgdorfeii
In the United States, Lyme disease is caused by the tick-borne spirochete Borrelia burgdorfeii (Bb). Sixty per cent of untreated patients develop arthritis that can last for several years, mainly in large joints such as the knee [84]. These patients have high titres of Bb-specific antibodies, and Bb DNA can be detected in the joint fluid by PCR [85]. Treatment of these patients with antibiotics usually ameliorates the arthritis, which indicates that bystander inflammatory response to the spirochete is responsible for early Lyme arthritis [86]. A subset of patients will progress from acute to chronic arthritis despite treatment with antibiotics and lack of detectable Bb DNA in synovial fluid [85–87]. Antibiotic-resistant Lyme arthritis is associated with the MHC class II alleles human leucocyte antigen (HLA)-DRB1*0401, *0101 and *0404, indicating that its mechanism is T cell-mediated and distinct from acute Lyme arthritis [88]. Cellular and humoral responses to outer surface protein A (OspA) of Bb develop in around 70% of patients with antibiotic-resistant Lyme arthritis, often at the beginning of prolonged arthritic episodes [89–92]. T cell and humoral responses to OspA, but not to other spirochete antigens, were found to correlate with the presence or severity of arthritis [92,93]. Specifically, antibiotic-resistant patients responded preferentially to the T cell epitope OspA165–173, and T cells responsive to this epitope were expanded in the joint fluid compared with peripheral blood in HLA-DRB1*0401-positive patients [89,94,95]. An initial computer algorithm search identified lymphocyte function-associated antigen (LFA)1αL332–340, a peptide derived from the light chain of human leucocyte adhesion molecule, as homologous to OspA165–173, and able to bind HLA-DRB1*0401 [96]. Synovial fluid mononuclear cells from patients with antibiotic-resistant arthritis produced IFN-γ in response to both OspA165–173 and LFA1αL332–340, suggesting that mimicry between these two proteins may cause the inflammation associated with arthritis. LFA-1α has also been identified in the synovia of patients with antibiotic-resistant Lyme arthritis [97]. However, other studies showed that in treatment-resistant patients, LFA1αL332–340 was a weak agonist for OspA165–173-specific T cells and mainly induced the Th2-type cytokine IL-13 [98]. LFA1αL332–340 binds well to HLA-DRB*0401, but not to the more commonly associated allele HLA-DRB1*0101 [99]. In addition, although cross-reactive T cells were identified in the majority of patients in one study, there was no correlation between T cell response to LFA1αL332–340 and clinical status [100]. These studies weaken the argument that LFA1αL332–340 cross-reactivity is important in the pathology of antibiotic-resistant Lyme arthritis. On the other hand, Maier et al. identified 15 other human and murine self-peptides that could stimulate an OspA165–173-specific T cell hybridoma [101], so other peptides may prove to be more important in disease pathology.
There are several rodent models in which arthritis is induced upon infection with Bb[102–105]. In C3H mice, joints are infiltrated with neutrophils 10–14 days after infection and, at the peak of arthritis (3–5 weeks), synovial lesions show leucocyte infiltration with mononuclear cells [103]. C57BL/6-beige mice, which have impaired macrophage motility and chemotaxis, develop severe arthritis [106], whereas C57BL/6 mice develop minimal arthritis unless deficient in IL-10 and IL-6 [107,108]. These studies indicate that macrophage-derived anti-inflammatory cytokines protect these mice from severe joint inflammation. Transferring Bb-specific T cells alone in the absence of B cells will exacerbate and accelerate the onset of arthritis in C57/BL6-SCID mice [109]. Rodent models are helpful only in studying acute Lyme arthritis, as the arthritis resolves within a few weeks and is not antibiotic-resistant.
Neurological complications, including myelitis and peripheral neuropathy, can occur in 10–12% of untreated patients infected with Bb and can arise even after antibiotic treatment [110]. Patients with chronic neuroborreliosis have been reported to have antibodies reactive to nerve axons in their serum [111], as well as antibodies and T cells specific for myelin basic protein (MBP) in spinal fluid [112,113]. Patient serum that was reactive to axons and neuroblastoma cells was also cross-reactive with Bb flagellin [111,114]. Next, it was discovered that a mAb for flagellin was cross-reactive with human heat shock protein 60 and with neuroblastoma cell lines [115,116] and slowed neurite outgrowth in culture [117]. Antibody cross-reactivity has also been described between human central nervous system (CNS) proteins and Bb OspA [118]. Several host neural peptides were identified as cross-reactive with Bb-specific T cells from CSF of a patient with chronic neuroborreliosis using peptide libraries and biometric data analysis [119]. However, studies such as those in non-human primates suggest that bystander inflammatory responses to the persistently infective pathogen may explain more clearly the CNS complications of this disease [120–122].
Herpes simplex virus
Herpetic stromal keratitis (HSK) is caused by corneal infection by herpes simplex virus (HSV) and can lead to blindness [123,124]. Whereas progression from epithelial infection to stromal keratitis is not prevented by anti-viral drugs, the symptoms of HSK can be alleviated with immunosuppressive drugs such as corticosteroids [125], indicating that HSK is an autoimmune disease. Because of the difficulties associated with studying the disease in humans, much of the characterization of HSK has utilized murine infection with HSV-1. Within 72 h of infection proinflammatory cytokines IL-1 and IL-6 are produced, which leads to influx of neutrophils into the corneal stroma [126–129]. Significantly, SCID mice reconstituted with CD4+ T cells and depleted of neutrophils exhibit a lower incidence and severity of HSK [130]. Macrophage and natural killer (NK) cell influx follows subsequently in the cornea and may contribute to disease pathology directly or through the production of inflammatory cytokines [131–134]. Starting around 10 days after infection, a second wave of infiltration occurs, consisting mainly of neutrophils and CD4+ T cells, which is heavily dependent on local production of IFN-γ[135,136]. Interestingly, the peak of HSK (day 14 post-infection) is 5–7 days after the infectious virus is typically detectable, suggesting that HSK pathology does not require the presence of the replicating virus [137,138]. However, viral DNA has been detected 37 or more days post-infection and could stimulate DCs and macrophages to activate T cells through bystander activation or the presentation of cryptic epitopes [139–141]. It was discovered early on that CD4+ T cells were necessary for the development of HSK [135,136], and molecular mimicry has been postulated in addition to the mechanisms mentioned above. Cornea-specific T cell clones that cross-reacted with an epitope in the immunoglobulin (Ig)H locus (which was shown to defer susceptibility to HSK) were also found to recognize the HSV-1-derived protein UL6 [142,143]. Transfer of these cross-reactive T cells induced HSK lesions in nude mice, and HSV-1 viral mutants lacking the UL6 peptide did not induce HSK lesions in susceptible mice. However, in other studies employing a different susceptible mouse strain, infection failed to produce T cells reactive to either to UL6 or IgH [142,143]. In addition, T cell lines isolated from the cornea of HSK patients did not show reactivity to UL6 or other human corneal antigens [144–146]. This suggests that, in humans, T cells may cause pathology via bystander destruction.
Uveitis
Uveitis is a group of intra-ocular inflammatory diseases that are potentially blinding [147]. It is believed that many subgroups of this disease are autoimmune-mediated, in part because of the strong association with certain HLA alleles [148]. Humoral and cellular responses to the retinal antigens interphotoreceptor retinoid binding protein and S-antigen are well characterized in humans [149,150] and animal models in rodents and primates (experimental autoimmune uveitis, EAU) are based on injecting these proteins in complete Freund's adjuvant. Singh et al. identified a CD4+ T cell epitope in human S-antigen and several virus and Escherichia coli-derived peptides with sequence similarity [151,152]. Clinical and histological manifestations typical of EAU, including inflammatory infiltrates in affected eyes, were seen in Lewis rats immunized with these mimics. In addition, proliferation assays performed from lymph nodes demonstrated cross-reactive responses between the mimics and the retinal autoantigen. Starting with a different S-antigen CD4+ T cell epitope, Wildner and Diedrichs-Mohring found mimics derived from rotavirus and bovine milk casein [153]. In the same study, patients with uveitis were found to have an increased T cell and antibody response to S-antigen and the two identified mimics compared with healthy donors. Aside for a report of an outbreak of uveitis in children after echovirus infection, no pathogen has yet to be associated epidemiologically with uveitis [154].
Diabetes
Type I diabetes (T1D) results from autoimmune destruction of pancreatic cells by autoreactive T cells and/or inflammatory cytokines. Although there is a definite genetic component to T1D, the concordance rate in monozygotic twins is only approximately 40% [155,156], and epidemiological evidence suggests that pathogens play a role in development. Many different viruses have been associated with T1D development [157]. Studies showed a higher incidence of T1D in people with congenital rubella [158] and antibodies to pancreatic islet cells in rubella-infected patients [159]. Similarly, cytomegalovirus (CMV) was isolated from T1D patients [160] and antibodies to pancreatic islet cells detected in CMV-infected patients [161]. It was also noted that mumps infection often preceded the onset of T1D in children [162,163]. A convincing study showed that CVB4 isolated from the pancreas of acute-onset patients could induce diabetes upon transfer into susceptible mice [163]. CVB4-specific IgM antibodies could be detected in children newly diagnosed with T1D [164,165]. There is some evidence that CVB4 may cause T1D via molecular mimicry. T cells isolated from T1D patients reacted with both glutamic acid decarboxylase (GAD-65) (an identified autoantigen in T1D) and protein 2C in CVB4. However, another study did not observe similar T cell cross-reactivity [166], and yet another showed that cross-reactivity was observed in both diseased patients and healthy controls [167]. In vitro studies suggest that rubella virus may act by producing antibodies and CTLs cross-reactive with islets [168,169]. There is also evidence that CMV can induce cross-reactive antibodies and Th cells [161,170]. In vitro studies showed that the mumps virus could infect and replicate in human cell lines, induce the release of IL-1 and IL-6 and up-regulate expression of MHC class I and class II antigens [171–173]. As the virus has also been shown to replicate in exocrine pancreas [174], it is possible that cytokine release and HLA up-regulation following mumps virus infection may lead to autoimmunity.
The non-obese diabetic (NOD) mouse model develops diabetes through the spontaneous destruction of pancreatic β cells. Similar to human T1D, the T cell response to GAD-65 appears to be important in disease pathogenesis, and epitope spreading may then result in responses to other autoantigens such as insulin [175]. Although in one study immunization of NOD mice with the CVB4-derived 2C protein induced T cells cross-reactive with GAD-65 (supporting the mimicry hypothesis) [176], in another study CVB4 infection did not induce cross-reactive T cells [177]. In a study where CVB4 accelerated the onset of diabetes, it was found that a threshold level of β cell-specific T cells needed to already be present for disease acceleration to occur [178]. Thus, bystander activation may be a more likely explanation than molecular mimicry in the NOD model. BDC2·5 mice are transgenic for a diabetogenic TCR. These mice develop diabetes similar to that seen following CVB4 infection after treatment with streptozotocin (which damages the pancreas) but not after treatment with poly I:C (a Toll-like receptor-3 agonist). This suggests that in this model, the release of cryptic antigens following viral infection may be the mechanism of diabetes induction [179]. Infecting diabetes-resistant BB (DR-BB) rats with Kilham's rat virus (KRV) induces diabetes in about 30% of these animals and insulitis without diabetes in an additional 30% [180]. Interestingly, unlike CVB4 in mice, KRV is not trophic for the pancreas but rather for lymphoid organs such as the spleen, thymus and lymph nodes. It is not very well understood how this virus causes diabetes without infecting β cells, but inactivating macrophages prevents diabetes in KRV-infected DR-BB rats [181]. There are also data that the virus may trigger previously quiescent β cell-specific T cells in DR-BB rats [182]. Finally, CVB4 was found to produce abnormalities in glucose tolerance tests and impaired insulin secretion in patas monkeys [183].
Guillain–Barré syndrome
Guillain–Barré syndrome (GBS) is a paralytic illness affecting both myelin and axons of the peripheral nervous system [184]. Several studies have demonstrated anti-glycolipid antibodies in the serum of a proportion of patients [185]. There are different clinical variants of the disease, which can correlate with the specific type of glycolipid targeted by the antibodies. Glycolipids found most commonly in neural tissues include the gangliosides and cerebrosides. Onset of GBS occurs days or weeks following an infection or immunization [186]. Although several microorganisms have been associated with GBS development, Campylobacter jejuni is the most extensively studied pathogen as it is a common antecedent to GBS. In addition, there is mounting evidence suggesting that lipopolysaccharide (LPS) on the outer core of the bacteria can mimic host gangliosides. LPS from C. jejuni serotypes associated with GBS were shown to resemble human gangliosides structurally [187,188], and priming of mice, rats and rabbits with the above-mentioned LPS produced corresponding anti-ganglioside antibodies [189–191]. Several studies have shown that C. jejuni serotypes associated with GBS are more likely to contain ganglioside-like epitopes compared with serotypes isolated from C. jejuni-infected patients with gastroenteritis but no neurological symptoms, with one study linking ganglioside mimicry to specific GBS clinical subtypes [192,193]. Furthermore, Yuki et al. reported that rabbits immunized with C. jejuni LPS developed flaccid limb weakness that was associated with antibodies to the ganglioside GM1 and peripheral nerve pathology identical to that seen in GBS [194].
Patients infected with Mycoplasma pneumoniae prior to the development of GBS often have antibodies to galactocerebroside. These antibodies can cross-react with glycolipids on M. pneumoniae[195,196]. Associated antibodies to GM1 have also been reported [197]. Similar to what occurs following C. jejuni infection, patients infected with Haemophilus influenzae can develop antibodies to bacterial LPS that are cross-reactive with ganglioside [198]. The presence of a ganglioside-like structure on the surface of H. influenzae suggests that molecular mimicry may explain its association with GBS induction [199,200].
Multiple sclerosis
Multiple sclerosis (MS) is characterized by a loss of the myelin sheath surrounding axons in the CNS [201]. Demyelination is associated with elevated levels of CD4+ T cells specific for major myelin proteins, and the disease is generally thought to be autoimmune [202–204]. Although it is not known precisely what triggers the development of MS, it is well established that relapses or disease flares in patients diagnosed with the relapsing–remitting form of MS are often associated with exogenous infections, particular upper respiratory infections. In total, more than 24 viral agents have been linked to MS [205,206]. Most of the associations have been circumstantial, but some studies have found evidence of specific pathogens in human tissue. Antigens from herpesvirus type 6 were found in MS plaques but not from tissues from other neurological disorders [207]. Similarly, compared with CSF from patients with other neurological diseases, CSF from MS patients was shown to have higher levels of the bacteria Chlamydia pneumoniae[208]. In vitro studies have also provided evidence linking MS and infectious agents. MS patients have activated T cells specific for MBP [209–211]. Eight pathogen-derived peptides, including epitopes from HSV, adenovirus and human papillomavirus, were identified that are able to activate MBP-specific T cell clones derived from MS patients [212]. Significantly, these peptides were found to be presented most efficiently by subtypes of HLA-DR2 that are associated with susceptibility to MS. Despite the difficulty in linking MS to any one pathogen, the amount of epidemiological evidence reported over the years shows that environmental factors play a strong role in disease development, and suggests that a cumulative lifetime exposure to certain microorganisms can influence disease development [213–216]. In addition, a recent study showed that the degree of concordance for monozygotic twins (generally reported at 40% or less) was influenced by environmental factors [217].
There are numerous rodent models of demyelination which, although not identical to the human disease, are used to study MS. The major infectious models in mice are Theiler's murine encephalomyelitis virus (TMEV), murine hepatitis virus (MHV) and Semliki Forest virus (SFV). Each has distinct immunopathological mechanisms and illustrate the various potential ways pathogens may induce MS. There are two strains of TMEV (TMEV-DA and TMEV-BeAn) which cause an initial acute grey matter disease followed by a chronic progressive demyelination in the white matter of the spinal chord known as TMEV-induced demyelinating disease (TMEV-IDD) [205,218,219]. Although the two strains induce slightly different diseases, the key characteristics of TMEV-IDD (abnormal gait and spastic hindlimb paralysis) remain the same. Intracerebral (i.c.) injection of virus leads to persistent CNS infection; the level of infectious virus is low during the chronic phase, but abundant amounts of viral RNA and viral antigen can be detected throughout the lifetime of the mouse [220–222]. The immune response is initiated by the presentation of persistent viral antigens by CNS-resident APCs to Th1-type CD4+ T cells, but reactivity to myelin does not appear until after the onset of clinical symptoms (30–35 days post-infection) [223–226]. Thus, TMEV-IDD is caused by epitope spreading from viral determinants to self-myelin determinants. Interestingly, in SJL mice, reactivity appears to multiple myelin peptides starting with the immunodominant epitope and spreading at later time-points to other subdominant myelin determinants in a hierarchical manner [226,227]. In contrast to TMEV, mice inoculated with neurotropic strains of MHV will have a single major symptomatic episode (ataxia, hindlimb paresis, paralysis) from which the majority will recover [228]. CNS infection results in an influx of immune cells that for the most part will clear the virus, although virus does persist in low amounts [229]. Demyelination begins about 1 week post-infection and peaks at week 3, after which lesion repair and remyelination generally occurs [230–232]. The exact mechanism of demyelination in this model is somewhat controversial, but appears to be bystander myelin destruction by the immune response recruited initially to the CNS to control viral infection. There is no evidence of self-specific immunity in the CNS of MHV-infected mice [233]. T and B-cell deficient RAG1−/− mice, which were resistant to demyelination, developed histological disease after adoptive transfer with splenoctyes from MHV-inoculated mice, which involved the recruitment of activated macrophages/microglia to sites of demyelination in the spinal cord [234]. Chemokine receptor knock-out mice (CCR5−/−) showed reduced demyelination that correlated with reduced macrophage but not T cell infiltration into the CNS of MHV-infected mice [235]. CD4-deficient mice showed less severe disease than CD8-deficient mice [236,237]. Collectively, these studies suggest that macrophages are responsible primarily for myelin destruction in the MHV model, but that T cells are required to recruit macrophages into the CNS. Like MHV, SFV leads to a transient clinical disease [238,239]. The virus is, for the most part, cleared from the CNS by day 6 post-infection, while demyelination peaks at day 14 and then wanes [240,241]. Demyelination is not seen in nude or SCID mice, demonstrating that it is T cell-mediated [240,242]. In BALB/c mice it is thought that demyelination is due to cytolytic damage of virus-infected oligodendrocytes, although this has not been proved definitively. Depletion of CD8+ T cells virtually abolished lesions of demyelination, whereas depletion of CD4+ T cells did not have that effect [243]. Other studies in BALB/c mice have shown that Th1-type cytokines are involved in viral clearance but not demyelination [244,245]. In C57/Bl6 mice, molecular mimicry may also play a role in demyelination. Infected mice have MBP-reactive T cells [246], and antibodies reactive to MBP and myelin oligodendrocyte protein (MOG) [247]. Computer algorithms uncovered homology between an epitope in the SFV surface protein E2 and MOG18–32[248]. Mice primed with either peptide develop paralytic symptoms with histopathology resembling that of mice infected with SFV. The authors of that study concluded that the cross-reactive antibody response was mainly responsible for the demyelinating lesions.
Summary and perspectives
The immune system has evolved checks and balances to prevent the destruction of host tissue. It is perhaps not surprising that a strong immune response to an invading pathogen could disrupt this regulation and lead to autoimmunity. As outlined above, there is significant evidence suggesting that different classes of pathogens (bacteria, viruses and parasites) are involved in triggering or propagating self-reactive immune responses. However, the evidence for a definitive link for infection-induced autoimmunity is stronger for certain diseases than for others.
The argument for infection-induced pathology is much stronger for diseases associated with one or two specific pathogens than for diseases with multiple causal associations. For example, the fact that infection with C. jejuni is a common antecedent to GBS makes a strong argument that this disease is infection-triggered. In contrast, for diseases such as TID and MS that have been associated with dozens of pathogens, but none in particular, much more needs to be done to make a convincing case. The most compelling proof would be the disappearance of symptoms with the clearance of the infection. This is the case in Lyme disease, where treatment with antibiotics alleviates acute arthritis. However, as outlined previously in this paper, there are many ways a pathogen can cause disease even after the infection has been cleared. In these cases, epidemiological studies showing that people infected with a particular agent have an increased incidence of these diseases compared with people never infected, while not wholly definitive, would certainly strengthen the infection-induced autoimmunity argument.
In human autoimmune diseases, where direct evidence for a role for a particular pathogen is weak, it is all the more important to have supporting animal models. The strongest support comes from animal models in which infection with the agent thought to induce disease in humans causes similar symptoms in animals, as exemplified by induction of heart disease in mice infected with T. cruzi and CVB and arthritis in mice infected with Bb. In other animal models, disease can be shown to be induced by priming with a pathogen-derived antigen, thus strengthening the argument for the involvement of that pathogen in the human disease. The ability to induce heart disease in rats primed with Streptococcal M protein is strong evidence that S. pyogenes causes heart disease in humans via molecular mimicry. Although the link between S. pyogenes infection and neurological disorders in humans is uncertain, at best, the fact that movement and behaviour disorders can be induced in mice primed with S. pyogenes homogenate also lends credibility to that theory. In cases where it is uncertain whether a disease pathology is actually autoimmune (such as uveitis and myocarditis following CVB infection), animal models have played a crucial role in elucidating the potential mechanisms of disease induction.
The heterogeneity of the human population, rather than the weakness of the data, may be in play in instances where the evidence linking infection and autoimmunity is tenuous or even conflicting. It is not difficult to imagine that some people may be more susceptible to developing autoimmune disease following a particular infection than others, or that mimic peptides derived from different infectious agents may be able to trigger a particular autoimmune disease depending on the ability of the infected individual to present various epitopes in the context of their various HLA molecules. Defining the genetic markers that predispose patients to different autoimmune diseases with a suspected infectious trigger would be an important contribution to defining the underlying disease pathogenesis.
References
- 1.Selgrade MK, Cooper GS, Germolec DR, Heindel JJ. Linking environmental agents and autoimmune disease: an agenda for future research. Environ Health Perspect. 1999;107(Suppl. 5):811–3. doi: 10.1289/ehp.99107s5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woodruff JF. Viral myocarditis. Am J Pathol. 1980;101:425–84. A review. [PMC free article] [PubMed] [Google Scholar]
- 3.Bowles NE, Richardson PJ, Olsen EG, Archard LC. Detection of Coxsackie-B-virus-specific RNA sequences in myocardial biopsy samples from patients with myocarditis and dilated cardiomyopathy. Lancet. 1986;1:1120–3. doi: 10.1016/s0140-6736(86)91837-4. [DOI] [PubMed] [Google Scholar]
- 4.Pauschinger M, Doerner A, Kuehl U, et al. Enteroviral RNA replication in the myocardium of patients with left ventricular dysfunction and clinically suspected myocarditis. Circulation. 1999;99:889–95. doi: 10.1161/01.cir.99.7.889. [DOI] [PubMed] [Google Scholar]
- 5.Fairweather D, Kaya Z, Shellam GR, Lawson CM, Rose NR. From infection to autoimmunity. J Autoimmun. 2001;16:175–86. doi: 10.1006/jaut.2000.0492. [DOI] [PubMed] [Google Scholar]
- 6.Herzum M, Ruppert V, Kuytz B, Jomaa H, Nakamura I, Maisch B. Coxsackievirus B3 infection leads to cell death of cardiac myocytes. J Mol Cell Cardiol. 1994;26:907–13. doi: 10.1006/jmcc.1994.1108. [DOI] [PubMed] [Google Scholar]
- 7.McManus BM, Chow LH, Wilson JE, et al. Direct myocardial injury by enterovirus: a central role in the evolution of murine myocarditis. Clin Immunol Immunopathol. 1993;68:159–69. doi: 10.1006/clin.1993.1113. [DOI] [PubMed] [Google Scholar]
- 8.Chow LH, Beisel KW, McManus BM. Enteroviral infection of mice with severe combined immunodeficiency. Lab Invest. 1992;66:24–31. Evidence for direct viral pathogenesis of myocardial injury. [PubMed] [Google Scholar]
- 9.Si X, Wang Y, Wong J, Zhang J, McManus BM, Luo H. Dysregulation of the ubiquitin–proteasome system by curcumin suppresses coxsackievirus B3 replication. J Virol. 2007;81:3142–50. doi: 10.1128/JVI.02028-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo H, Zhang J, Cheung C, Suarez A, McManus BM, Yang D. Proteasome inhibition reduces coxsackievirus B3 replication in murine cardiomyocytes. Am J Pathol. 2003;163:381–5. doi: 10.1016/S0002-9440(10)63667-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo H, Zhang J, Dastvan F, et al. Ubiquitin-dependent proteolysis of cyclin D1 is associated with coxsackievirus-induced cell growth arrest. J Virol. 2003;77:1–9. doi: 10.1128/JVI.77.1.1-9.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woodruff JF, Woodruff JJ. Involvement of T lymphocytes in the pathogenesis of coxsackie virus B3 heart disease. J Immunol. 1974;113:1726–34. [PubMed] [Google Scholar]
- 13.Hashimoto I, Komatsu T. Myocardial changes after infection with Coxsackie virus B3 in nude mice. Br J Exp Pathol. 1978;59:13–20. [PMC free article] [PubMed] [Google Scholar]
- 14.Guthrie M, Lodge PA, Huber SA. Cardiac injury in myocarditis induced by Coxsackievirus group B, type 3 in Balb/c mice is mediated by Lyt 2 + cytolytic lymphocytes. Cell Immunol. 1984;88:558–67. doi: 10.1016/0008-8749(84)90188-6. [DOI] [PubMed] [Google Scholar]
- 15.Huber SA. Coxsackievirus-induced myocarditis is dependent on distinct immunopathogenic responses in different strains of mice. Lab Invest. 1997;76:691–701. [PubMed] [Google Scholar]
- 16.Henke A, Huber S, Stelzner A, Whitton JL. The role of CD8+ T lymphocytes in coxsackievirus B3-induced myocarditis. J Virol. 1995;69:6720–8. doi: 10.1128/jvi.69.11.6720-6728.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gauntt CJ, Arizpe HM, Higdon AL, et al. Molecular mimicry, anti-coxsackievirus B3 neutralizing monoclonal antibodies, and myocarditis. J Immunol. 1995;154:2983–95. [PubMed] [Google Scholar]
- 18.Gauntt CJ, Higdon AL, Arizpe HM, et al. Epitopes shared between coxsackievirus B3 (CVB3) and normal heart tissue contribute to CVB3-induced murine myocarditis. Clin Immunol Immunopathol. 1993;68:129–34. doi: 10.1006/clin.1993.1108. [DOI] [PubMed] [Google Scholar]
- 19.Huber SA. Autoimmunity in myocarditis: relevance of animal models. Clin Immunol Immunopathol. 1997;83:93–102. doi: 10.1006/clin.1997.4342. [DOI] [PubMed] [Google Scholar]
- 20.Huber SA, Moraska A, Cunningham M. Alterations in major histocompatibility complex association of myocarditis induced by coxsackievirus B3 mutants selected with monoclonal antibodies to group A streptococci. Proc Natl Acad Sci USA. 1994;91:5543–7. doi: 10.1073/pnas.91.12.5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rose NR, Hill SL. The pathogenesis of postinfectious myocarditis. Clin Immunol Immunopathol. 1996;80:S92–9. doi: 10.1006/clin.1996.0146. [DOI] [PubMed] [Google Scholar]
- 22.Tam PE, Fontana DR, Messner RP. Coxsackievirus B1-induced chronic inflammatory myopathy: differences in induction of autoantibodies to muscle and nuclear antigens by cloned myopathic and amyopathic viruses. J Lab Clin Med. 2003;142:196–204. doi: 10.1016/S0022-2143(03)00108-2. [DOI] [PubMed] [Google Scholar]
- 23.Whitton JL, Feuer R. Myocarditis, microbes and autoimmunity. Autoimmunity. 2004;37:375–86. doi: 10.1080/08916930410001713089. [DOI] [PubMed] [Google Scholar]
- 24.Schwimmbeck PL, Bigalke B, Schulze K, Pauschinger M, Kuhl U, Schultheiss HP. The humoral immune response in viral heart disease: characterization and pathophysiological significance of antibodies. Med Microbiol Immunol. 2004;193:115–9. doi: 10.1007/s00430-003-0217-7. [DOI] [PubMed] [Google Scholar]
- 25.Bengtsson E, Birke G, Wingstrand H. Acute non-specific myocarditis in scarlet fever and acute haemolytic tonsillitis A clinical investigation of 3,069 cases of scarlet fever, 798 cases of acute tonsillitis, and 333 cases of haemolytic streptococcus carriers. Cardiologia. 1951;18:360–74. doi: 10.1159/000165039. [DOI] [PubMed] [Google Scholar]
- 26.Friedman I, Laufer A, Ron N, Davies AM. Experimental myocarditis: in vitro and in vivo studies of lymphocytes sensitized to heart extracts and group A streptococci. Immunology. 1971;20:225–32. [PMC free article] [PubMed] [Google Scholar]
- 27.Cunningham MW, McCormack JM, Talaber LR, et al. Human monoclonal antibodies reactive with antigens of the group A Streptococcus and human heart. J Immunol. 1988;141:2760–6. [PubMed] [Google Scholar]
- 28.Adderson EE, Shikhman AR, Ward KE, Cunningham MW. Molecular analysis of polyreactive monoclonal antibodies from rheumatic carditis: human anti-N-acetylglucosamine/anti-myosin antibody V region genes. J Immunol. 1998;161:2020–31. [PubMed] [Google Scholar]
- 29.Mertens NM, Galvin JE, Adderson EE, Cunningham MW. Molecular analysis of cross-reactive anti-myosin/anti-streptococcal mouse monoclonal antibodies. Mol Immunol. 2000;37:901–13. doi: 10.1016/s0161-5890(01)00007-4. [DOI] [PubMed] [Google Scholar]
- 30.Cunningham MW, McCormack JM, Fenderson PG, Ho MK, Beachey EH, Dale JB. Human and murine antibodies cross-reactive with streptococcal M protein and myosin recognize the sequence GLN-LYS-SER-LYS-GLN in M protein. J Immunol. 1989;143:2677–83. [PubMed] [Google Scholar]
- 31.Fenderson PG, Fischetti VA, Cunningham MW. Tropomyosin shares immunologic epitopes with group A streptococcal M proteins. J Immunol. 1989;142:2475–81. [PubMed] [Google Scholar]
- 32.Galvin JE, Hemric ME, Ward K, Cunningham MW. Cytotoxic mAb from rheumatic carditis recognizes heart valves and laminin. J Clin Invest. 2000;106:217–24. doi: 10.1172/JCI7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ellis NM, Li Y, Hildebrand W, Fischetti VA, Cunningham MW. T cell mimicry and epitope specificity of cross-reactive T cell clones from rheumatic heart disease. J Immunol. 2005;175:5448–56. doi: 10.4049/jimmunol.175.8.5448. [DOI] [PubMed] [Google Scholar]
- 34.Fae KC, da Silva DD, Oshiro SE, et al. Mimicry in recognition of cardiac myosin peptides by heart-intralesional T cell clones from rheumatic heart disease. J Immunol. 2006;176:5662–70. doi: 10.4049/jimmunol.176.9.5662. [DOI] [PubMed] [Google Scholar]
- 35.Raizada V, Williams RC, Jr., Chopra P, et al. Tissue distribution of lymphocytes in rheumatic heart valves as defined by monoclonal anti-T cell antibodies. Am J Med. 1983;74:90–6. doi: 10.1016/0002-9343(83)91124-5. [DOI] [PubMed] [Google Scholar]
- 36.Guilherme L, Oshiro SE, Fae KC, et al. T-cell reactivity against streptococcal antigens in the periphery mirrors reactivity of heart-infiltrating T lymphocytes in rheumatic heart disease patients. Infect Immun. 2001;69:5345–51. doi: 10.1128/IAI.69.9.5345-5351.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cunningham MW, Antone SM, Smart M, Liu R, Kosanke S. Molecular analysis of human cardiac myosin-cross-reactive B- and T-cell epitopes of the group A streptococcal M5 protein. Infect Immun. 1997;65:3913–23. doi: 10.1128/iai.65.9.3913-3923.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quinn A, Kosanke S, Fischetti VA, Factor SM, Cunningham MW. Induction of autoimmune valvular heart disease by recombinant streptococcal m protein. Infect Immun. 2001;69:4072–8. doi: 10.1128/IAI.69.6.4072-4078.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy GE, Swift HF. The induction of rheumatic-like cardiac lesions in rabbits by repeated focal infections with group A streptococci; comparison with the cardiac lesions of serum disease. J Exp Med. 1950;91:485–98. doi: 10.1084/jem.91.5.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cromartie WJ, Craddock JG. Rheumatic-like cardiac lesions in mice. Science. 1966;154:285–7. doi: 10.1126/science.154.3746.285. [DOI] [PubMed] [Google Scholar]
- 41.Singer HS, Giuliano JD, Hansen BH, et al. Antibodies against human putamen in children with Tourette syndrome. Neurology. 1998;50:1618–24. doi: 10.1212/wnl.50.6.1618. [DOI] [PubMed] [Google Scholar]
- 42.Pavone P, Bianchini R, Parano E, et al. Anti-brain antibodies in PANDAS versus uncomplicated streptococcal infection. Pediatr Neurol. 2004;30:107–10. doi: 10.1016/S0887-8994(03)00413-2. [DOI] [PubMed] [Google Scholar]
- 43.Kurlan R. Tourette's syndrome and ‘PANDAS’: will the relation bear out? Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infection. Neurology. 1998;50:1530–4. doi: 10.1212/wnl.50.6.1530. [DOI] [PubMed] [Google Scholar]
- 44.Snider LA, Swedo SE. Post-streptococcal autoimmune disorders of the central nervous system. Curr Opin Neurol. 2003;16:359–65. doi: 10.1097/01.wco.0000073938.19076.31. [DOI] [PubMed] [Google Scholar]
- 45.Bronze MS, Dale JB. Epitopes of streptococcal M proteins that evoke antibodies that cross-react with human brain. J Immunol. 1993;151:2820–8. [PubMed] [Google Scholar]
- 46.Husby G, van de Rijn I, Zabriskie JB, Abdin ZH, Williams RC., Jr. Antibodies reacting with cytoplasm of subthalamic and caudate nuclei neurons in chorea and acute rheumatic fever. J Exp Med. 1976;144:1094–110. doi: 10.1084/jem.144.4.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kirvan CA, Swedo SE, Heuser JS, Cunningham MW. Mimicry and autoantibody-mediated neuronal cell signaling in Sydenham chorea. Nat Med. 2003;9:914–20. doi: 10.1038/nm892. [DOI] [PubMed] [Google Scholar]
- 48.Kirvan CA, Swedo SE, Snider LA, Cunningham MW. Antibody-mediated neuronal cell signaling in behavior and movement disorders. J Neuroimmunol. 2006;179:173–9. doi: 10.1016/j.jneuroim.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 49.Kirvan CA, Cox CJ, Swedo SE, Cunningham MW. Tubulin is a neuronal target of autoantibodies in Sydenham's chorea. J Immunol. 2007;178:7412–21. doi: 10.4049/jimmunol.178.11.7412. [DOI] [PubMed] [Google Scholar]
- 50.Hoffman KL, Hornig M, Yaddanapudi K, Jabado O, Lipkin WI. A murine model for neuropsychiatric disorders associated with group A beta-hemolytic streptococcal infection. J Neurosci. 2004;24:1780–91. doi: 10.1523/JNEUROSCI.0887-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soares MB, Santos RR. Immunopathology of cardiomyopathy in the experimental Chagas disease. Mem Inst Oswaldo Cruz. 1999;94(Suppl. 1):257–62. doi: 10.1590/s0074-02761999000700043. [DOI] [PubMed] [Google Scholar]
- 52.Leon JS, Engman DM. The significance of autoimmunity in the pathogenesis of Chagas heart disease. Front Biosci. 2003;8:e315–22. doi: 10.2741/1023. [DOI] [PubMed] [Google Scholar]
- 53.Cunha-Neto E, Bilate AM, Hyland KV, Fonseca SG, Kalil J, Engman DM. Induction of cardiac autoimmunity in Chagas heart disease: a case for molecular mimicry. Autoimmunity. 2006;39:41–54. doi: 10.1080/08916930500485002. [DOI] [PubMed] [Google Scholar]
- 54.Sartori AM, Shikanai-Yasuda MA, Amato Neto V, Lopes MH. Follow-up of 18 patients with human immunodeficiency virus infection and chronic Chagas' disease, with reactivation of Chagas' disease causing cardiac disease in three patients. Clin Infect Dis. 1998;26:177–9. doi: 10.1086/516257. [DOI] [PubMed] [Google Scholar]
- 55.Pereira JB, Wilcox HP, Coura JR. The evolution of chronic chagasic cardiopathy. Rev Soc Bras Med Trop. 1992;25:101–8. doi: 10.1590/s0037-86821992000200003. I. The influence of parasitemia. [DOI] [PubMed] [Google Scholar]
- 56.Elias FE, Vigliano CA, Laguens RP, Levin MJ, Berek C. Analysis of the presence of Trypanosoma cruzi in the heart tissue of three patients with chronic Chagas' heart disease. Am J Trop Med Hyg. 2003;68:242–7. [PubMed] [Google Scholar]
- 57.Higuchi ML, De Morais CF, Pereira Barreto AC, et al. The role of active myocarditis in the development of heart failure in chronic Chagas' disease: a study based on endomyocardial biopsies. Clin Cardiol. 1987;10:665–70. doi: 10.1002/clc.4960101113. [DOI] [PubMed] [Google Scholar]
- 58.Abel LC, Rizzo LV, Ianni B, et al. Chronic Chagas' disease cardiomyopathy patients display an increased IFN-gamma response to Trypanosoma cruzi infection. J Autoimmun. 2001;17:99–107. doi: 10.1006/jaut.2001.0523. [DOI] [PubMed] [Google Scholar]
- 59.Reis MM, Higuchi Mde L, Benvenuti LA, et al. An in situ quantitative immunohistochemical study of cytokines and IL-2R+ in chronic human chagasic myocarditis: correlation with the presence of myocardial Trypanosoma cruzi antigens. Clin Immunol Immunopathol. 1997;83:165–72. doi: 10.1006/clin.1997.4335. [DOI] [PubMed] [Google Scholar]
- 60.Cunha-Neto E, Dzau VJ, Allen PD, et al. Cardiac gene expression profiling provides evidence for cytokinopathy as a molecular mechanism in Chagas' disease cardiomyopathy. Am J Pathol. 2005;167:305–13. doi: 10.1016/S0002-9440(10)62976-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Teixeira AR, Teixeira G, Macedo V, Prata A. Trypanosoma cruzi-sensitized T-lymphocyte mediated 51CR release from human heart cells in Chagas' disease. Am J Trop Med Hyg. 1978;27:1097–107. doi: 10.4269/ajtmh.1978.27.1097. [DOI] [PubMed] [Google Scholar]
- 62.Todd CW, Todd NR, Guimaraes AC. Do lymphocytes from Chagasic patients respond to heart antigens? Infect Immun. 1983;40:832–5. doi: 10.1128/iai.40.2.832-835.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mosca W, Plaja J, Hubsch R, Cedillos R. Longitudinal study of immune response in human Chagas' disease. J Clin Microbiol. 1985;22:438–41. doi: 10.1128/jcm.22.3.438-441.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gruber A, Zingales B. Trypanosoma cruzi: characterization of two recombinant antigens with potential application in the diagnosis of Chagas' disease. Exp Parasitol. 1993;76:1–12. doi: 10.1006/expr.1993.1001. [DOI] [PubMed] [Google Scholar]
- 65.Cunha-Neto E, Duranti M, Gruber A, et al. Autoimmunity in Chagas disease cardiopathy: biological relevance of a cardiac myosin-specific epitope crossreactive to an immunodominant Trypanosoma cruzi antigen. Proc Natl Acad Sci USA. 1995;92:3541–5. doi: 10.1073/pnas.92.8.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cunha-Neto E, Coelho V, Guilherme L, Fiorelli A, Stolf N, Kalil J. Autoimmunity in Chagas' disease. J Clin Invest. 1996;98:1709–12. doi: 10.1172/JCI118969. Identification of cardiac myosin-B13 Trypanosoma cruzi protein crossreactive T cell clones in heart lesions of a chronic Chagas' cardiomyopathy patient. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abel LC, Kalil J, Cunha Neto E. Molecular mimicry between cardiac myosin and Trypanosoma cruzi antigen B13: identification of a B13-driven human T cell clone that recognizes cardiac myosin. Braz J Med Biol Res. 1997;30:1305–8. doi: 10.1590/s0100-879x1997001100007. [DOI] [PubMed] [Google Scholar]
- 68.Lannes-Vieira J. Trypanosoma cruzi-elicited CD8+ T cell-mediated myocarditis: chemokine receptors and adhesion molecules as potential therapeutic targets to control chronic inflammation? Mem Inst Oswaldo Cruz. 2003;98:299–304. doi: 10.1590/s0074-02762003000300002. [DOI] [PubMed] [Google Scholar]
- 69.Rizzo LV, Cunha-Neto E, Teixeira AR. Autoimmunity in Chagas' disease: specific inhibition of reactivity of CD4+ T cells against myosin in mice chronically infected with Trypanosoma cruzi. Infect Immun. 1989;57:2640–4. doi: 10.1128/iai.57.9.2640-2644.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.dos Santos RR, Rossi MA, Laus JL, Silva JS, Savino W, Mengel J. Anti-CD4 abrogates rejection and reestablishes long-term tolerance to syngeneic newborn hearts grafted in mice chronically infected with Trypanosoma cruzi. J Exp Med. 1992;175:29–39. doi: 10.1084/jem.175.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ribeiro-Dos-Santos R, Mengel JO, Postol E, et al. A heart-specific CD4+ T-cell line obtained from a chronic chagasic mouse induces carditis in heart-immunized mice and rejection of normal heart transplants in the absence of Trypanosoma cruzi. Parasite Immunol. 2001;23:93–101. doi: 10.1046/j.1365-3024.2001.00368.x. [DOI] [PubMed] [Google Scholar]
- 72.Girones N, Rodriguez CI, Carrasco-Marin E, Hernaez RF, de Rego JL, Fresno M. Dominant T- and B-cell epitopes in an autoantigen linked to Chagas' disease. J Clin Invest. 2001;107:985–93. doi: 10.1172/JCI10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oliveira MF, Bijovsky AT, Carvalho TU, de Souza W, Alves MJ, Colli W. A monoclonal antibody to Trypanosoma cruzi trypomastigotes recognizes a myosin tail epitope. Parasitol Res. 2001;87:1043–9. doi: 10.1007/s004360100465. [DOI] [PubMed] [Google Scholar]
- 74.Giordanengo L, Guinazu N, Stempin C, Fretes R, Cerban F, Gea S. Cruzipain, a major Trypanosoma cruzi antigen, conditions the host immune response in favor of parasite. Eur J Immunol. 2002;32:1003–11. doi: 10.1002/1521-4141(200204)32:4<1003::AID-IMMU1003>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 75.McCormick TS, Rowland EC. Trypanosoma cruzi: cross-reactive anti-heart autoantibodies produced during infection in mice. Exp Parasitol. 1989;69:393–401. doi: 10.1016/0014-4894(89)90088-x. [DOI] [PubMed] [Google Scholar]
- 76.Leon JS, Daniels MD, Toriello KM, Wang K, Engman DM. A cardiac myosin-specific autoimmune response is induced by immunization with Trypanosoma cruzi proteins. Infect Immun. 2004;72:3410–7. doi: 10.1128/IAI.72.6.3410-3417.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Leon JS, Godsel LM, Wang K, Engman DM. Cardiac myosin autoimmunity in acute Chagas' heart disease. Infect Immun. 2001;69:5643–9. doi: 10.1128/IAI.69.9.5643-5649.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meneghelli UG. Chagasic enteropathy. Rev Soc Bras Med Trop. 2004;37:252–60. doi: 10.1590/s0037-86822004000300012. [DOI] [PubMed] [Google Scholar]
- 79.Van Voorhis WC, Eisen H. FL-160: a surface antigen of Trypanosoma cruzi that mimics mammalian nervous tissue. J Exp Med. 1989;169:641–52. doi: 10.1084/jem.169.3.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Van Voorhis WC, Barrett L, Koelling R, Farr AG. FL-160 proteins of Trypanosoma cruzi are expressed from a multigene family and contain two distinct epitopes that mimic nervous tissues. J Exp Med. 1993;178:681–94. doi: 10.1084/jem.178.2.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Van Voorhis WC, Schlekewy L, Trong HL. Molecular mimicry by Trypanosoma cruzi: the F1-160 epitope that mimics mammalian nerve can be mapped to a 12-amino acid peptide. Proc Natl Acad Sci USA. 1991;88:5993–7. doi: 10.1073/pnas.88.14.5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Petry K, Eisen H. Chemical characterization of epitopes common to Trypanosoma cruzi and mammalian nervous cells. Mem Inst Oswaldo Cruz. 1988;83(Suppl. 1):498–501. doi: 10.1590/s0074-02761988000500057. [DOI] [PubMed] [Google Scholar]
- 83.Gea S, Ordonez P, Cerban F, Iosa D, Chizzolini C, Vottero-Cima E. Chagas' disease cardioneuropathy: association of anti-Trypanosoma cruzi and anti-sciatic nerve antibodies. Am J Trop Med Hyg. 1993;49:581–8. doi: 10.4269/ajtmh.1993.49.581. [DOI] [PubMed] [Google Scholar]
- 84.Steere AC, Schoen RT, Taylor E. The clinical evolution of Lyme arthritis. Ann Intern Med. 1987;107:725–31. doi: 10.7326/0003-4819-107-5-725. [DOI] [PubMed] [Google Scholar]
- 85.Nocton JJ, Dressler F, Rutledge BJ, Rys PN, Persing DH, Steere AC. Detection of Borrelia burgdorferi DNA by polymerase chain reaction in synovial fluid from patients with Lyme arthritis. N Engl J Med. 1994;330:229–34. doi: 10.1056/NEJM199401273300401. [DOI] [PubMed] [Google Scholar]
- 86.Steere AC, Levin RE, Molloy PJ, Kalish RA, Abraham JH, 3rd, Liu NY, Schmid CH. Treatment of Lyme arthritis. Arthritis Rheum. 1994;37:878–88. doi: 10.1002/art.1780370616. [DOI] [PubMed] [Google Scholar]
- 87.Carlson D, Hernandez J, Bloom BJ, Coburn J, Aversa JM, Steere AC. Lack of Borrelia burgdorferi DNA in synovial samples from patients with antibiotic treatment-resistant Lyme arthritis. Arthritis Rheum. 1999;42:2705–9. doi: 10.1002/1529-0131(199912)42:12<2705::AID-ANR29>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 88.Malawista SE. Resolution of Lyme arthritis, acute or prolonged: a new look. Inflammation. 2000;24:493–504. doi: 10.1023/a:1007079705485. [DOI] [PubMed] [Google Scholar]
- 89.Chen J, Field JA, Glickstein L, Molloy PJ, Huber BT, Steere AC. Association of antibiotic treatment-resistant Lyme arthritis with T cell responses to dominant epitopes of outer surface protein A of Borrelia burgdorferi. Arthritis Rheum. 1999;42:1813–22. doi: 10.1002/1529-0131(199909)42:9<1813::AID-ANR4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 90.Lengl-Janssen B, Strauss AF, Steere AC, Kamradt T. The T helper cell response in Lyme arthritis: differential recognition of Borrelia burgdorferi outer surface protein A in patients with treatment-resistant or treatment-responsive Lyme arthritis. J Exp Med. 1994;180:2069–78. doi: 10.1084/jem.180.6.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kalish RA, Leong JM, Steere AC. Association of treatment-resistant chronic Lyme arthritis with HLA-DR4 and antibody reactivity to OspA and OspB of Borrelia burgdorferi. Infect Immun. 1993;61:2774–9. doi: 10.1128/iai.61.7.2774-2779.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kalish RA, Leong JM, Steere AC. Early and late antibody responses to full-length and truncated constructs of outer surface protein A of Borrelia burgdorferi in Lyme disease. Infect Immun. 1995;63:2228–35. doi: 10.1128/iai.63.6.2228-2235.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Akin E, McHugh GL, Flavell RA, Fikrig E, Steere AC. The immunoglobulin (IgG) antibody response to OspA and OspB correlates with severe and prolonged Lyme arthritis and the IgG response to P35 correlates with mild and brief arthritis. Infect Immun. 1999;67:173–81. doi: 10.1128/iai.67.1.173-181.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Meyer AL, Trollmo C, Crawford F, et al. Direct enumeration of Borrelia-reactive CD4 T cells ex vivo by using MHC class II tetramers. Proc Natl Acad Sci USA. 2000;97:11433–8. doi: 10.1073/pnas.190335897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Steere AC, Gross D, Meyer AL, Huber BT. Autoimmune mechanisms in antibiotic treatment-resistant lyme arthritis. J Autoimmun. 2001;16:263–8. doi: 10.1006/jaut.2000.0495. [DOI] [PubMed] [Google Scholar]
- 96.Gross DM, Forsthuber T, Tary-Lehmann M, et al. Identification of LFA-1 as a candidate autoantigen in treatment-resistant Lyme arthritis. Science. 1998;281:703–6. doi: 10.1126/science.281.5377.703. [DOI] [PubMed] [Google Scholar]
- 97.Akin E, Aversa J, Steere AC. Expression of adhesion molecules in synovia of patients with treatment-resistant lyme arthritis. Infect Immun. 2001;69:1774–80. doi: 10.1128/IAI.69.3.1774-1780.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Trollmo C, Meyer AL, Steere AC, Hafler DA, Huber BT. Molecular mimicry in Lyme arthritis demonstrated at the single cell level: LFA-1 alpha L is a partial agonist for outer surface protein A-reactive T cells. J Immunol. 2001;166:5286–91. doi: 10.4049/jimmunol.166.8.5286. [DOI] [PubMed] [Google Scholar]
- 99.Steere AC, Falk B, Drouin EE, Baxter-Lowe LA, Hammer J, Nepom GT. Binding of outer surface protein A and human lymphocyte function-associated antigen 1 peptides to HLA-DR molecules associated with antibiotic treatment-resistant Lyme arthritis. Arthritis Rheum. 2003;48:534–40. doi: 10.1002/art.10772. [DOI] [PubMed] [Google Scholar]
- 100.Kalish RS, Wood JA, Golde W, et al. Human T lymphocyte response to Borrelia burgdorferi infection: no correlation between human leukocyte function antigen type 1 peptide response and clinical status. J Infect Dis. 2003;187:102–8. doi: 10.1086/346059. [DOI] [PubMed] [Google Scholar]
- 101.Maier B, Molinger M, Cope AP, et al. Multiple cross-reactive self-ligands for Borrelia burgdorferi-specific HLA-DR4-restricted T cells. Eur J Immunol. 2000;30:448–57. doi: 10.1002/1521-4141(200002)30:2<448::AID-IMMU448>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 102.Barthold SW, Moody KD, Terwilliger GA, Duray PH, Jacoby RO, Steere AC. Experimental Lyme arthritis in rats infected with Borrelia burgdorferi. J Infect Dis. 1988;157:842–6. doi: 10.1093/infdis/157.4.842. [DOI] [PubMed] [Google Scholar]
- 103.Barthold SW, Beck DS, Hansen GM, Terwilliger GA, Moody KD. Lyme borreliosis in selected strains and ages of laboratory mice. J Infect Dis. 1990;162:133–8. doi: 10.1093/infdis/162.1.133. [DOI] [PubMed] [Google Scholar]
- 104.Ma Y, Seiler KP, Eichwald EJ, Weis JH, Teuscher C, Weis JJ. Distinct characteristics of resistance to Borrelia burgdorferi-induced arthritis in C57BL/6N mice. Infect Immun. 1998;66:161–8. doi: 10.1128/iai.66.1.161-168.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schaible UE, Kramer MD, Museteanu C, Zimmer G, Mossmann H, Simon MM. The severe combined immunodeficiency (scid) mouse. A laboratory model for the analysis of Lyme arthritis and carditis. J Exp Med. 1989;170:1427–32. doi: 10.1084/jem.170.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Barthold SW, de Souza M. Exacerbation of Lyme arthritis in beige mice. J Infect Dis. 1995;172:778–84. doi: 10.1093/infdis/172.3.778. [DOI] [PubMed] [Google Scholar]
- 107.Anguita J, Rincon M, Samanta S, Barthold SW, Flavell RA, Fikrig E. Borrelia burgdorferi-infected, interleukin-6-deficient mice have decreased Th2 responses and increased lyme arthritis. J Infect Dis. 1998;178:1512–5. doi: 10.1086/314448. [DOI] [PubMed] [Google Scholar]
- 108.Brown JP, Zachary JF, Teuscher C, Weis JJ, Wooten RM. Dual role of interleukin-10 in murine Lyme disease: regulation of arthritis severity and host defense. Infect Immun. 1999;67:5142–50. doi: 10.1128/iai.67.10.5142-5150.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McKisic MD, Redmond WL, Barthold SW. Cutting edge: T cell-mediated pathology in murine Lyme borreliosis. J Immunol. 2000;164:6096–9. doi: 10.4049/jimmunol.164.12.6096. [DOI] [PubMed] [Google Scholar]
- 110.Sigal LH. Lyme disease: a review of aspects of its immunology and immunopathogenesis. Annu Rev Immunol. 1997;15:63–92. doi: 10.1146/annurev.immunol.15.1.63. [DOI] [PubMed] [Google Scholar]
- 111.Sigal LH, Tatum AH. Lyme disease patients' serum contains IgM antibodies to Borrelia burgdorferi that cross-react with neuronal antigens. Neurology. 1988;38:1439–42. doi: 10.1212/wnl.38.9.1439. [DOI] [PubMed] [Google Scholar]
- 112.Wang WZ, Fredrikson S, Sun JB, Link H. Lyme neuroborreliosis: evidence for persistent up-regulation of Borrelia burgdorferi-reactive cells secreting interferon-gamma. Scand J Immunol. 1995;42:694–700. doi: 10.1111/j.1365-3083.1995.tb03713.x. [DOI] [PubMed] [Google Scholar]
- 113.Garcia-Monco JC, Coleman JL, Benach JL. Antibodies to myelin basic protein in Lyme disease. J Infect Dis. 1988;158:667–8. doi: 10.1093/infdis/158.3.667. [DOI] [PubMed] [Google Scholar]
- 114.Sigal LH. Cross-reactivity between Borrelia burgdorferi flagellin and a human axonal 64,000 molecular weight protein. J Infect Dis. 1993;167:1372–8. doi: 10.1093/infdis/167.6.1372. [DOI] [PubMed] [Google Scholar]
- 115.Dai Z, Lackland H, Stein S, et al. Molecular mimicry in Lyme disease: monoclonal antibody H9724 to B. Biochim Biophys Acta. 1993;1181:97–100. doi: 10.1016/0925-4439(93)90096-j. burgdorferi flagellin specifically detects chaperonin-HSP60. [DOI] [PubMed] [Google Scholar]
- 116.Fikrig E, Berland R, Chen M, Williams S, Sigal LH, Flavell RA. Serologic response to the Borrelia burgdorferi flagellin demonstrates an epitope common to a neuroblastoma cell line. Proc Natl Acad Sci USA. 1993;90:183–7. doi: 10.1073/pnas.90.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sigal LH, Williams S. A monoclonal antibody to Borrelia burgdorferi flagellin modifies neuroblastoma cell neuritogenesis in vitro: a possible role for autoimmunity in the neuropathy of Lyme disease. Infect Immun. 1997;65:1722–8. doi: 10.1128/iai.65.5.1722-1728.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Alaedini A, Latov N. Antibodies against OspA epitopes of Borrelia burgdorferi cross-react with neural tissue. J Neuroimmunol. 2005;159:192–5. doi: 10.1016/j.jneuroim.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 119.Hemmer B, Gran B, Zhao Y, et al. Identification of candidate T-cell epitopes and molecular mimics in chronic Lyme disease. Nat Med. 1999;5:1375–82. doi: 10.1038/70946. [DOI] [PubMed] [Google Scholar]
- 120.Pachner AR, Cadavid D, Shu G, et al. Central and peripheral nervous system infection, immunity, and inflammation in the NHP model of Lyme borreliosis. Ann Neurol. 2001;50:330–8. [PubMed] [Google Scholar]
- 121.Ramesh G, Alvarez AL, Roberts ED, et al. Pathogenesis of Lyme neuroborreliosis: Borrelia burgdorferi lipoproteins induce both proliferation and apoptosis in rhesus monkey astrocytes. Eur J Immunol. 2003;33:2539–50. doi: 10.1002/eji.200323872. [DOI] [PubMed] [Google Scholar]
- 122.Steiner I. Treating post Lyme disease: trying to solve one equation with too many unknowns. Neurology. 2003;60:1888–9. doi: 10.1212/01.wnl.0000077046.67754.60. [DOI] [PubMed] [Google Scholar]
- 123.Liesegang TJ. Epidemiology of ocular herpes simplex. Arch Ophthalmol. 1989;107:1160–5. doi: 10.1001/archopht.1989.01070020226030. Natural history in Rochester, Minn, 1950 through 1982. [DOI] [PubMed] [Google Scholar]
- 124.Liesegang TJ. Classification of herpes simplex virus keratitis and anterior uveitis. Cornea. 1999;18:127–43. doi: 10.1097/00003226-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 125.Heiligenhaus A, Steuhl KP. Treatment of HSV-1 stromal keratitis with topical cyclosporin A: a pilot study. Graefes Arch Clin Exp Ophthalmol. 1999;237:435–8. doi: 10.1007/s004170050257. [DOI] [PubMed] [Google Scholar]
- 126.Fenton RR, Molesworth-Kenyon S, Oakes JE, Lausch RN. Linkage of IL-6 with neutrophil chemoattractant expression in virus-induced ocular inflammation. Invest Ophthalmol Vis Sci. 2002;43:737–43. [PubMed] [Google Scholar]
- 127.Lausch RN, Chen SH, Tumpey TM, Su YH, Oakes JE. Early cytokine synthesis in the excised mouse cornea. J Interferon Cytokine Res. 1996;16:35–40. doi: 10.1089/jir.1996.16.35. [DOI] [PubMed] [Google Scholar]
- 128.Staats HF, Lausch RN. Cytokine expression in vivo during murine herpetic stromal keratitis. Effect of protective antibody therapy. J Immunol. 1993;151:277–83. [PubMed] [Google Scholar]
- 129.Tumpey TM, Chen SH, Oakes JE, Lausch RN. Neutrophil-mediated suppression of virus replication after herpes simplex virus type 1 infection of the murine cornea. J Virol. 1996;70:898–904. doi: 10.1128/jvi.70.2.898-904.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Thomas J, Gangappa S, Kanangat S, Rouse BT. On the essential involvement of neutrophils in the immunopathologic disease: herpetic stromal keratitis. J Immunol. 1997;158:1383–91. [PubMed] [Google Scholar]
- 131.Hendricks RL, Tumpey TM, Finnegan A. IFN-gamma and IL-2 are protective in the skin but pathologic in the corneas of HSV-1-infected mice. J Immunol. 1992;149:3023–8. [PubMed] [Google Scholar]
- 132.Bauer D, Mrzyk S, Van Rooijen N, Steuhl KP, Heiligenhaus A. Incidence and severity of herpetic stromal keratitis: impaired by the depletion of lymph node macrophages. Exp Eye Res. 2001;72:261–9. doi: 10.1006/exer.2000.0947. [DOI] [PubMed] [Google Scholar]
- 133.Habu S, Akamatsu K, Tamaoki N, Okumura K. In vivo significance of NK cell on resistance against virus (HSV-1) infections in mice. J Immunol. 1984;133:2743–7. [PubMed] [Google Scholar]
- 134.Zisman B, Hirsch MS, Allison AC. Selective effects of anti-macrophage serum, silica and anti-lymphocyte serum on pathogenesis of herpes virus infection of young adult mice. J Immunol. 1970;104:1155–9. [PubMed] [Google Scholar]
- 135.Bouley DM, Kanangat S, Wire W, Rouse BT. Characterization of herpes simplex virus type-1 infection and herpetic stromal keratitis development in IFN-gamma knockout mice. J Immunol. 1995;155:3964–71. [PubMed] [Google Scholar]
- 136.Tang Q, Hendricks RL. Interferon gamma regulates platelet endothelial cell adhesion molecule 1 expression and neutrophil infiltration into herpes simplex virus-infected mouse corneas. J Exp Med. 1996;184:1435–47. doi: 10.1084/jem.184.4.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Streilein JW, Dana MR, Ksander BR. Immunity causing blindness: five different paths to herpes stromal keratitis. Immunol Today. 1997;18:443–9. doi: 10.1016/s0167-5699(97)01114-6. [DOI] [PubMed] [Google Scholar]
- 138.Halford WP, Gebhardt BM, Carr DJ. Persistent cytokine expression in trigeminal ganglion latently infected with herpes simplex virus type 1. J Immunol. 1996;157:3542–9. [PubMed] [Google Scholar]
- 139.Gangappa S, Babu JS, Thomas J, Daheshia M, Rouse BT. Virus-induced immunoinflammatory lesions in the absence of viral antigen recognition. J Immunol. 1998;161:4289–300. [PubMed] [Google Scholar]
- 140.Maggs DJ, Chang E, Nasisse MP, Mitchell WJ. Persistence of herpes simplex virus type 1 DNA in chronic conjunctival and eyelid lesions of mice. J Virol. 1998;72:9166–72. doi: 10.1128/jvi.72.11.9166-9172.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lundberg P, Welander P, Han X, Cantin E. Herpes simplex virus type 1 DNA is immunostimulatory in vitro and in vivo. J Virol. 2003;77:11158–69. doi: 10.1128/JVI.77.20.11158-11169.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhao ZS, Granucci F, Yeh L, Schaffer PA, Cantor H. Molecular mimicry by herpes simplex virus-type 1: autoimmune disease after viral infection. Science. 1998;279:1344–7. doi: 10.1126/science.279.5355.1344. [DOI] [PubMed] [Google Scholar]
- 143.Panoutsakopoulou V, Sanchirico ME, Huster KM, et al. Analysis of the relationship between viral infection and autoimmune disease. Immun. 2001;15:137–47. doi: 10.1016/s1074-7613(01)00172-8. [DOI] [PubMed] [Google Scholar]
- 144.Verjans GM, Remeijer L, Mooy CM, Osterhaus AD. Herpes simplex virus-specific T cells infiltrate the cornea of patients with herpetic stromal keratitis: no evidence for autoreactive T cells. Invest Ophthalmol Vis Sci. 2000;41:2607–12. [PubMed] [Google Scholar]
- 145.Koelle DM, Reymond SN, Chen H, et al. Tegument-specific, virus-reactive CD4 T cells localize to the cornea in herpes simplex virus interstitial keratitis in humans. J Virol. 2000;74:10930–8. doi: 10.1128/jvi.74.23.10930-10938.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Babu JS, Thomas J, Kanangat S, Morrison LA, Knipe DM, Rouse BT. Viral replication is required for induction of ocular immunopathology by herpes simplex virus. J Virol. 1996;70:101–7. doi: 10.1128/jvi.70.1.101-107.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Durrani OM, Meads CA, Murray PI. Uveitis: a potentially blinding disease. Ophthalmologica. 2004;218:223–36. doi: 10.1159/000078612. [DOI] [PubMed] [Google Scholar]
- 148.Martin TM, Kurz DE, Rosenbaum JT. Genetics of uveitis. Ophthalmol Clin North Am. 2003;16:555–65. doi: 10.1016/s0896-1549(03)00071-3. [DOI] [PubMed] [Google Scholar]
- 149.de Smet MD, Yamamoto JH, Mochizuki M, et al. Cellular immune responses of patients with uveitis to retinal antigens and their fragments. Am J Ophthalmol. 1990;110:135–42. doi: 10.1016/s0002-9394(14)76981-8. [DOI] [PubMed] [Google Scholar]
- 150.Doekes G, van der Gaag R, Rothova A, et al. Humoral and cellular immune responsiveness to human S-antigen in uveitis. Curr Eye Res. 1987;6:909–19. doi: 10.3109/02713688709034859. [DOI] [PubMed] [Google Scholar]
- 151.Singh VK, Yamaki K, Abe T, Shinohara T. Molecular mimicry between uveitopathogenic site of retinal S-antigen and Escherichia coli protein: induction of experimental autoimmune uveitis and lymphocyte cross-reaction. Cell Immunol. 1989;122:262–73. doi: 10.1016/0008-8749(89)90166-4. [DOI] [PubMed] [Google Scholar]
- 152.Singh VK, Kalra HK, Yamaki K, Abe T, Donoso LA, Shinohara T. Molecular mimicry between a uveitopathogenic site of S-antigen and viral peptides. J Immunol. 1990;144:1282–7. Induction of experimental autoimmune uveitis in Lewis rats. [PubMed] [Google Scholar]
- 153.Wildner G, Diedrichs-Mohring M. Autoimmune uveitis induced by molecular mimicry of peptides from rotavirus, bovine casein and retinal S-antigen. Eur J Immunol. 2003;33:2577–87. doi: 10.1002/eji.200324058. [DOI] [PubMed] [Google Scholar]
- 154.Lashkevich VA, Koroleva GA, Lukashev AN, Denisova EV, Katargina LA. Enterovirus uveitis. Rev Med Virol. 2004;14:241–54. doi: 10.1002/rmv.433. [DOI] [PubMed] [Google Scholar]
- 155.Kaprio J, Tuomilehto J, Koskenvuo M, et al. Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia. 1992;35:1060–7. doi: 10.1007/BF02221682. [DOI] [PubMed] [Google Scholar]
- 156.Kyvik KO, Green A, Beck-Nielsen H. Concordance rates of insulin dependent diabetes mellitus: a population based study of young Danish twins. BMJ. 1995;311:913–7. doi: 10.1136/bmj.311.7010.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jun HS, Yoon JW. A new look at viruses in type 1 diabetes. Diabetes Metab Res Rev. 2003;19:8–31. doi: 10.1002/dmrr.337. [DOI] [PubMed] [Google Scholar]
- 158.McIntosh ED, Menser MA. A fifty-year follow-up of congenital rubella. Lancet. 1992;340:414–5. doi: 10.1016/0140-6736(92)91483-o. [DOI] [PubMed] [Google Scholar]
- 159.Ginsberg-Fellner F, Witt ME, Yagihashi S, et al. Congenital rubella syndrome as a model for type 1 (insulin-dependent) diabetes mellitus: increased prevalence of islet cell surface antibodies. Diabetologia. 1984;27(Suppl.):87–9. doi: 10.1007/BF00275655. [DOI] [PubMed] [Google Scholar]
- 160.Pak CY, Eun HM, McArthur RG, Yoon JW. Association of cytomegalovirus infection with autoimmune type 1 diabetes. Lancet. 1988;2:1–4. doi: 10.1016/s0140-6736(88)92941-8. [DOI] [PubMed] [Google Scholar]
- 161.Pak CY, Cha CY, Rajotte RV, McArthur RG, Yoon JW. Human pancreatic islet cell specific 38 kilodalton autoantigen identified by cytomegalovirus-induced monoclonal islet cell autoantibody. Diabetologia. 1990;33:569–72. doi: 10.1007/BF00404146. [DOI] [PubMed] [Google Scholar]
- 162.Gamble DR. Relation of antecedent illness to development of diabetes in children. BMJ. 1980;281:99–101. doi: 10.1136/bmj.281.6233.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Helmke K, Otten A, Willems W. Islet cell antibodies in children with mumps infection. Lancet. 1980;2:211–2. doi: 10.1016/s0140-6736(80)90105-1. [DOI] [PubMed] [Google Scholar]
- 164.Frisk G, Fohlman J, Kobbah M, et al. High frequency of Coxsackie-B-virus-specific IgM in children developing type I diabetes during a period of high diabetes morbidity. J Med Virol. 1985;17:219–27. doi: 10.1002/jmv.1890170303. [DOI] [PubMed] [Google Scholar]
- 165.Hyoty H, Hiltunen M, Knip M, et al. A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Diabetes. 1995;44:652–7. doi: 10.2337/diab.44.6.652. Childhood Diabetes in Finland (DiMe) Study Group. [DOI] [PubMed] [Google Scholar]
- 166.Schloot NC, Willemen SJ, Duinkerken G, Drijfhout JW, de Vries RR, Roep BO. Molecular mimicry in type 1 diabetes mellitus revisited: T-cell clones to GAD65 peptides with sequence homology to Coxsackie or proinsulin peptides do not crossreact with homologous counterpart. Hum Immunol. 2001;62:299–309. doi: 10.1016/s0198-8859(01)00223-3. [DOI] [PubMed] [Google Scholar]
- 167.Sarugeri E, Dozio N, Meschi F, Pastore MR, Bonifacio E. T cell responses to type 1 diabetes related peptides sharing homologous regions. J Mol Med. 2001;79:213–20. doi: 10.1007/s001090100194. [DOI] [PubMed] [Google Scholar]
- 168.Ou D, Mitchell LA, Metzger DL, Gillam S, Tingle AJ. Cross-reactive rubella virus and glutamic acid decarboxylase (65 and 67) protein determinants recognised by T cells of patients with type I diabetes mellitus. Diabetologia. 2000;43:750–62. doi: 10.1007/s001250051373. [DOI] [PubMed] [Google Scholar]
- 169.Karounos DG, Wolinsky JS, Thomas JW. Monoclonal antibody to rubella virus capsid protein recognizes a beta-cell antigen. J Immunol. 1993;150:3080–5. [PubMed] [Google Scholar]
- 170.Hiemstra HS, Schloot NC, van Veelen PA, et al. Cytomegalovirus in autoimmunity: T cell crossreactivity to viral antigen and autoantigen glutamic acid decarboxylase. Proc Natl Acad Sci USA. 2001;98:3988–91. doi: 10.1073/pnas.071050898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Cavallo MG, Baroni MG, Toto A, et al. Viral infection induces cytokine release by beta islet cells. Immunology. 1992;75:664–8. [PMC free article] [PubMed] [Google Scholar]
- 172.Prince GA, Jenson AB, Billups LC, Notkins AL. Infection of human pancreatic beta cell cultures with mumps virus. Nature. 1978;271:158–61. doi: 10.1038/271158a0. [DOI] [PubMed] [Google Scholar]
- 173.Parkkonen P, Hyoty H, Koskinen L, Leinikki P. Mumps virus infects beta cells in human fetal islet cell cultures upregulating the expression of HLA class I molecules. Diabetologia. 1992;35:63–9. doi: 10.1007/BF00400853. [DOI] [PubMed] [Google Scholar]
- 174.Vuorinen T, Nikolakaros G, Simell O, Hyypia T, Vainionpaa R. Mumps and Coxsackie B3 virus infection of human fetal pancreatic islet-like cell clusters. Pancreas. 1992;7:460–4. doi: 10.1097/00006676-199207000-00007. [DOI] [PubMed] [Google Scholar]
- 175.Tisch R, Yang XD, Singer SM, Liblau RS, Fugger L, McDevitt HO. Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic mice. Nature. 1993;366:72–5. doi: 10.1038/366072a0. [DOI] [PubMed] [Google Scholar]
- 176.Tian J, Lehmann PV, Kaufman DL. T cell cross-reactivity between coxsackievirus and glutamate decarboxylase is associated with a murine diabetes susceptibility allele. J Exp Med. 1994;180:1979–84. doi: 10.1084/jem.180.5.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lawson CM. Evidence for mimicry by viral antigens in animal models of autoimmune disease including myocarditis. Cell Mol Life Sci. 2000;57:552–60. doi: 10.1007/PL00000717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Serreze DV, Ottendorfer EW, Ellis TM, Gauntt CJ, Atkinson MA. Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T-cells in pancreatic islets. Diabetes. 2000;49:708–11. doi: 10.2337/diabetes.49.5.708. [DOI] [PubMed] [Google Scholar]
- 179.Horwitz MS, Ilic A, Fine C, Rodriguez E, Sarvetnick N. Presented antigen from damaged pancreatic beta cells activates autoreactive T cells in virus-mediated autoimmune diabetes. J Clin Invest. 2002;109:79–87. doi: 10.1172/JCI11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Guberski DL, Thomas VA, Shek WR, et al. Induction of type I diabetes by Kilham's rat virus in diabetes-resistant BB/Wor rats. Science. 1991;254:1010–3. doi: 10.1126/science.1658938. [DOI] [PubMed] [Google Scholar]
- 181.Chung YH, Jun HS, Kang Y, et al. Role of macrophages and macrophage-derived cytokines in the pathogenesis of Kilham rat virus-induced autoimmune diabetes in diabetes-resistant BioBreeding rats. J Immunol. 1997;159:466–71. [PubMed] [Google Scholar]
- 182.Ellerman KE, Richards CA, Guberski DL, Shek WR, Like AA. Kilham rat triggers T-cell-dependent autoimmune diabetes in multiple strains of rat. Diabetes. 1996;45:557–62. doi: 10.2337/diab.45.5.557. [DOI] [PubMed] [Google Scholar]
- 183.Yoon JW, London WT, Curfman BL, Brown RL, Notkins AL. Coxsackie virus B4 produces transient diabetes in nonhuman primates. Diabetes. 1986;35:712–6. doi: 10.2337/diab.35.6.712. [DOI] [PubMed] [Google Scholar]
- 184.Willison HJ, Yuki N. Peripheral neuropathies and anti-glycolipid antibodies. Brain. 2002;125:2591–625. doi: 10.1093/brain/awf272. [DOI] [PubMed] [Google Scholar]
- 185.Ilyas AA, Willison HJ, Quarles RH, et al. Serum antibodies to gangliosides in Guillain–Barré syndrome. Ann Neurol. 1988;23:440–7. doi: 10.1002/ana.410230503. [DOI] [PubMed] [Google Scholar]
- 186.Joseph SA, Tsao CY. Guillain–Barré syndrome. Adolesc Med. 2002;13:487–94. [PubMed] [Google Scholar]
- 187.Aspinall GO, Fujimoto S, McDonald AG, Pang H, Kurjanczyk LA, Penner JL. Lipopolysaccharides from Campylobacter jejuni associated with Guillain–Barré syndrome patients mimic human gangliosides in structure. Infect Immun. 1994;62:2122–5. doi: 10.1128/iai.62.5.2122-2125.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yuki N, Taki T, Takahashi M, et al. Penner's serotype 4 of Campylobacter jejuni has a lipopolysaccharide that bears a GM1 ganglioside epitope as well as one that bears a GD1 a epitope. Infect Immun. 1994;62:2101–3. doi: 10.1128/iai.62.5.2101-2103.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ang CW, Endtz HP, Jacobs BC, et al. Campylobacter jejuni lipopolysaccharides from Guillain–Barré syndrome patients induce IgG anti-GM1 antibodies in rabbits. J Neuroimmunol. 2000;104:133–8. doi: 10.1016/s0165-5728(99)00279-9. [DOI] [PubMed] [Google Scholar]
- 190.Goodyear CS, O'Hanlon GM, Plomp JJ, et al. Monoclonal antibodies raised against Guillain–Barré syndrome-associated Campylobacter jejuni lipopolysaccharides react with neuronal gangliosides and paralyze muscle-nerve preparations. J Clin Invest. 1999;104:697–708. doi: 10.1172/JCI6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Wirguin I, Briani C, Suturkova-Milosevic L, et al. Induction of anti-GM1 ganglioside antibodies by Campylobacter jejuni lipopolysaccharides. J Neuroimmunol. 1997;78:138–42. doi: 10.1016/s0165-5728(97)00095-7. [DOI] [PubMed] [Google Scholar]
- 192.Ang CW, Laman JD, Willison HJ, et al. Structure of Campylobacter jejuni lipopolysaccharides determines antiganglioside specificity and clinical features of Guillain–Barré and Miller Fisher patients. Infect Immun. 2002;70:1202–8. doi: 10.1128/IAI.70.3.1202-1208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Nachamkin I, Liu J, Li M, et al. Campylobacter jejuni from patients with Guillain–Barré syndrome preferentially expresses a GD(1a)-like epitope. Infect Immun. 2002;70:5299–303. doi: 10.1128/IAI.70.9.5299-5303.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Yuki N, Susuki K, Koga M, et al. Carbohydrate mimicry between human ganglioside GM1 and Campylobacter jejuni lipooligosaccharide causes Guillain–Barré syndrome. Proc Natl Acad Sci USA. 2004;101:11404–9. doi: 10.1073/pnas.0402391101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kusunoki S, Shiina M, Kanazawa I. Anti-Gal-C antibodies in GBS subsequent to mycoplasma infection: evidence of molecular mimicry. Neurology. 2001;57:736–8. doi: 10.1212/wnl.57.4.736. [DOI] [PubMed] [Google Scholar]
- 196.Ang CW, Tio-Gillen AP, Groen J, et al. Cross-reactive anti-galactocerebroside antibodies and Mycoplasma pneumoniae infections in Guillain–Barré syndrome. J Neuroimmunol. 2002;130:179–83. doi: 10.1016/s0165-5728(02)00209-6. [DOI] [PubMed] [Google Scholar]
- 197.Susuki K, Odaka M, Mori M, Hirata K, Yuki N. Acute motor axonal neuropathy after Mycoplasma infection: evidence of molecular mimicry. Neurology. 2004;62:949–56. doi: 10.1212/01.wnl.0000115123.42929.fd. [DOI] [PubMed] [Google Scholar]
- 198.Koga M, Yuki N, Tai T, Hirata K. Miller Fisher syndrome and Haemophilus influenzae infection. Neurology. 2001;57:686–91. doi: 10.1212/wnl.57.4.686. [DOI] [PubMed] [Google Scholar]
- 199.Mori M, Kuwabara S, Miyake M, et al. Haemophilus influenzae infection and Guillain–Barré syndrome. Brain. 2000;123:2171–8. doi: 10.1093/brain/123.10.2171. [DOI] [PubMed] [Google Scholar]
- 200.Mori M, Kuwabara S, Miyake M, et al. Haemophilus influenzae has a GM1 ganglioside-like structure and elicits Guillain–Barré syndrome. Neurology. 1999;52:1282–4. doi: 10.1212/wnl.52.6.1282. [DOI] [PubMed] [Google Scholar]
- 201.Steinman L. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell. 1996;85:299–302. doi: 10.1016/s0092-8674(00)81107-1. [DOI] [PubMed] [Google Scholar]
- 202.Sun JB, Olsson T, Wang WZ, et al. Autoreactive T and B cells responding to myelin proteolipid protein in multiple sclerosis and controls. Eur J Immunol. 1991;21:1461–8. doi: 10.1002/eji.1830210620. [DOI] [PubMed] [Google Scholar]
- 203.Ota K, Matsui M, Milford EL, Mackin GA, Weiner HL, Hafler DA. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature. 1990;346:183–7. doi: 10.1038/346183a0. [DOI] [PubMed] [Google Scholar]
- 204.Bernard CC, de Rosbo NK. Immunopathological recognition of autoantigens in multiple sclerosis. Acta Neurol (Napoli) 1991;13:171–8. [PubMed] [Google Scholar]
- 205.Olson JK, Croxford JL, Miller SD. Virus-induced autoimmunity: potential role of viruses in initiation, perpetuation, and progression of T-cell-mediated autoimmune disease. Viral Immunol. 2001;14:227–50. doi: 10.1089/088282401753266756. [DOI] [PubMed] [Google Scholar]
- 206.Fujinami RS. Can virus infections trigger autoimmune disease? J Autoimmun. 2001;16:229–34. doi: 10.1006/jaut.2000.0484. [DOI] [PubMed] [Google Scholar]
- 207.Challoner PB, Smith KT, Parker JD, et al. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci USA. 1995;92:7440–4. doi: 10.1073/pnas.92.16.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sriram S, Stratton CW, Yao S, et al. Chlamydia pneumoniae infection of the central nervous system in multiple sclerosis. Ann Neurol. 1999;46:6–14. [PubMed] [Google Scholar]
- 209.Zhang J, Raus J. Myelin basic protein-reactive T cells in multiple sclerosis: pathologic relevance and therapeutic targeting. Cytotechnology. 1994;16:181–7. doi: 10.1007/BF00749906. [DOI] [PubMed] [Google Scholar]
- 210.Wucherpfennig KW, Zhang J, Witek C, et al. Clonal expansion and persistence of human T cells specific for an immunodominant myelin basic protein peptide. J Immunol. 1994;152:5581–92. [PubMed] [Google Scholar]
- 211.Allegretta M, Nicklas JA, Sriram S, Albertini RJ. T cells responsive to myelin basic protein in patients with multiple sclerosis. Science. 1990;247:718–21. doi: 10.1126/science.1689076. [DOI] [PubMed] [Google Scholar]
- 212.Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kurtzke JF. Epidemiologic evidence for multiple sclerosis as an infection. Clin Microbiol Rev. 1993;6:382–427. doi: 10.1128/cmr.6.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kurtzke JF. Multiple sclerosis in time and space – geographic clues to cause. J Neurovirol. 2000;6(Suppl. 2):S134–40. [PubMed] [Google Scholar]
- 215.Kurtzke JF, Gudmundsson KR, Bergmann S. Multiple sclerosis in Iceland: 1. Evidence of a postwar epidemic. Neurology. 1982;32:143–50. doi: 10.1212/wnl.32.2.143. [DOI] [PubMed] [Google Scholar]
- 216.Kurtzke JF, Hyllested K. Multiple sclerosis in the Faroe Islands: I. Clinical and epidemiological features. Ann Neurol. 1979;5:6–21. doi: 10.1002/ana.410050104. [DOI] [PubMed] [Google Scholar]
- 217.Islam T, Gauderman WJ, Cozen W, Hamilton AS, Burnett ME, Mack TM. Differential twin concordance for multiple sclerosis by latitude of birthplace. Ann Neurol. 2006;60:56–64. doi: 10.1002/ana.20871. [DOI] [PubMed] [Google Scholar]
- 218.Oleszak EL, Chang JR, Friedman H, Katsetos CD, Platsoucas CD. Theiler's virus infection: a model for multiple sclerosis. Clin Microbiol Rev. 2004;17:174–207. doi: 10.1128/CMR.17.1.174-207.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kim BS, Palna JP. Immune mechanisms of Theiler's virus-induced demyelination. Exp Mol Med. 1999;31:115–21. doi: 10.1038/emm.1999.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Dal Canto MC, Lipton HL. Ultrastructural immunohistochemical localization of virus in acute and chronic demyelinating Theiler's virus infection. Am J Pathol. 1982;106:20–9. [PMC free article] [PubMed] [Google Scholar]
- 221.Lipton HL, Twaddle G, Jelachich ML. The predominant virus antigen burden is present in macrophages in Theiler's murine encephalomyelitis virus-induced demyelinating disease. J Virol. 1995;69:2525–33. doi: 10.1128/jvi.69.4.2525-2533.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Trottier M, Kallio P, Wang W, Lipton HL. High numbers of viral RNA copies in the central nervous system of mice during persistent infection with Theiler's virus. J Virol. 2001;75:7420–8. doi: 10.1128/JVI.75.16.7420-7428.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Barbano RL, Dal Canto MC. Serum and cells from Theiler's virus-infected mice fail to injure myelinating cultures or to produce in vivo transfer of disease. J Neurol Sci. 1984;66:283–93. doi: 10.1016/0022-510x(84)90017-0. The pathogenesis of Theiler's virus-induced demyelination appears to differ from that of EAE. [DOI] [PubMed] [Google Scholar]
- 224.Miller SD, Clatch RJ, Pevear DC, Trotter JL, Lipton HL. Class II-restricted T cell responses in Theiler's murine encephalomyelitis virus (TMEV)-induced demyelinating disease. J Immunol. 1987;138:3776–84. I. Cross-specificity among TMEV substrains and related picornaviruses, but not myelin proteins. [PubMed] [Google Scholar]
- 225.Clatch RJ, Lipton HL, Miller SD. Characterization of Theiler's murine encephalomyelitis virus (TMEV)-specific delayed-type hypersensitivity responses in TMEV-induced demyelinating disease: correlation with clinical signs. J Immunol. 1986;136:920–7. [PubMed] [Google Scholar]
- 226.Miller SD, Vanderlugt CL, Begolka WS, et al. Persistent infection with Theiler's virus leads to CNS autoimmunity via epitope spreading. Nat Med. 1997;3:1133–6. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]
- 227.Katz-Levy Y, Neville KL, Padilla J, et al. Temporal development of autoreactive Th1 responses and endogenous presentation of self myelin epitopes by central nervous system-resident APCs in Theiler's virus-infected mice. J Immunol. 2000;165:5304–14. doi: 10.4049/jimmunol.165.9.5304. [DOI] [PubMed] [Google Scholar]
- 228.Lavi E, Gilden DH, Wroblewska Z, Rorke LB, Weiss SR. Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology. 1984;34:597–603. doi: 10.1212/wnl.34.5.597. [DOI] [PubMed] [Google Scholar]
- 229.Knobler RL, Lampert PW, Oldstone MB. Virus persistence and recurring demyelination produced by a temperature-sensitive mutant of MHV-4. Nature. 1982;298:279–80. doi: 10.1038/298279a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Wang FI, Hinton DR, Gilmore W, Trousdale MD, Fleming JO. Sequential infection of glial cells by the murine hepatitis virus JHM strain (MHV-4) leads to a characteristic distribution of demyelination. Lab Invest. 1992;66:744–54. [PubMed] [Google Scholar]
- 231.Takahashi K, Goto N, Matsubara Y, Fujiwara K. Postinflammatory remyelination in the spinal cord of mice infected with mouse hepatitis virus, JHM strain. Jpn J Exp Med. 1987;57:145–51. [PubMed] [Google Scholar]
- 232.Erlich SS, Fleming JO, Stohlman SA, Weiner LP. Experimental neuropathology of chronic demyelination induced by a JHM virus variant (DS) Arch Neurol. 1987;44:839–42. doi: 10.1001/archneur.1987.00520200043016. [DOI] [PubMed] [Google Scholar]
- 233.Stohlman SA, Hinton DR. Viral induced demyelination. Brain Pathol. 2001;11:92–106. doi: 10.1111/j.1750-3639.2001.tb00384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Wu GF, Perlman S. Macrophage infiltration, but not apoptosis, is correlated with immune-mediated demyelination following murine infection with a neurotropic coronavirus. J Virol. 1999;73:8771–80. doi: 10.1128/jvi.73.10.8771-8780.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Glass WG, Liu MT, Kuziel WA, Lane TE. Reduced macrophage infiltration and demyelination in mice lacking the chemokine receptor CCR5 following infection with a neurotropic coronavirus. Virology. 2001;288:8–17. doi: 10.1006/viro.2001.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wu GF, Dandekar AA, Pewe L, Perlman S. CD4 and CD8 T cells have redundant but not identical roles in virus-induced demyelination. J Immunol. 2000;165:2278–86. doi: 10.4049/jimmunol.165.4.2278. [DOI] [PubMed] [Google Scholar]
- 237.Lane TE, Liu MT, Chen BP, et al. A central role for CD4(+) T cells and RANTES in virus-induced central nervous system inflammation and demyelination. J Virol. 2000;74:1415–24. doi: 10.1128/jvi.74.3.1415-1424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Fazakerley JK, Pathak S, Scallan M, Amor S, Dyson H. Replication of the A7(74) strain of Semliki Forest virus is restricted in neurons. Virology. 1993;195(7):627–37. doi: 10.1006/viro.1993.1414. [DOI] [PubMed] [Google Scholar]
- 239.Balluz IM, Glasgow GM, Killen HM, Mabruk MJ, Sheahan BJ, Atkins GJ. Virulent and avirulent strains of Semliki Forest virus show similar cell tropism for the murine central nervous system but differ in the severity and rate of induction of cytolytic damage. Neuropathol Appl Neurobiol. 1993;19:233–9. doi: 10.1111/j.1365-2990.1993.tb00433.x. [DOI] [PubMed] [Google Scholar]
- 240.Amor S, Scallan MF, Morris MM, Dyson H, Fazakerley JK. Role of immune responses in protection and pathogenesis during Semliki Forest virus encephalitis. J Gen Virol. 1996;77:281–91. doi: 10.1099/0022-1317-77-2-281. [DOI] [PubMed] [Google Scholar]
- 241.Mokhtarian F, Swoveland P. Predisposition to EAE induction in resistant mice by prior infection with Semliki Forest virus. J Immunol. 1987;138:3264–8. [PubMed] [Google Scholar]
- 242.Fazakerley JK, Amor S, Webb HE. Reconstitution of Semliki forest virus infected mice, induces immune mediated pathological changes in the CNS. Clin Exp Immunol. 1983;52:115–20. [PMC free article] [PubMed] [Google Scholar]
- 243.Subak-Sharpe I, Dyson H, Fazakerley J. In vivo depletion of CD8+ T cells prevents lesions of demyelination in Semliki Forest virus infection. J Virol. 1993;67:7629–33. doi: 10.1128/jvi.67.12.7629-7633.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Keogh B, Atkins GJ, Mills KH, Sheahan BJ. Avirulent Semliki Forest virus replication and pathology in the central nervous system is enhanced in IL-12-defective and reduced in IL-4-defective mice: a role for Th1 cells in the protective immunity. J Neuroimmunol. 2002;125:15–22. doi: 10.1016/s0165-5728(02)00014-0. [DOI] [PubMed] [Google Scholar]
- 245.Keogh B, Atkins GJ, Mills KH, Sheahan BJ. Role of interferon-gamma and nitric oxide in the neuropathogenesis of avirulent Semliki Forest virus infection. Neuropathol Appl Neurobiol. 2003;29:553–62. doi: 10.1046/j.1365-2990.2003.00492.x. [DOI] [PubMed] [Google Scholar]
- 246.Mokhtarian F, Shi Y, Zhu PF, Grob D. Immune responses, and autoimmune outcome, during virus infection of the central nervous system. Cell Immunol. 1994;157:195–210. doi: 10.1006/cimm.1994.1216. [DOI] [PubMed] [Google Scholar]
- 247.Smith-Norowitz TA, Sobel RA, Mokhtarian F. B cells and antibodies in the pathogenesis of myelin injury in Semliki Forest virus encephalomyelitis. Cell Immunol. 2000;200:27–35. doi: 10.1006/cimm.2000.1613. [DOI] [PubMed] [Google Scholar]
- 248.Mokhtarian F, Zhang Z, Shi Y, Gonzales E, Sobel RA. Molecular mimicry between a viral peptide and a myelin oligodendrocyte glycoprotein peptide induces autoimmune demyelinating disease in mice. J Neuroimmunol. 1999;95:43–54. doi: 10.1016/s0165-5728(98)00254-9. [DOI] [PubMed] [Google Scholar]