

Abstract
Disulfide-rich peptides represent a megadiverse group of natural products with very promising therapeutic potential. To accelerate their functional characterization, high-throughput chemical synthesis and folding methods are required, including efficient mapping of multiple disulfide bridges. Here, we describe a novel approach for such mapping and apply it to a three-disulfide bridged conotoxin, μ-SxIIIA (from the venom of Conus striolatus) whose discovery is also reported here for the first time. μ-SxIIIA was chemically synthesized with three cysteine residues labeled 100% with 15N/13C, while the remaining three cysteine residues were incorporated using a mixture of 70%:30% unlabeled:labeled Fmoc-protected residues. After oxidative folding, the major product was analyzed by NMR spectroscopy. Sequence-specific resonance assignments for the isotope-enriched Cys residues were determined with 2D versions of standard triple resonance (1H,13C,15N) NMR experiments and 2D [13C,1H] HSQC. Disulfide patterns were directly determined with cross-disulfide NOEs confirming that the oxidation product had the disulfide connectivities characteristic of μ-conotoxins. μ-SxIIIA was found to be a potent blocker of the sodium channel subtype NaV1.4 (IC50 = 7 nM). These results suggest that differential incorporation of isotope-labeled cysteine residues is an efficient strategy to map disulfides and should facilitate the discovery and structure-function studies of many bioactive peptides.
Introduction
Small, disulfide-rich peptides include toxins derived from venomous animals such as spiders, scorpions, mollusks, plant-derived cyclotides or proteinase inhibitors, many of which have therapeutic applications 1-5. Although these megadiverse groups of disulfide-rich peptides may comprise millions of distinct sequences, only a small fraction has been discovered and characterized to-date 1,6,7. There is a considerable interest in accelerating the exploration of these unique natural product libraries 8. Examples of disulfide scaffolds found in these peptides are shown in Figure 1. A major challenge for study of disulfide-rich peptides is synthesis of native-folded molecules, and then determination of their disulfide bonding patterns (Table 1). Currently, chemical synthesis of peptides containing more than two disulfide bridges utilizes direct oxidation methods 9,10, but this can result in the formation of both native and non-native disulfide bridges depending on the reactivity of the cysteine thiolates and thermodynamic stability of the native conformation. In many instances, oxidative folding of disulfide-rich peptides results in multiple species and very low yield of native-folded scorpion toxins or certain conotoxins 11,12. Knowledge of the disulfide patterns in the resulting oxidation products is critical for a rigorous interpretation of the bioactivity data.
Figure 1.

Representative disulfide patterns of cysteine-rich peptides.
Table 1.
Sequences of μ-conotoxins reported to date. Of these peptides, only five were isolated directly from the venoms (GIIIA, GIIIB, GIIIC, PIIIA and TIIIA), and the remaining structures were deduced from cDNA sequences.
| Conotoxin | Sequence | References |
|---|---|---|
| SxIIIA | RCCTGKKGSCSGRACKNL–KCCA# | This work |
| SxIIIB | ZKCCTGKKGSCSGRACKNL–RCCA# | This work |
| GIIIA | RDCCT–OOKKCKDRQCKOQ–RCCA# | 43 |
| GIIIB | RDCCT–OORKCKDRRCKOM–KCCA# | 43 |
| GIIIC | RDCCT–OOKKCKDRQCKOL–KCCA# | 43 |
| PIIIA | ZRLCCG–FOKSCRSRQCKOH–RCC# | 44 |
| SmIIIA | ZRCCN–GRRGCSSRWCRDHSRCC# | 38 |
| SIIIA | ZNCCN–––GGCSSKWCRDHARCC# | 45 |
| KIIIA | CCN–––––CSSKWCRDHSRCC# | 45 |
| CnIIIA | GRCCD–VPNACSGRWCRDHAQCC# | 46 |
| CnIIIB | ZGCCG–EPNLCFTRWCRNNARCCRQQ | 46 |
| MIIIA | ZGCCN–VPNGCSGRWCRDHAQCC# | 46 |
| CIIIA | GRCCE–GPNGCSSRWCKDHARCC# | 46 |
| TIIIA | RHGCCK–GOKGCSSRECROQ–HCC# | 26 |
Z, pyroglutamate; O, 4-hydroxyproline;
, amidated C-terminus
Determination of disulfide bridge connectivity in short (<40 AA), disulfide-rich peptides remains a challenging task. Mass spectrometric fragmentation is still of a limited use for disulfide mapping (peptide bonds are significantly more stable than disulfides), despite tremendous progress in various analytical applications 13-16. Combining enzymatic digest/mass spectrometry analysis is also not efficient for many bioactive peptides, in particular for those containing adjacent cysteine residues, since short peptides often lack convenient proteolytic or chemical cleavage sites (Figure 1). A commonly used method to determine disulfide bridge connectivities was introduced by Gray 17,18. Using phosphine-based compounds, such as tris(2-carboxyethyl)phosphine (TCEP), the folded peptides are partially reduced at low pH, followed by an HPLC-purification of the unfolding intermediates and their subsequent alkylation. Each of the alkylated intermediates is then subjected to complete reduction, followed by a second alkylation and the Edman’s sequencing. In addition to being laborious (most of the described above steps require optimizations), this time-consuming strategy has a further limitation in that intramolecular rearrangements of disulfide bridges can occur during the partial reduction and/or alkylation. A modification of Gray’s strategy was introduced by the Watson group; the partially unfolded intermediates were cyanylated followed by cleavage in aqueous ammonia 19-21. There is a continuous need for new methods that allow rapid and unambiguous determination of the bridging pattern in disulfide-rich peptides 8.
One group of the disulfide-rich peptides currently being explored for potential therapeutics are sodium channel blocking μ-conotoxins. Two members of the μ-conotoxin family, namely μ-KIIIA and μ-SIIIA, possess very potent analgesic activity following systemic administration 22,23. As shown in Table 1, μ-conotoxins are relatively short (16-25 AA) and contain three disulfide bridges that stabilize the native conformation. NMR studies of μ-conotoxins showed the following disulfide bridging connectivity: CysI-CysIV, CysII-CysV and CysIII-CysVI 24-27. To produce a synthetic μ-conotoxin, the reduced/linear peptide is subjected to direct oxidation in which all six cysteines are allowed to form disulfide bonds simultaneously 28. The oxidation products are HPLC-purified and tested for biological activity. For proper interpretation of bioactivity results, it is necessary to ensure that the synthetic peptide contains the native pattern of disulfide bridges.
In this report, we applied a novel disulfide mapping approach to characterize a μ-conotoxin from Conus striolatus, μ-SxIIIA, that was discovered by concerted efforts that employ molecular cloning, phylogeny and exogenomics strategy 4,7. μ-SxIIIA was chemically synthesized with three Cys residues labeled with 100% 15N/13C, while the remaining three Cys residues were incorporated using a mixture of 70%:30% unlabeled:labeled Fmoc-protected residues. NMR assignments were made with fast, 2D versions of triple resonance experiments, and the disulfide pattern was then directly determined from cross-disulfide Hα/Hβ2/Hβ3 NOESY crosspeaks. This NMR-based strategy for analyzing synthetic disulfide-rich peptides should be broadly applicable to the very large and structurally-diverse libraries of peptidic natural products, and to SAR studies of these peptides.
Results
Molecular cloning and the chemical synthesis
Using the exogenomics strategy 4, two new conotoxin sequences belonging to the M-superfamily were identified from Conus striolatus. The cDNA-derived sequences of the peptides, μ-SxIIIA and μ-SxIIIB, were very similar to each other and were predicted to have C-terminal amidation (Table 1). The Arg residue in the second intercysteine loop and the number of amino acid residues in all three intercysteine loops suggested that they belonged to μ-conotoxins. However, the sequences within the intercysteine loops of these peptides were significantly different from those of all previously characterized μ-conotoxin (Table 1), and this provided a motivation to more closely examine their structures and functions. One of these peptides, μ-SxIIIA, was chemically synthesized on a solid support using the standard Fmoc protocol. All Cys residues were protected with a trityl group. After cleavage and HPLC purification of the reduced peptide, several direct oxidation conditions were tested. As illustrated in Figure 2, folding yields increased from 27 ± 3 % under the standard folding conditions (ambient temperature, no salt) to 46 ± 4 % when the peptide was oxidized at high ionic strength and at low temperature. The major folding species was purified by reversed-phase HPLC and the identity of the final product confirmed by mass spectrometry (MWexp/calc = 2271.4/2271.8).
Figure 2.
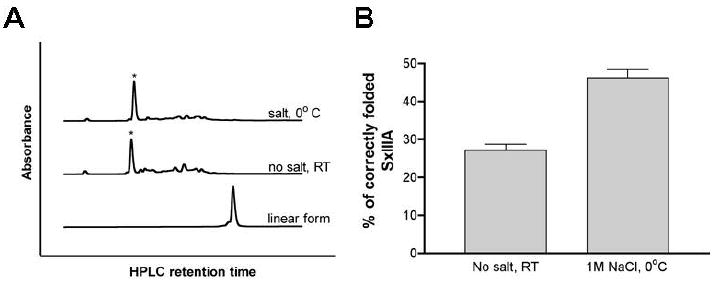
The oxidative folding of μ-SxIIIA (A) Folding was carried out for two hours in the presence of reduced and oxidized glutathione, followed by an HPLC separation using reverse-phase C18 column. Correctly folded species are indicated by asterisks. Top HPLC trace shows the folding reaction carried out at 0 °C and 1 M NaCl, whereas the middle HPLC trace shows the same folding reaction at room temperature and with no salt. The former folding conditions produced fewer low-populated folding species. Folding yields shown in panel B were obtained by integrating the HPLC peaks. (B) Bar graphs represent folding yields of μ-SxIIIA at two different conditions: no salt at room temperature and 1 M NaCl at 0 °C. Error bars represent standard error calculated from three experiments.
NMR-based mapping of the disulfide bridges
To confirm synthetic μ-SxIIIA has the canonical disulfide pattern found in other μ-conotoxins, we developed a new NMR-based strategy that relies on preparing peptides with different levels of 13C/15N isotope enrichment at selected cysteine residues. In this application we prepared conotoxin μ-SxIIIA with 100% isotope (13C/15N) enrichment of cysteines C2, C3, and C10 and 30% enrichment of C15, C20, and C21 (Figure 3A). Cysteine specific resonance assignments were achieved using intensities of the 2D [ 13C,1H] HSQC signals to initially resolve the two classes of cysteines, 30% and 100%, and then triple resonance methods to identity and order the two cys-cys dipeptides, C2C3 and C20C21. The remaining and isolated cysteines, C10 and C15, were identified based on intensity alone. This resulted in unambiguous sequence-specific resonance assignments. Following assignment, 2D F2-13C-edited [1H,1H] NOESY was recorded to identify NOEs across disulfide bonds.
Figure 3.
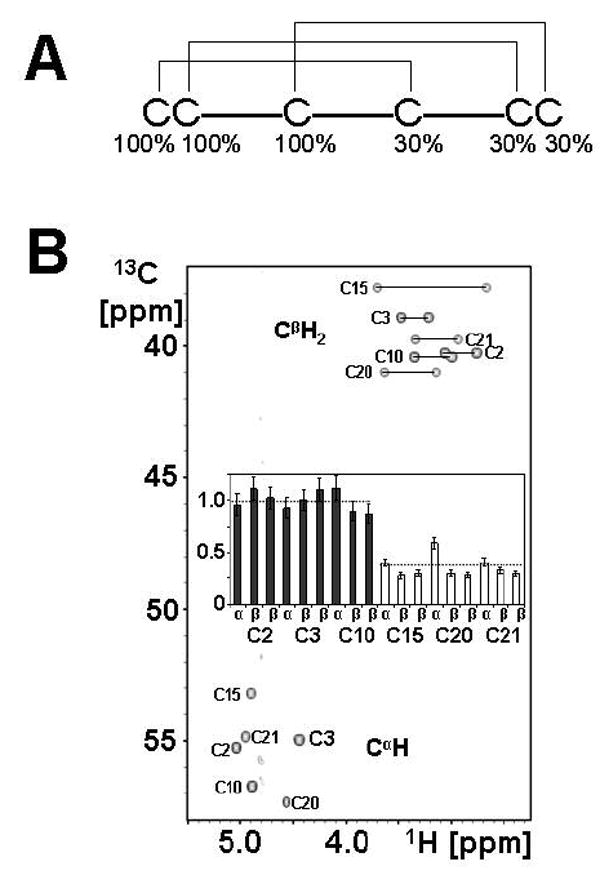
(A) The chemically-synthesized μ-SxIIIA contains three cysteines labeled 100% with 15N/13C, and a mixture of 70%:30% 14N/12C:15N/13C for the remaining three cysteines. (B) 2D [13C,1H] HSQC spectrum of C2,C3,C10,C15,C20,C21 15N-,13C-enriched μ-SxIIIA. Resonance assignments of methine (lower) and methylene (upper) μ-SxIIIA signals. The methylene Hβ2/Hβ3 signals are connected with a solid line. Signal intensities are plotted in a bar graph and are normalized to an average of the same signals (methine or methylene) in 100% labeled C2, C3, and C10 residues. The dashed lines are the arithmetic means of the 30% (0.32 ± 0.10) and 100% (1.0 ± 0.09) labeled groups. The error bars are derived from propagation of error in quadrature. The standard error of ~ 0.1 for both isotope classes suggests enrichment levels differing by ~ 25% could be reliably distinguished. Regular and constant time 2D [13C,1H] HSQC data were analyzed and both gave similar results; the constant time version is shown here.
The two cysteine labeling classes were first identified with 2D [13C,1H] HSQC. The methine and methylene signals of the six cysteines are completely resolved, and as expected, the group with 100% enrichment have signal intensities ~ 3x larger than those from the group enriched at 30% (Figure 3B). The average of the normalized signal intensity of the two groups is shown as dashed lines in Figure 3B (1.0 ± 0.09 and 0.32 ± 0.10 for 100% and 30% labeled groups, respectively). This demonstrates the strategy can be used to distinguish these differently enriched cysteines. The 2D [15N, 1H] HSQC spectrum of the molecule is also of high quality but shows only five of the expected six amide signals and the signal intensity shows a wide intensity range. The amide of C2 is significantly exchange broadened in our solution conditions (not shown).
After separating the two classes of cysteine residues, the cys-cys dipeptides were identified in a straight forward manner using HNCACB and CBCA(CO)NH (Figure 4). Dipeptides C2C3 and C20C21 show a characteristic single pair of Cα and Cβ signals in the C3 and C21 CBCA(CO)NH strips plots (Figure 4, correlations boxed in C3 and C21 A strips) and two pairs of Cα and Cβ signals in the corresponding HNCACB (Figure 4, B strips). Note the absence of CBCA(CO)NH signals in C10, C15, and C20; also consistent with having unlabeled amino acids at residues 9, 14, and 19. The Hα and Hβ assignments from HBHA(CO)NH and HNHA are also consistent with the two dipeptide sequences (not shown). The resulting Cα,β/Hα,β chemical shifts were then mapped on the 2D [13C,1H] HSQC and their relative signal intensities used to confirm C2C3 and C20C21 dipeptide fragments. The two remaining cysteine residues, C10 and C15, are isolated in sequence and also differently enriched — C10 (100%) and C15 (30%). Isolated cysteines will not show inter-residue signals in CBCA(CO)NH (see A strips for C10 and C15 in Figure 4) and only intra-cysteine Cα and Cβ signals in HNCACB (B strips for C10 and C15). The two isolated cysteines were consistent with this pattern, and could then be assigned based on signal intensities in the 2D [13C,1H] HSQC spectrum (Figure 3). This strategy resulted in sequence specific assignment for all cysteine CH and NH pairs with exception of the exchange broadened amide of C2 (Supplemental Table S1). Independent confirmation of C2, C3, and C15 resonance assignments was also performed using two independently prepared and isotope labeled peptides, 1) uniform 13C- and 15N-labeled C2/C15 and 2) C2/C3/C15 μ-SxIIIA. An overlay of 2D [13C,1H] HSQC for these peptides with the fully labeled μ-SxIIIA described here (Figure 3A) confirms these assignments (Supplemental Figure S1).
Figure 4.
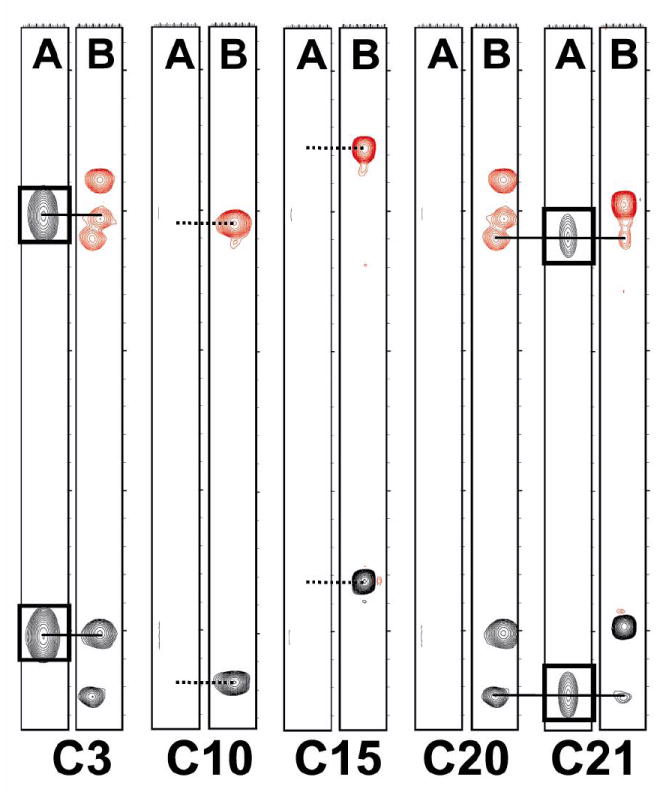
Sequence-Specific Resonance Assignments. Amide NH strips for μ-SxIIIA showing the CBCA(CO)NH (A) and HNCACB (B) signals of the five observable amide NH signals. HNCACB Cα crosspeaks are shown in black and Cβ crosspeaks are shown in red while CBCA(CO)NH Cα and Cβ crosspeaks are both shown in black.
Following cysteine assignment, a 2D F2-13C-edited NOESY was recorded to identify the NOEs expected across each disulfide bond (Figure 5); the underlying hypothesis being that interproton distances across the disulfide bond are sufficiently short to give a defining NOE network for each disulfide. The most intense NOE signals observed in Figure 5 arise from intra-cysteine methylene Hβ2/Hβ3 NOEs in the 2.6 – 3.8 ppm region and Hα/Hβ2/3 NOEs in the 4.4 – 5.1 ppm region. These confirmed our through-bond resonance assignments. In addition to the above expected intra-cysteine NOEs, several other inter-cysteine NOEs define the disulfide bridge. Disulfide C2-C15 is defined with NOEs #1 – 6, C3-C20 with NOEs #7 – 9, and C10-C21 with NOE #10 (see Figure 5 legend for details). There are fewer inter-cysteine NOEs observed across the C10-C21 disulfide due to Hβ methylene resonance overlap. This disulfide pattern is identical to that found in all other μ-conotoxins characterized to date (Table 1).
Figure 5.
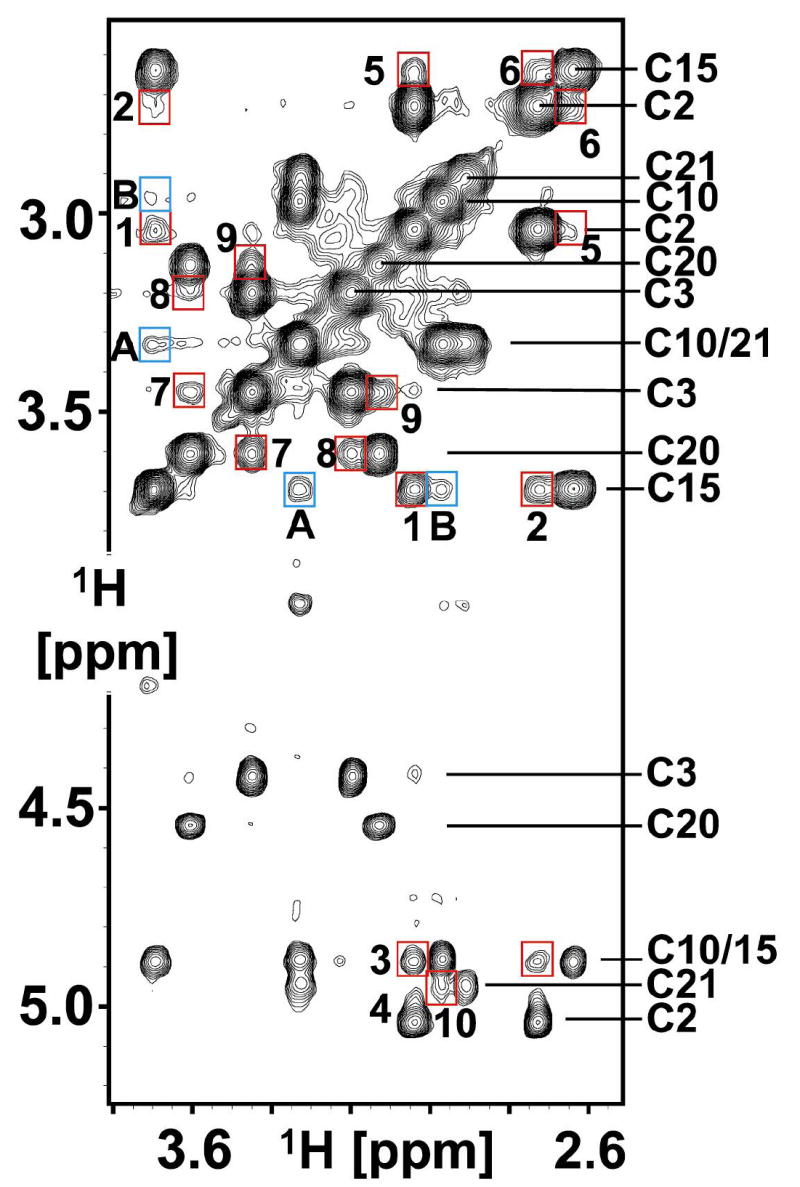
μ-SxIIIA NOESY Assignments. 2D F2-13C-edited [1H,1H] NOESY spectrum of μ-SxIIIA recorded at 15 °C with a mixing time of 250 ms. Cysteine Hα/Hβ assignments are labeled on right side of spectrum (also Supplemental Table 1). Intra-cysteine NOEs are unlabeled. Inter-cysteine NOEs are boxed in red with the following assignments: 1, C2-C15; 2, C2-C15; 3, C2-C15; 4, C2-C15; 5, C2-C15; 6, C2-C15; 7, C3-C20; 8, C3-C20; 9, C3-C20; 10, C10-C21. Boxed in blue are inter-cystine NOEs.
There are also NOEs arising from proximal cystines. In particular NOEs A and B (Figure 5) connect C10 with C15, in apparent conflict with the C10-C21 and C2-C15 disulfide bond identification. This potential problem was successfully resolved using our sparse NOE dataset to calculate a fold for μ-SxIIIA. The converged fold is only consistent with the C2-C15, C3-C20, and C10-C21 disulfides and the NOEs linking C10 with C15 are clearly inter-cystine (Supplemental Figure S2 and S3).
Bioactivity
Since μ-SxIIIA belonged to the μ-conotoxin family, we examined whether it could block sodium channels. When injected intraperitoneally or subcutaneously in mice, μ-SxIIIA caused motor impairment/paralysis, and was lethal at doses as low as 0.6 nmoles per mouse. Since similar behavioral effects are observed with other μ-conotoxins, such as μ-GIIIA, this paralytic activity indicated that μ-SxIIIA could be a potent blocker of skeletal muscle sodium channel subtype, Nav1.4. As shown in Figure 6, 10 nM μ-SxIIIA significantly, yet reversibly blocked rat Nav1.4 expressed in Xenopus oocytes. Dose-response analysis showed it had an IC50 of 7 nM, making μ-SxIIIA an even more potent blocker of the skeletal muscle sodium channel than the previously characterized μ-GIIIA, which has an IC50 of 19 nM against rat NaV1.4 expressed in oocytes 29. A more detailed neurobiological characterization of μ-SxIIIA will be described elsewhere.
Figure 6.
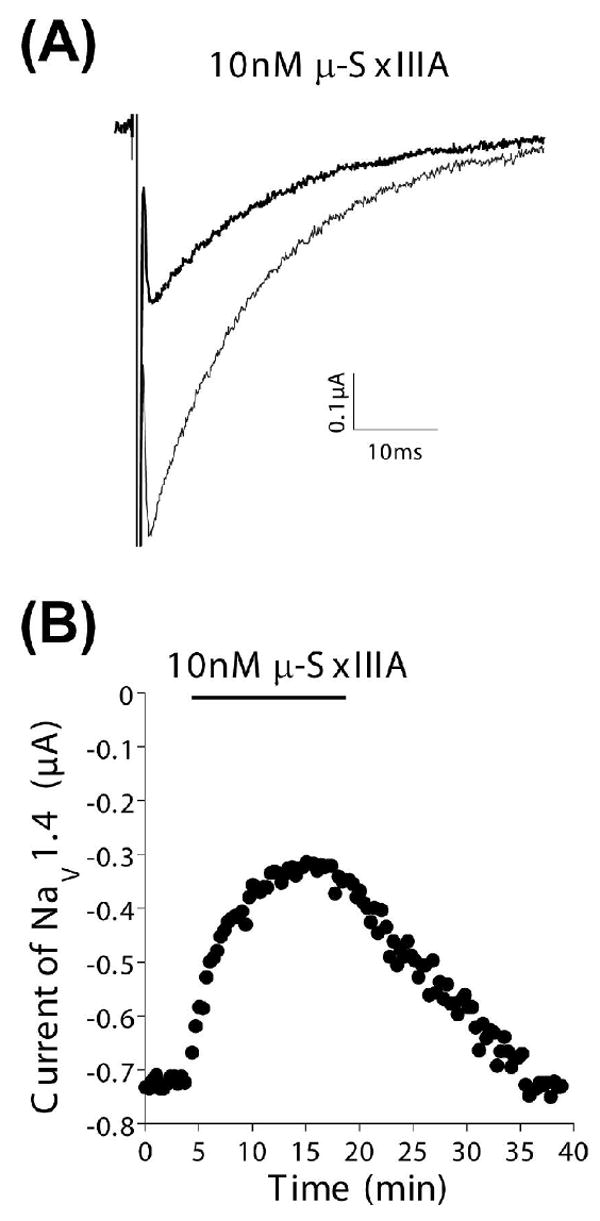
μ-SxIIIA Blocks Skeletal Muscle Sodium Channels. Cloned Nav1.4 was expressed in Xenopus oocytes, which were voltage clamped as described in Methods. (A) Representative recordings of the sodium current in control solution (large trace) and in the presence of 10 nM μ-SxIIIA (attenuated trace). (B) Time course of the block of NaV1.4 by 10 nM μ-SxIIIA (bar indicates when peptide was present) and following its washout. Same oocyte as in (A). Recovery of the current during peptide-washout indicates the block was reversible.
Discussion
We used molecular cloning techniques to discover a novel μ-conotoxin from the cone snail Conus striolatus. This discovery represents an example of how to successfully mine the diverse natural product libraries comprising hundreds of thousands of unique disulfide-rich peptides 1,6,7. Chemical synthesis and the oxidative folding of μ-SxIIIA illustrates one challenge in the characterization of such peptides; namely, how to rapidly and unambiguously determine the disulfide bridging patterns in a synthetic, cysteine-rich peptide. As a proof-of-concept, we performed a novel, NMR-based assessment of the disulfide bridges in μ-SxIIIA using differential incorporation of isotope-labeled Cys residues. Although NMR methods have been extensively applied to conotoxins to study their structure, dynamics and posttranslational modifications 30-35, this is the first report of using selectively labeled peptides to rapidly determine disulfide bridging patterns. Our NMR analysis of the peptides also differs from the previously reported use of 13C chemical shifts to verify whether cysteines residues were in the reduced or oxidized state 36. The main advantage of the NMR method described here is the rapid verification of the disulfide patterns in the oxidized peptides that contain multiple disulfide bridges. This information is critical for a proper interpretation of any functional data (and in particular negative data) for either newly-discovered peptides or SAR analogs. By labeling only the cysteine residues of this 21 residue peptide, the NMR spectra are dramatically simplified compared to an unlabeled sample. The availability of the adjacent cysteines, C2-C3 and C20-C21, in μ-SxIIIA allowed us to rapidly collect 2D versions of powerful 3D triple resonance experiments, and make unambiguous assignments. In contrast to the heavily overlapped 2D NOESY for an unlabeled sample, the selectively cysteine labeled 13C-edited 2D NOESY allowed for rapid and complete assignment.
The disulfide bridging pattern in synthetic μ-SxIIIA was directly determined based on cross-disulfide Hα/Hβ2/Hβ3 NOESY crosspeaks and confirmed that the major oxidation product indeed had the disulfide connectivities characteristic of all μ-conotoxins characterized to date (Table 1). This finding further validated our assumption that for closely related conotoxins identified from cDNA, the disulfide patterns are conserved, increasing the odds of predicting which cysteine residues should be isotope-labeled for mapping their connectivities. There are, however, few instances where predicting the native-like connectivity of disulfide bridges in conotoxins identified solely from cDNA sequencing appeared challenging (for a relevant review see 11), and requires a more judicious design of the differential incorporation of isotope-labeled cysteine, and perhaps even a synthesis of more than one labeled peptide.
The NMR-based mapping of the disulfide bridges may offer both economic and time-saving advantages, compared to a standard, chemical-based peptide mapping 17-19,21. For both methods, comparable amounts of the synthetic peptide are needed (300 nmoles of a synthetic peptide yields peptide solution > 1 mM in a 250 μl NMR tube). Producing NMR-quantities of the μ-SxIIIA containing the differentially-labeled cysteine residues required only 133 μmoles (~78 mgs) of the labeled Fmoc-protected cysteine derivative (based on the 25 μmoles synthesis scale with a two-fold excess of the Fmoc-protected intermediate during a coupling reaction). Therefore, the costs of producing the labeled peptide may become even several fold lower, as compared to total costs associated with multiple mass spectrometry analyses and at least several Edman’s sequencing reactions that are critical during the chemical-based peptide mapping. With respect to a time-saving aspect, it took 3-4 days to determine the disulfide pattern in μ-SxIIIA (2-3 days of recording NMR spectra and one day of the data analysis), whereas the standard peptide mapping procedure can take from one-two weeks or even months to obtain unambiguous information about the disulfide connectivity.
The NMR-based method does have some limitations, although we think it is generally superior to chemistry-based methods. The 2D F2-13C- NOESY used to simplify the NOESY spectrum detects only on those 1H signals directly attached to 13C and this means cysteines labeled at 30% will have a similar reduction in the signal/noise ratio of NOE signals. This did not present a problem in the current study using a highly sensitive cryogenic probe and 1 mM sample concentration. Resonance overlap will hamper observation and interpretation of NOEs. In the current scheme there was overlap of a C10 and C21 Hβ resonance that precluded observation of Hβ/Hβ NOEs for the C10-C21 disulfide bond (Figure 5). Two or more disulfides that closely approach one another in the molecules tertiary structure could make interpretation of disulfide bonds based on NOEs ambiguous. We observed two cross-cystine NOEs that suggests the potential problems that might be seen with other conotoxin peptides, however we were able to assign these NOE cross-peaks for μ-SxIIIA. This limitation can be generally addressed by using the sparse NOE dataset to calculate the peptide fold (Supplemental Figure S2).
This methodology can also be extended to other disulfide-rich peptides, for example to those containing the so-called inhibitory cystine knot (ICK) motif (Figs. 1 and 7). This motif is commonly found in peptides derived from animals, plants, fungi or viruses 35. To synthesize the labeled ICK peptide, three cysteine residues would be 100% 13C/15N labeled, whereas the remaining three cysteines introduced with enrichment levels of 25%, 50%, and 75% labeled:unlabeled amino acid as shown in Figure 7. The labeled C-III and C-IV which are next to each other could be identified using 2D versions of HNCACB and CBCA(CO)NH experiments as shown for μ-SxIIIA. Measuring the intensity of each crosspeak using a 2D [13C, 1H] HSQC spectrum, one should be able to confirm the enrichment levels for each cysteine residue. From Figure 3, we conservatively estimate the limit of isotopic enrichment resolution is about 25%. Subsequently, a 2D F2-13C-edited NOESY experiment would verify formation of the disulfide bridges. The NOE patterns in this spectrum would then indicate the disulfide framework of the peptide. Yet another route to access natively folded ICK-type peptides would be to use regioselective folding as recently described for conotoxin μO-MrVIB combined with a simple variant of our NMR disulfide assay 37. In an ICK-type peptide, one pair of Trt-protected cysteines would be labeled with 13C/15N, whereas the second pair would be protected with the Acm groups. After the first oxidation step, only three potential folding intermediates can be formed, and only one would contain the native-like pattern of two disulfide bridges. This species could be easily detected using cross-disulfide NOEs. The native-like folding intermediate would then be subjected to the second oxidation step, resulting in the correctly folded final product. Nearly any cysteine-rich peptide, even those lacking consecutive Cys residues, could be unambiguously mapped using two labeled samples having different patterns and levels of isotopic enrichment. An additional strategy for peptides lacking consecutive cysteine residues would be to introduce additional 15N- and/or 15N, 13C-labeled amino acids adjacent to labeled cysteines. This would expand the assignment capability.
Figure 7.
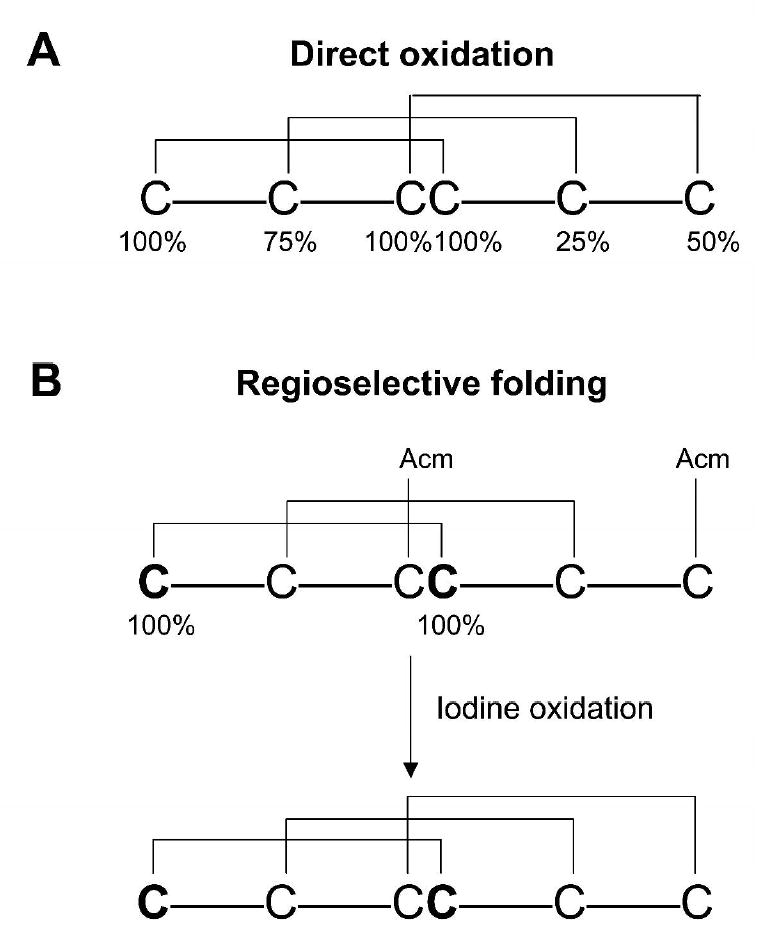
General strategies for NMR-based mapping of disulfide bridges in inhibitory cystine knot (ICK) peptides. (A) one-step folding strategy with labeling levels as indicated: CI-100%, CII-75%, CIII/CIV- 100%, CV-25%, and CVI-50% 15N/13C. The 100% sites are differentiated using triple resonance NMR connectivities while the remaining isolated sites are differentiated by enrichment levels. An alternative strategy would be to introduce 15N- and/or 15N,13C- labeled amino acids adjacent to isolated cysteines expanding the NMR capability. (B) two-step folding strategy with labeled cysteines: 100% labeled with 15N/13C and orthogonal cysteine protection. Since the first folding step involves the direct oxidation of four cysteines, three products are possible, only one having two native disulfide bridges. The 15N/13C labeled pair of cysteine residues that form one of the native disulfide bridges allows a rapid selection of the correctly folded isomer to be further oxidized.
In summary, we have demonstrated that differential incorporation of isotope-labeled cysteine residues in a disulfide-rich peptide allows for rapid determination of the disulfide bridging pattern. This approach will facilitate structural and functional characterization of bioactive disulfide-rich peptides.
Methods
Cloning of conotoxins
PCR amplification, construction of cDNA libraries, and cloning of conotoxins were performed as described previously 38,39.
Peptide Synthesis
μ-SxIIIA was synthesized on a solid support using standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry, as described previously. The cysteine residues were protected with the trityl group. Fmoc-protected 15N/13C cysteine residue (U-13C3, 97-99%; 15N, 97-99%) was obtained from Cambridge Isotope Laboratories, Inc. (Andover, MA). The enrichment level for individual cysteine residues was controlled by an appropriate mixing (w/w) of the labeled and unlabeled Fmoc-protected cysteine residues, prior to the coupling. The peptide was removed from the resin by a 4-hr treatment with reagent K (trifluoroacetic acid (TFA)/ water/ ethanedithiol/ phenol/ thioanisole; 90/5/2.5/7.5/5 by volume), and subsequently filtered and precipitated with methyl-tert butyl ether (MTBE). The linear, reduced peptide was purified by reversed-phase HPLC using a preparative C18 Vydac column (218TP1022, 10 mm × 250 mm, 5 μm particle size). To elute the peptide, buffer A (0.1% TFA in water) and buffer B (0.1% TFA, v/v, in 90% aqueous acetonitrile) were used to produce a linear gradient from 5 to 30% of buffer B over 25 min. The flow rate was 5 ml/min and the elution was monitored by measuring absorbance at 220 nm. Oxidative folding was carried out in buffered solution (0.1 M Tris-HCl, pH 7.5) containing 1 mM EDTA, 1 mM reduced and 1 mM oxidized glutathione. After completion of folding (2 hrs), the reaction was quenched by acidification with formic acid (8% final concentration). Folded μ-SxIIIA was purified by semi-preparative HPLC. Folding yields were determined by integration of HPLC peaks, following separations of the folding reactions quenched at steady-state (at 2 hrs).
NMR spectroscopy
Purified μ-SxIIIA (1 mM) was dissolved in an NMR buffer containing 40 mM sodium phosphate (pH 6.2), 50 mM sodium chloride, 90% H2O and 10% D2O. Standard triple resonance NMR experiments were recorded at 25 °C on a Varian Inova 600 MHz NMR spectrometer equipped with a cryogenic triple-resonance probe and z-axis pulsed-field gradients40. Data were processed with FELIX 2004 (Accelrys, San Diego), and resonances assigned using standard approaches within the SPARKY program (T.D. Goddard and D.G. Kneller, University of California, San Francisco).
Fold Calculations
NOEs were obtained from a 2D F2-13C-edited NOESY (250 msec mix time). NOEs were manually assigned and intensities measured using the tools in SPARKY, and then exported to CYANA for fold calculation 41. Twelve NOEs (Figure 5) were used in calculations and ten coordinate sets of the lowest energy were superimposed using Cα, Cβ heavy atoms of the six cysteines (Supplemental Figures S2 and S3). Structures were visualized, superimposed, and figures made using PYMOL (Delano Scientific).
Biological assays
SxIIIA was tested for its ability to block the cloned mammalian sodium channel Nav1.4 expressed in Xenopus oocytes using methods recently described 22. Briefly, oocytes were two-electrode voltage-clamped, and sodium currents were acquired using a holding potential of −80 mV and stepping to 0 mV for 40 ms every 20 seconds. SxIIIA exposure was carried out in a static bath to conserve peptide. Recordings were conducted at room temperature (~20 °C). For in vivo bioassays, conotoxins were delivered to Swiss Webster mice (25-30 g) by intraperitoneal or subcutaneous injection, as described previously 42. The lyophilized peptides were dissolved in normal saline solution and injected using a 29-gauge insulin syringe.
Supplementary Material
Supplemental Table S1 summarizes chemical shifts. Supplemental Figure S1 shows the overlaid 2D [ 13C, 1H] HSQC spectra of C2, C15 and C2,C3,C10,C15,C20,C21 15N-,13C-enriched μ-SxIIIA. Supplemental Figure S2 shows a stereoview of a representative μ-SxIIIA structure calculated from the twelve NOEs listed in Figure 5, and the same molecule superimposed on the cysteine heavy atoms of μ-SmIIIA coordinates (1Q2J.pdb). Supplemental Figure S3 is a bar graph showing the average Cα-Cα (white) and Cβ-Cβ (grey) distances for all possible cystines in ten calculated μ-SxIIIA structures. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Drs. Robert Schackmann and Scott Endicott for their help with the chemical synthesis of peptides. This work was supported by the funding from the National Institute of Health: R21 NS055845(GB) and Program Project GM 48677 (BMO).
Footnotes
This work was supported by the NIH grants: R21 NS055845(GB) and Program Project GM 48677 (BMO)
References
- 1.Beeton CE, Gutman GA, Chandy KG. In: Handbook of Biologically Active Peptides. Kastin AJ, editor. Academic Press; 2005. pp. 403–414. [Google Scholar]
- 2.Craik DJ, Clark RJ, Daly NL. Expert Opin Investig Drugs. 2007;16:595–604. doi: 10.1517/13543784.16.5.595. [DOI] [PubMed] [Google Scholar]
- 3.Livett BG, Gayler KR, Khalil Z. Curr Med Chem. 2004;11:1715–23. doi: 10.2174/0929867043364928. [DOI] [PubMed] [Google Scholar]
- 4.Olivera BM. J Biol Chem. 2006;281:31173–7. doi: 10.1074/jbc.R600020200. [DOI] [PubMed] [Google Scholar]
- 5.Norton RS, Olivera BM. Toxicon. 2006;48:780–98. doi: 10.1016/j.toxicon.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 6.Sollod BL, Wilson D, Zhaxybayeva O, Gogarten JP, Drinkwater R, King GF. Peptides. 2005;26:131–9. doi: 10.1016/j.peptides.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 7.Olivera BM, Teichert RW. Mol Interv. 2007;7:251–60. doi: 10.1124/mi.7.5.7. [DOI] [PubMed] [Google Scholar]
- 8.Bulaj G. Curr Opin Chem Biol. 2008 [Google Scholar]
- 9.Moroder L, Besse D, Musiol HJ, Rudolph-Bohner S, Siedler F. Biopolymers. 1996;40:207–34. doi: 10.1002/(sici)1097-0282(1996)40:2<207::aid-bip2>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 10.Moroder L, Musiol HJ, Gotz M, Renner C. Biopolymers. 2005;80:85–97. doi: 10.1002/bip.20174. [DOI] [PubMed] [Google Scholar]
- 11.Bulaj G, Olivera BM. Antioxid Redox Signal. 2008;10:141–56. doi: 10.1089/ars.2007.1856. [DOI] [PubMed] [Google Scholar]
- 12.Altamirano MM, Garcia C, Possani LD, Fersht AR. Nat Biotechnol. 1999;17:187–91. doi: 10.1038/6192. [DOI] [PubMed] [Google Scholar]
- 13.Jones MD, Patterson SD, Lu HS. Anal Chem. 1998;70:136–43. doi: 10.1021/ac9707693. [DOI] [PubMed] [Google Scholar]
- 14.Jakubowski JA, Sweedler JV. Anal Chem. 2004;76:6541–7. doi: 10.1021/ac0494376. [DOI] [PubMed] [Google Scholar]
- 15.Zhang M, Kaltashov IA. Anal Chem. 2006;78:4820–9. doi: 10.1021/ac060132w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borges CR, Qi J, Wu W, Torng E, Hinck AP, Watson JT. Anal Biochem. 2004;329:91–103. doi: 10.1016/j.ab.2004.01.037. [DOI] [PubMed] [Google Scholar]
- 17.Gray WR. Protein Sci. 1993;2:1749–55. doi: 10.1002/pro.5560021018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gray WR. Protein Sci. 1993;2:1732–48. doi: 10.1002/pro.5560021017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu J, Watson JT. Protein Sci. 1997;6:391–8. doi: 10.1002/pro.5560060215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu W, Huang W, Qi J, Chou YT, Torng E, Watson JT. J Proteome Res. 2004;3:770–7. doi: 10.1021/pr049961t. [DOI] [PubMed] [Google Scholar]
- 21.Wu J, Watson JT. Methods Mol Biol. 2002;194:1–22. doi: 10.1385/1-59259-181-7:001. [DOI] [PubMed] [Google Scholar]
- 22.Zhang MM, Green BR, Catlin P, Fiedler B, Azam L, Chadwick A, Terlau H, McArthur JR, French RJ, Gulyas J, Rivier JE, Smith BJ, Norton RS, Olivera BM, Yoshikami D, Bulaj G. J Biol Chem. 2007;282:30699–706. doi: 10.1074/jbc.M704616200. [DOI] [PubMed] [Google Scholar]
- 23.Green BR, Catlin P, Zhang MM, Fiedler B, Bayudan W, Morrison A, Norton RS, Smith BJ, Yoshikami D, Olivera BM, Bulaj G. Chem Biol. 2007;14:399–407. doi: 10.1016/j.chembiol.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 24.Keizer DW, West PJ, Lee EF, Yoshikami D, Olivera BM, Bulaj G, Norton RS. J Biol Chem. 2003;278:46805–13. doi: 10.1074/jbc.M309222200. [DOI] [PubMed] [Google Scholar]
- 25.Nielsen KJ, Watson M, Adams DJ, Hammarstrom AK, Gage PW, Hill JM, Craik DJ, Thomas L, Adams D, Alewood PF, Lewis RJ. J Biol Chem. 2002;277:27247–55. doi: 10.1074/jbc.M201611200. [DOI] [PubMed] [Google Scholar]
- 26.Lewis RJ, Schroeder CI, Ekberg J, Nielsen KJ, Loughnan M, Thomas L, Adams DA, Drinkwater R, Adams DJ, Alewood PF. Mol Pharmacol. 2007;71:676–85. doi: 10.1124/mol.106.028225. [DOI] [PubMed] [Google Scholar]
- 27.Hill JM, Alewood PF, Craik DJ. Biochemistry. 1996;35:8824–35. doi: 10.1021/bi960073o. [DOI] [PubMed] [Google Scholar]
- 28.Fuller E, Green BR, Catlin P, Buczek O, Nielsen JS, Olivera BM, Bulaj G. FEBS J. 2005;272:1727–38. doi: 10.1111/j.1742-4658.2005.04602.x. [DOI] [PubMed] [Google Scholar]
- 29.Safo P, Rosenbaum T, Shcherbatko A, Choi DY, Han E, Toledo-Aral JJ, Olivera BM, Brehm P, Mandel G. J Neurosci. 2000;20:76–80. doi: 10.1523/JNEUROSCI.20-01-00076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marx UC, Daly NL, Craik DJ. Magn Reson Chem. 2006;44 Spec No:S41–50. doi: 10.1002/mrc.1821. [DOI] [PubMed] [Google Scholar]
- 31.Mitchell SS, Shon KJ, Olivera BM, Ireland CM. J Nat Toxins. 1996;5:191–208. [Google Scholar]
- 32.Pisarewicz K, Mora D, Pflueger FC, Fields GB, Mari F. J Am Chem Soc. 2005;127:6207–15. doi: 10.1021/ja050088m. [DOI] [PubMed] [Google Scholar]
- 33.Goldenberg DP, Koehn RE, Gilbert DE, Wagner G. Protein Sci. 2001;10:538–50. doi: 10.1110/ps.30701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Atkinson RA, Kieffer B, Dejaegere A, Sirockin F, Lefevre JF. Biochemistry. 2000;39:3908–19. doi: 10.1021/bi992651h. [DOI] [PubMed] [Google Scholar]
- 35.Pallaghy PK, Nielsen KJ, Craik DJ, Norton RS. Protein Sci. 1994;3:1833–9. doi: 10.1002/pro.5560031022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma D, Rajarathnam K. J Biomol NMR. 2000;18:165–71. doi: 10.1023/a:1008398416292. [DOI] [PubMed] [Google Scholar]
- 37.Bulaj G, Zhang MM, Green BR, Fiedler B, Layer RT, Wei S, Nielsen JS, Low SJ, Klein BD, Wagstaff JD, Chicoine L, Harty TP, Terlau H, Yoshikami D, Olivera BM. Biochemistry. 2006;45:7404–14. doi: 10.1021/bi060159+. [DOI] [PubMed] [Google Scholar]
- 38.West PJ, Bulaj G, Garrett JE, Olivera BM, Yoshikami D. Biochemistry. 2002;41:15388–93. doi: 10.1021/bi0265628. [DOI] [PubMed] [Google Scholar]
- 39.Corpuz GP, Jacobsen RB, Jimenez EC, Watkins M, Walker C, Colledge C, Garrett JE, McDougal O, Li W, Gray WR, Hillyard DR, Rivier J, McIntosh JM, Cruz LJ, Olivera BM. Biochemistry. 2005;44:8176–86. doi: 10.1021/bi047541b. [DOI] [PubMed] [Google Scholar]
- 40.Cavanagh J, Fairbrother WJ, Palmer AGr, Skelton NJ. Protein NMR Spectroscopy, Principles and Practice. Academic Press; San Diego: 1996. [Google Scholar]
- 41.Guntert P. Methods Mol Biol. 2004;278:353–78. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 42.Bulaj G, DeLaCruz R, Azimi-Zonooz A, West P, Watkins M, Yoshikami D, Olivera BM. Biochemistry. 2001;40:13201–8. doi: 10.1021/bi010683a. [DOI] [PubMed] [Google Scholar]
- 43.Cruz LJ, Gray WR, Olivera BM, Zeikus RD, Kerr L, Yoshikami D, Moczydlowski E. J Biol Chem. 1985;260:9280–8. [PubMed] [Google Scholar]
- 44.Shon KJ, Olivera BM, Watkins M, Jacobsen RB, Gray WR, Floresca CZ, Cruz LJ, Hillyard DR, Brink A, Terlau H, Yoshikami D. J Neurosci. 1998;18:4473–81. doi: 10.1523/JNEUROSCI.18-12-04473.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bulaj G, West PJ, Garrett JE, Watkins M, Zhang MM, Norton RS, Smith BJ, Yoshikami D, Olivera BM. Biochemistry. 2005;44:7259–65. doi: 10.1021/bi0473408. [DOI] [PubMed] [Google Scholar]
- 46.Zhang MM, Fiedler B, Green BR, Catlin P, Watkins M, Garrett JE, Smith BJ, Yoshikami D, Olivera BM, Bulaj G. Biochemistry. 2006;45:3723–32. doi: 10.1021/bi052162j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1 summarizes chemical shifts. Supplemental Figure S1 shows the overlaid 2D [ 13C, 1H] HSQC spectra of C2, C15 and C2,C3,C10,C15,C20,C21 15N-,13C-enriched μ-SxIIIA. Supplemental Figure S2 shows a stereoview of a representative μ-SxIIIA structure calculated from the twelve NOEs listed in Figure 5, and the same molecule superimposed on the cysteine heavy atoms of μ-SmIIIA coordinates (1Q2J.pdb). Supplemental Figure S3 is a bar graph showing the average Cα-Cα (white) and Cβ-Cβ (grey) distances for all possible cystines in ten calculated μ-SxIIIA structures. This material is available free of charge via the Internet at http://pubs.acs.org.