

Abstract
Array comparative genomic hybridization (CGH) is useful to assess genome-wide chromosomal imbalance, but the requirement for relatively large amounts of DNA can be a limitation, in particular for samples extracted from small tumor areas on paraffin sections. Whole genome amplification (WGA) can be performed before array CGH to obtain sufficient DNA, but the possibility of artifacts attributable to biased amplification cannot be excluded. We optimized the WGA protocol to generate sufficient DNA with minimum amplification bias. Using formalin-fixed, paraffin-embedded histological sections of tumors carrying known TP53 mutations, LOH 1p, LOH 10q, LOH 19q, and EGFR amplification, we first optimized the protocol so that these genetic alterations were detected after WGA. We found that a ligation step before WGA is important because it allows a short reaction time with Phi29 to generate WGA-DNA with greatly decreased amplification bias. Using template >150 ng of DNA, a ligation step before WGA, and a short reaction time with Phi29 DNA polymerase (<1.5 hours), we obtained WGA-DNA (>4 μg) with minimum amplification bias (less than threefold). Using this protocol, we performed array CGH (Agilent 105K) before and after WGA. Pearson correlation analysis indicated a significant positive correlation in array CGH results between DNA before and after WGA (P < 0.0001). These results suggest that genetic analyses are possible using WGA-DNA extracted from paraffin sections, but that they should be performed with a carefully optimized and controlled protocol.
Array comparative genomic hybridization (CGH) is a useful method to detect amplifications and deletions at the whole-genome level and has contributed to the detection of novel gene loci associated with pathogenesis of various human neoplasms.1,2,3 However, array CGH requires a relatively large amount of DNA (>2 μg), and this has been a major limitation impeding application of array CGH to certain samples, in particular DNA extracted from small tumor areas or from single-cell, laser-captured samples on formalin-fixed, paraffin-embedded sections. To overcome this limitation, whole genome amplification (WGA) methods have been developed. Previously, most WGA methods were polymerase chain reaction (PCR)-based (using Taq polymerase), such as degenerated oligonucleotide-primed,4,5 ligation-mediated,6 and primer extension preamplification7 PCR procedures. However, several studies have suggested that these methods may generate significant sequence representation bias and artifacts during the WGA process.8
More recently, a non-PCR-based isothermal method, multiple displacement amplification, has been applied to small DNA samples, leading to the synthesis of DNA with less sequence representation bias.9,10 This method is based on the annealing of random hexamers to denatured DNA, followed by strand-displacement synthesis at a constant temperature, resulting in DNA products of high molecular weight.11 The reaction is catalyzed by Phi29 DNA polymerase or a large fragment of the Bst DNA polymerase, which results in error rates 100 times lower than with Taq polymerase.12,13 Spits and colleagues14 compared Phi29 and Bst DNA polymerases, and showed that the Phi29 generated accurately sufficient DNA, whereas the Bst showed low efficiency and high error rate during WGA. Most of the WGA studies recently published use Phi29.8,10,14,15,16,17 Several studies have demonstrated that WGA using Phi29 DNA polymerase yielded significantly less amplification bias than PCR-based methods,8,10,15 and validated the use of Phi29 DNA polymerase for WGA on high-quality genomic DNA extracted from 1 to 1000 cells.14,16,17
In contrast, only a few studies have successfully applied multiple displacement amplification-based WGA with DNA from formalin-fixed, paraffin-embedded tissues,18,19,20 in which the DNA is degraded because of formalin fixation, containing strand breaks, base damage, and DNA-protein crosslinks.21,22 BAC array CGH showed similar results before and after eightfold WGA with Klenow enzyme, when 50 ng of template DNA extracted from paraffin sections was used,19 but the amounts of amplified DNA were too small to be applied to recent commercially available platforms. In one study with 10 ng of template DNA extracted from paraffin sections, WGA using Bst DNA polymerase produced a median 990-fold DNA amplification, but 27 K BAC array CGH showed that most of the deletions observed in non-WGA DNA were not reproduced after WGA, although representational distortion of threefold was achieved by quantitative PCR analysis in six genes.18 Bredel and colleagues20 reported that WGA-DNA from paraffin sections could be used for array CGH using Phi29, but normalization using amplified reference DNA was necessary because of significant amplification bias.
The objective of the present study was to establish a Phi29-based WGA method using small amounts of DNA extracted from formalin-fixed, paraffin-embedded tissues for array CGH. We have optimized several critical steps including DNA extraction, amount of template DNA, and reaction time with Phi29. Because one of the key factors leading to failure of unbiased WGA on DNA extracted from paraffin sections may be the presence of highly degraded DNA with short fragments, we also assessed whether a ligation step before WGA improves the results. To validate WGA, we used tumor samples on paraffin sections, in which known TP53 mutations, LOH 1p, LOH 10q, LOH 19q, and EGFR amplification were present.
Materials and Methods
Tumor Samples
Genetic alterations in two tumor samples (glioblastomas) on formalin-fixed, paraffin-embedded histological sections were previously reported.23 One sample (tumor 1) had LOH 1p, EGFR amplification, and a TP53 mutation (GTT→CTT, Val274Leu in exon 8) and another (tumor 2) had LOH 1p, LOH 10q, LOH 19q, and a TP53 mutation (ATG→ATA, Met237Ile in exon 7). Both tumor samples were fixed in buffered formalin and embedded in paraffin more than 15 years ago. Paraffin sections were cut to 4 to 5 μm thickness 5 years ago, and stored at room temperature. Using these samples, we assessed several critical steps mentioned below, to ensure that these genetic alterations were reproducibly detected after WGA.
DNA Extraction
Paraffin sections were deparaffinized in xylene for 15 minutes and then put into 100%, 95%, and 70% ethanol for 5 minutes each and then into distilled water. After air-drying, tumor areas on the sections were scraped off the slide and washed with PBS (pH 7.4) solution twice, suspended in 400 μl of 1 N NaSCN solution, and incubated at 37°C overnight. Samples were then suspended in 400 μl of DNA extraction buffer (mixture of 360 μl of ATL buffer and 40 μl of proteinase K; DNeasy mini kit, Qiagen, Valencia, CA), and were incubated at 55°C overnight. Additional proteinase K (80 μl) was added and incubated for a total of 60 hours. After reaction with 8 μl of RNase (100 mg/ml) for 2 minutes at room temperature, the samples were made up with 420 μl of ATL to a total volume of 900 μl and separated into two parts (450 μl each). Each part was mixed with 450 μl of AL buffer and 450 μl of 100% ethanol, and incubated at room temperature for 5 minutes. The samples were then loaded onto DNeasy mini spin columns (DNeasy mini kit, Qiagen, CA). The column was washed with AW1 buffer and the column membrane dried with 80% ethanol, after which the purified genomic DNA was eluted with 25 μl of nuclease-free water. The DNA concentration was determined by spectrophotometric absorption at 230, 260, and 280nm and the quality was calculated as the A260/A230 and A260/A280 ratio.
Phi29-Based Amplification of Genomic DNA
Genomic DNA (10, 50, 150, 300 ng) samples were ligated at 24°C for 30 minutes with ligase, and then amplified with Phi29 enzyme at 30°C for 30, 60, 90, or 180 minutes. Other samples were not ligated before WGA. Ligation and amplification with Phi29 enzyme were performed using components of a Qiagen FFPE amplification kit. Briefly, various amounts of purified genomic DNA in a total volume of 10 μl were heated to 95°C for 5 minutes for denaturation. After cooling on ice for 5 minutes, 8 μl of FFPE buffer, 1 μl of ligation enzyme, and 1 μl of FFPE enzyme were added, and then incubated at 24°C for 30 minutes, followed by heat inactivation at 95°C for 5 minutes. After ligation, 20-μl samples were mixed with 30 μl of a prepared reaction mixture (29 μl of reaction buffer and 1 μl of Midi Phi29 enzyme per reaction), and were incubated at 30°C for different reaction times (30 to 180 minutes). After amplification, the Phi29 enzyme was inactivated by heating at 95°C for 10 minutes.
Direct DNA Sequencing for TP53 Mutations
To identify TP53 mutations in exon 7 or exon 8 in WGA products, PCR amplification was performed as previously described.24 Briefly, PCR was performed in a total volume of 10 μl, consisting of 1 μl each of WGA products (concentration, ∼100 ng/μl), 0.5 U of Platinum TaqDNA polymerase (Invitrogen, Cergy Pontoise, France), 1 mmol/L MgCl2, 0.1 mmol/L of each dNTP, 0.2 mmol/L of each primer, 10 mmol/L Tris-HCl, pH 8.3, and 50 mmol/L KCl in a thermal cycler (Biometra, Archamps, France) with an initial denaturing step at 95°C for 5 minutes, followed by 37 cycles of denaturation at 95°C for 50 seconds, annealing at 60°C (exon 7) or 61°C (exon 8) for 60 seconds, extension at 72°C for 60 seconds, and a final extension at 72°C for 5 minutes. The sequencing reaction was performed using a Big Dye Terminator cycle sequencing kit (ABI Prism; Applied Biosystems, Foster City, CA) in an ABI 3100 Prism DNA sequencer (Applied Biosystems).
Loss of Heterozygosity (LOH) on Chromosomes 1p, 10q, and 19q
LOH analysis was performed using two microsatellite markers (D10S536, D10S1683) on chromosome 10q, two (D19S408, D10S596) on chromosome 19q, and two (D1S2736, D1S468) on chromosome 1p. For markers D1S2736 and D19S408, PCR reactions were performed in a total volume of 12.5 μl with 6.25 μl of 2× TaqMan Universal PCR Master Mix (Applied Biosystems), 4 μl of primer sets (1.25 μmol/L of each primer), 1.25 μl of 1.5 μmol/L probe [21-bp oligomer complementary to the microsatellite CA repeat: 5′,6-carboxyfluorescein (FAM)-TGTGTGTGTGTGTGTGTGTGT-3′,6-carboxy-tetramethylrhodamine], and 10 ng of DNA. For markers D10S536, D10S1683, D1S468, D19S596, PCR reactions were performed in a total volume of 18.75 μl with 9.375 μl of 2× AmpliTaq Gold PCR Master Mix (Roche, Basel, Switzerland), 6 μl of primer sets (2.5 μmol/L of each primer), 1.875 μl of 1.5 μmol/L of the same probe as above, and 20 ng of DNA, with cycling parameters as reported previously.25
PCR was performed for each individual DNA sample in triplicate on a 96-well optical plate with an ABI 7900HT PCR system (Applied Biosystems). Amplification of a pool of six reference loci served to normalize for differences in the amount of total input DNA, as described previously. To calculate the average δCt [δCT (normal)], DNA was isolated from 10 formalin-fixed, paraffin-embedded normal tissues. The Ct, δCt [Ct (microsatellite) − Ct (reference pool)], δδCt [δCt (tumor) − δCt (normal)] values, the relative copy number (2−δδCt), and the tolerance interval with confidence of 95% determined from the pooled SD of normal DNA for the loci were calculated as reported previously. On the basis of this tolerance interval, copy numbers <1.33 were considered to represent losses. Samples showing LOH 1p, 10q, and 19q with two markers were considered as LOH. As a control, different amounts (1, 2, 5, 10, and 20 ng) of unamplified genomic DNA (gDNA) were used to make a standard curve by real-time quantitative PCR to calculate the loci representation of the six loci in WGA products. This allows determination of whether results of LOH identification might be attributable to PCR amplification of native DNA or WGA products.
EGFR Amplification
EGFR amplification was detected by differential PCR as previously described with some modification26 using the cystic fibrosis (CF) sequence as a reference. The primer sequences were as follows: 5′-AGCCATGCCCGCATTAGCTC-3′ (sense) and 5′-AAAGGAATGCAACTTCCCAA-3′ (antisense) for EGFR and 5′-GGCACCATTAAAGAAAATATCATCTT-3′ (sense) and 5′-GTTGGCATGCTTTGATGACGCTTC-3′ (antisense) for the CF reference gene. The sizes of the PCR fragments were 110 bp for EGFR and 79 bp for CF. The mean EGFR/CF ratio using DNA from peripheral blood of healthy adults was 0.73, with a standard variation of 0.20. A threshold value of 1.79 was regarded as evidence of EGFR amplification, as previously reported.27
Locus Representation
Locus representation relative to the unamplified genomic DNA was analyzed with six loci in genomic DNA, including D10S536, D10S1683 on 10q, D1S2736, D1S468 on 1p, and D19S408, D19S596 on 19q. For the six loci, quantitative PCR reactions were performed in a total volume of 12.5 μl for markers D1S2736 and D19S408 or 18.75 μl for D10S536, D10S1683, D1S468, and D19S596, as described above.
Unamplified genomic DNA (gDNA) was used to generate a standard curve of 1, 2, 5, 10, and 20 ng to quantify the WGA products on the six loci. The standard curve was used for determination of locus copy numbers in the WGA product by determining the threshold cycle number by real-time PCR. Locus representation (WGA/gDNA) is reported as a percentage. Amplification bias between two loci is the ratio between the two locus representation values.10 The mean value of amplification bias in all tested loci was calculated to assess the amplification bias in the WGA products.
Array CGH
The WGA reaction mixture was purified with a NucleoTraPCR kit (Macherey-Nagel, UK), a MicroSpin G-50 column (GE Healthcare, Chalfont St Giles, UK), or agarose gel electrophoresis followed by NucleoTraPCR kit, before DNA labeling for array CGH. With the NucleoTraPCR kit, the volume of the reaction mixture was adjusted to 100 μl using PBS (pH 7.4), and then 400 μl of buffer NT was added. After mixture with thorough stirring, 10 μl of the NucleoTraPCR suspension was added to the reaction mix and was incubated at room temperature for 10 minutes. Samples were centrifuged at 10,000 × g for 30 seconds and supernatants were discarded. Pellets were washed with 400 μl of buffers NT2, NT3, and NT3, respectively. The silica matrix was dried at room temperature for 15 minutes. Nuclease-free water (30 μl) was added to the pellet, which was resuspended by stirring. The mixture was incubated at 55°C for 15 minutes. Samples were then centrifuged at 10,000 × g for 30 seconds and supernatants were transferred to clean tubes. With the MicroSpin G-50 column, 50 μl of reaction mixture was loaded onto the prepared column, and centrifuged for 2 minutes at 735 × g and the purified samples were collected in the bottom of the microcentrifuge tube.
For agarose gel electrophoresis followed by NucleoTraPCR kit, 50 μl of reaction mixture was loaded onto a 0.9% TBE agarose gel and electrophoresis was performed at 150 V for 30 minutes. After electrophoresis, gel slices containing the fragments (>500 bp) were excised, and NT1 buffer was added (300 μl/100 mg agarose gel). Then NucleoTraPCR suspension was added (4 μl/μg of DNA) and the sample was incubated at 50°C until the gel slices were dissolved. The sample was then centrifuged for 30 seconds at 10,000 × g and the supernatant was discarded. Pellets were washed with 400 μl of buffers NT2, NT3, and NT3, respectively. The silica matrix was dried at room temperature for 15 minutes. Nuclease-free water (30 μl) was added to the pellet, which was resuspended by stirring. The mixture was incubated at 55°C for 15 minutes. Samples were then centrifuged at 10,000 × g for 30 seconds and supernatants were transferred to clean tubes. The yield of WGA products before and after purification was determined by the PicoGreen dsDNA method (Invitrogen, CA) and UV absorption method.
The genomic profile changes of paired DNA samples before and after WGA were compared using a 105K CGH oligonucleotide microarrray (Agilent Technologies, Santa Clara, CA) according to the manufacturer's instructions. Briefly, 1 to 2 μg of sample and sex-matched reference DNA were chemically labeled, respectively, with ULS-Cy5 and ULS-Cy3 at 85°C for 30 minutes (Oligo aCGH labeling kit for FFPE samples, Agilent). Labeled samples were purified (Genomic DNA purification module, Agilent), combined, mixed with human Cot-1 DNA, and denatured at 95°C (Oligo aCGH hybridization kit). The mixture was applied to microarrays and hybridization was performed at 65°C for 40 hours. After hybridization, the microarrays were washed in Oligo aCGH wash buffer 1 at room temperature for 5 minutes and wash buffer 2 at 37°C for 1 minute. After drying, the microarrays were scanned using a DNA microarray scanner G2565BA (Agilent) and data (log2) were extracted from raw microarray image files using Feature Extraction software (version 9; Agilent). Data were analyzed by DNA Analytics software (version 4.0; Agilent) with default filter settings. The aberration detection method 2 (ADM2) algorithm with centralization and fuzzy zero correction was used to define aberrant intervals. Each of the arrays was independently analyzed and evaluated for genetic alterations before and after WGA amplification.
Statistical Analyses
To assess the similarity between DNA with or without WGA, Pearson's correlations of log2 ratios of probes from autosomes among tumor 1 and tumor 2 samples were performed with Graphpad Prism software (GraphPad, La Jolla, CA).
Results
Yield of WGA Products
WGA produced DNA of high molecular weight (0.5 ∼ 20 kb; Figure 1). The negative control (H2O) did not generate any DNA product when samples were incubated with Phi29 for <1.5 hours (Figure 1). A ligation step before WGA significantly affected the results. Samples treated with a ligation step, but not those without ligation, generated WGA products when the reaction time with Phi29 was less than 1.5 hours (Table 1, Figure 2).
Figure 1.
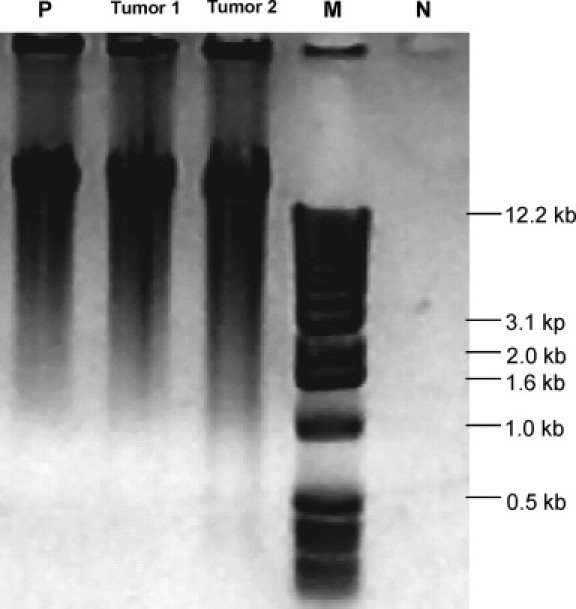
Agarose gel electrophoresis of WGA-DNA (0.9% agarose gel). WGA was performed with Phi29 (reaction time of 1.5 hours) and with 10 ng of template DNA for lambda DNA (P) and 300 ng for DNA extracted from tumors 1 and 2 on paraffin sections. For DNA from tumors 1 and 2, a ligation step was added before WGA. Note that WGA-DNA samples from tumor 1 and tumor 2 show similar amounts and similar molecular weights of DNA (0.5 ∼ 20 kb) with WGA-lambda DNA. M, 1-kb DNA ladder marker; N, negative control (H2O), showing that the negative control does not generate DNA product.
Table 1.
Genetic Alterations Detected after Whole Genome Amplification
| With ligation step |
Without ligation step |
|||||||
|---|---|---|---|---|---|---|---|---|
| 10 ng§ | 50 ng | 150 ng | 300 ng | 10 ng | 50 ng | 150 ng | 300 ng | |
| TP53 mutations* | ||||||||
| 0.5 hour† | +, + | +, + | +, + | +, + | No WGA product¶ | |||
| 1.0 hour | +, − | +, + | +, + | +, + | No WGA product | |||
| 1.5 hours | +, − | −, − | +, − | +, + | +, + | +, + | +, + | +, + |
| 2.0 hours | nd | nd | nd | nd | +, + | +, + | +, + | +, + |
| 3.0 hours | −, − | −, − | +, − | +, − | +, − | +, − | +, + | +, + |
| EGFR amplification | ||||||||
| 0.5 hour | nd | + | − | + | No WGA product | |||
| 1.0 hour | − | − | + | + | No WGA product | |||
| 1.5 hours | − | + | − | − | nd | + | + | + |
| 2.0 hours | nd | nd | nd | nd | − | + | + | + |
| 3.0 hours | − | + | − | + | − | + | + | + |
| LOH 1p‡ | ||||||||
| 0.5 hour | nd | +, − | +, + | +, + | No WGA product | |||
| 1.0 hour | −, − | −, − | +, + | +, + | No WGA product | |||
| 1.5 hours | −, − | +, + | −, − | +, − | −, − | −, − | +, + | +, + |
| 2.0 hours | nd | nd | nd | nd | −, − | +, − | +, + | +, + |
| 3.0 hours | −, − | +, − | +, − | +, − | −, − | +, + | +, − | +, − |
| LOH 10q | ||||||||
| 0.5 hour | nd | + | + | + | No WGA product | |||
| 1.0 hour | − | + | + | + | No WGA product | |||
| 1.5 hours | + | + | + | + | − | − | + | + |
| 2.0 hours | nd | nd | nd | nd | − | − | − | − |
| 3.0 hours | − | + | + | + | − | + | + | − |
| LOH 19q | ||||||||
| 0.5 hour | nd | + | + | + | No WGA product | |||
| 1.0 hour | − | + | + | + | No WGA product | |||
| 1.5 hours | + | + | − | + | − | + | + | + |
| 2.0 hours | nd | nd | nd | nd | + | − | + | + |
| 3.0 hours | − | + | + | + | − | + | + | + |
Results on TP53 mutations were scored as +, +, both TP53 mutations detected in exon 7 (ATG->ATA, codon 237) and in exon 8 (GTT->CTT, codon 274); +, −, one of the two mutations detected; −, −, none of the mutations detected.
Reaction time with Phi29.
Results on LOH were scored as +, +, LOH 1p (D1S2736/D1S468) detected in both tumors 1 and 2; +, −, one of the two LOH detected; −, −, none of the LOH detected.
Amount of template DNA before WGA.
Amount of DNA after WGA was not different from those before WGA in any sample irrespective of different amount of template DNA; nd, not determined.
Figure 2.
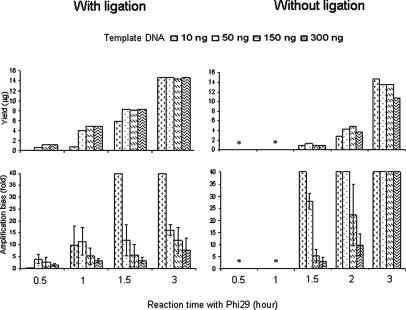
Top: Mean yield of Phi29-based WGA-amplified DNA. Note that the reaction time but not the amount of template DNA affects the yield. In samples without ligation, there was no WGA product when the reaction time with Phi29 was less than 1 hour, irrespective of the amount of template DNA. Bottom: Amplification bias in WGA products calculated from the data on copy number of six markers (D10S536 and D10S1683 at 10q, D1S2736 and D1S468 at 1p, and D19S408 and D19S596 at 19q) in WGA-amplified DNA (see Materials and Methods). Samples with a ligation step showed lower amplification bias in WGA products compared with samples without ligation. Error bars represent SD. Note that a higher amount of template DNA and shorter reaction time with Phi29 generate lower amplification bias. *No production of WGA-DNA.
WGA using 10 to 300 ng of DNA generated up to 15 μg of WGA-DNA, measured by the PicoGreen method (Figure 2). Samples without a ligation step, incubated with Phi29 for <1 hour, did not produce WGA-DNA, irrespective of the amount of template DNA (Figure 2). The ligation step did not significantly affect the yield of WGA-DNA after a 3-hour reaction with Phi29 (Figure 2). With the ligation step, template DNA (10 ng) gave a significantly lower yield of WGA products when incubated for <1.5 hours, but at 3 hours, all samples reached similar levels of WGA products (Figure 2). Fold changes depended on the amount of template DNA. Ten ng of template DNA gave changes of 14- to 1578-fold, whereas 300 ng of template DNA yielded only 3- to 50-fold changes.
Amplification Bias in WGA Products
Real-time quantitative PCR was performed at six chromosomal loci (1p36.2, 1p36.3, 10q23, 10q25, 19q13.2, 19q13.3) to assess amplification bias. The ligation step significantly reduced the level of amplification bias (Figure 2). Samples with >150 ng template DNA, with a ligation step, and with reaction times of <1.5 hours showed the least amplification bias (Figure 2), with a mean 26-fold (>4 μg of WGA product) with <3-fold amplification bias.
Detections of Genetic Alterations
TP53 Mutations
Before WGA, sample tumor 1 showed a TP53 mutation (GTT->CTT, codon 274, exon 8), and sample tumor 2 contained a TP53 mutation (ATG->ATA, codon 237 in exon 7). After WGA, TP53 mutations were detected in 92% (22 of 24) of samples without ligation, but in only 63% (20 of 32) of samples with ligation (Table 1). Correct results were obtained in WGA-DNA when samples with ligation were incubated with Phi29 for 0.5 to 1 hour, and when samples without ligation were incubated for 1.5 to 2.0 hours.
LOH
Before WGA, sample tumor 1 showed LOH 1p, and sample tumor 2 had LOH 1p, 10q, and 19q. After WGA, LOH was detected in samples with ligation (67%, 40 of 60), and in 50% (24 of 48) of samples without ligation (Table 1). The amount of template DNA and reaction time affected the results significantly. Correct results were obtained when samples with ligation (>150 ng of template DNA) were incubated with Phi29 for 0.5 to 1 hour. Loci representation of the six loci in WGA products determined by a standard curve derived from different amounts of unamplified genomic DNA shows that the results of LOH identification were not attributable to PCR amplification of native DNA.
EGFR Amplification
Before WGA, sample tumor 1 had EGFR amplification (7p12.3–12.1). After WGA, EGFR amplification was detected in samples with ligation (7 of 15, 47%) and samples without ligation (9 of 11, 82%; Table 1). The amount of template DNA affected the results significantly, but changing the reaction time with Phi29 did not affect the results. EGFR amplification was detected in most WGA products when we used >50 ng of template DNA. In summary, WGA-DNA obtained with sufficient template DNA (>150 ng), a ligation step, and a short reaction time with Phi29 (1 hour) gave correct results for all TP53 mutations, LOH 1p, LOH 10q, LOH 19q, and EGFR amplification (Table 1).
Array-CGH Analysis of WGA Products
Based on the results mentioned above, we chose the following conditions for WGA: 300 ng of template DNA, ligation before WGA, and reaction with Phi29 for 1 hour. After WGA, we obtained an ∼20-fold amount of DNA, with a mean value of amplification bias of 2.8-fold (Figure 2).
QC metrics, an indicator of baseline noise of log ratios of array CGH, indicated that the WGA samples from both tumors 1 and 2 had wide distribution of probe signals with DLRSpread of 0.55 to 0.7, suggestive of poor samples. WGA samples showed as many probes with high and low log ratios, thus making it difficult to calculate copy number changes, particularly in regions with low copy number changes, eg, LOH. The background noise signal was significantly reduced by application of agarose gel electrophoresis during the purification step before array CGH. However, different methods for purification of WGA-DNA before array CGH (see Materials and Methods) gave similar results on genome profiles (r = 0.8690, P < 0.0001).
Pearson correlation analysis showed a significant overall positive correlation in genome profiles (normal, gain, or loss) between samples before and after WGA for both tumors 1 and 2 (Table 2), with a mean value of r = 0.425 (P < 0.0001). Figure 3 shows array CGH results for representative chromosomes using DNA samples before and after WGA. In addition, array CGH using DNA samples both before and after WGA successfully detected known genetic alterations, ie, EGFR amplification (Figure 4) and LOH 10q23, 10q25, and 19q13.3.
Table 2.
Correlation of Genome Profiles Obtained by Array CGH between Samples Before and After WGA
| Correlation | Tumor 1 | Tumor 2 |
|---|---|---|
| Number of autosomal probes | 94,613 | 94,592 |
| Pearson r | 0.321 | 0.542 |
| 95% Confidence interval | 0.316 to 0.327 | 0.537 to 0.546 |
| P value (two-tailed) | <0.0001 | <0.0001 |
Pearson's correlations of log2 ratio values of autosomal probes among tumors 1 and 2 were calculated with Graphpad Prism software.
Figure 3.
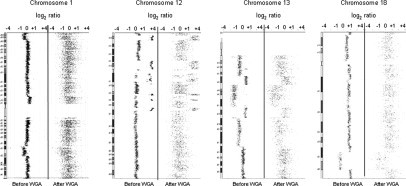
Array-CGH results using DNA before and after WGA. The genome profiles of selected chromosomes 1, 12, 13, and 18 of tumor 2 show similar gain and loss patterns in samples before and after WGA, despite the fact that samples after WGA show high noise.
Figure 4.
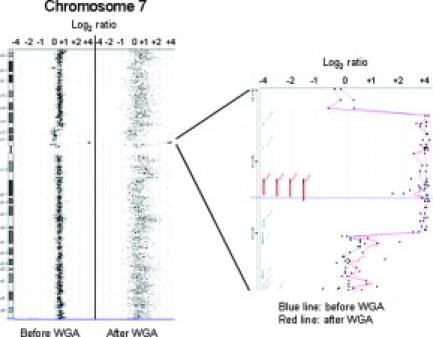
Array-CGH analysis in WGA-DNA reproduced results on EGFR amplification at 7p12.1 in tumor 1.
Discussion
DNA extracted from paraffin sections tends to be highly degraded and fragmented; the degree of damage depending on factors such as the length of formalin fixation and storage conditions.21,22 Accordingly, the products of WGA using DNA extracted from paraffin sections usually show such limitations in terms of higher amplification bias compared with samples obtained using high-quality DNA extracted from frozen tissue.17,20,28
In the present study, using tumor samples on histological sections that carry TP53 mutations, EGFR amplification, and LOH 1p, LOH 10q, LOH 19q, we optimized several critical steps of the WGA protocol to reproducibly detect these genetic alterations after WGA. We found that WGA using sufficient amounts of template DNA (>150 ng), with a ligation step before WGA, and a short reaction time with Phi29 DNA polymerase (<1.5 hours) could generate >4 μg of DNA (∼26-fold amplification; sufficient for array CGH analyses), with amplification bias of less than threefold. Compared with previous studies using paraffin DNA in which amplification bias was considerably higher,20 the present study achieved levels of amplification bias similar to those seen with Phi29-based WGA using high-quality DNA extracted from frozen tissues.10,17,29 With this protocol, we were able to detect most of the genetic alterations in the tumor samples (Table 1).
The key improvement in the present study was to add a ligation step before WGA, so as to generate sufficient WGA-DNA with shorter Phi29 reaction time (Figure 2) and greatly decreased amplification bias (Figure 2). In contrast to several studies that showed artificial DNA products in negative controls (H2O) during WGA after long reaction times (>6 hours) with Phi29,18,30 there was no such problem in negative controls in the present study (<1.5 hours of reaction time).
We found that although a low quantity of template DNA (eg, 10 ng) could generate sufficient WGA-DNA after long reaction times with Phi29, the amplification bias was too high to allow reproducible results (Table 1, Figure 2). It is thus recommended to start with >150 ng of template DNA, which can be easily extracted from paraffin sections (tumor areas of ∼3 to 5 mm in diameter from one to two paraffin sections with 3 to 4 μm thickness). However, the amount of DNA extracted by laser-captured single-cell microdissection on paraffin sections may be insufficient, leading to significant amplification bias with current protocols.
Array CGH analyses showed that WGA-DNA had higher background compared with template DNA (Figure 3), which makes the interpretation of the results difficult. A similar observation was reported by Bredel and colleagues,20 who used DNA from paraffin sections. Nevertheless, there was a significant positive correlation of array CGH results between DNA before and after WGA (Pearson's correlation, P < 0.001), suggesting that despite the high background, WGA-DNA samples may be used for array CGH analyses. Furthermore, array-CGH analysis confirmed most of the known genetic alterations in both samples before and after WGA.
The present study shows that genetic analyses for specific alterations and array CGH analyses for genome-wide chromosomal imbalance using WGA-DNA from paraffin sections are possible. However, they should be performed with a carefully optimized protocol, and verification of the results by other methods is recommended.
Footnotes
Supported by the International Agency for Research on Cancer (postdoctoral fellowship to J.H.) and the Foundation for Promotion of Cancer Research, Japan.
References
- 1.Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, Dairkee SH, Ljung BM, Gray JW, Albertson DG. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 2.Veltman JA, Fridlyand J, Pejavar S, Olshen AB, Korkola JE, DeVries S, Carroll P, Kuo WL, Pinkel D, Albertson D, Cordon-Cardo C, Jain AN, Waldman FM. Array-based comparative genomic hybridization for genome-wide screening of DNA copy number in bladder tumors. Cancer Res. 2003;63:2872–2880. [PubMed] [Google Scholar]
- 3.Paris PL, Albertson DG, Alers JC, Andaya A, Carroll P, Fridlyand J, Jain AN, Kamkar S, Kowbel D, Krijtenburg PJ, Pinkel D, Schroder FH, Vissers KJ, Watson VJ, Wildhagen MF, Collins C, van Dekken H. High-resolution analysis of paraffin-embedded and formalin-fixed prostate tumors using comparative genomic hybridization to genomic microarrays. Am J Pathol. 2003;162:763–770. doi: 10.1016/S0002-9440(10)63873-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daigo Y, Chin SF, Gorringe KL, Bobrow LG, Ponder BA, Pharoah PD, Caldas C. Degenerate oligonucleotide primed-polymerase chain reaction-based array comparative genomic hybridization for extensive amplicon profiling of breast cancers: a new approach for the molecular analysis of paraffin-embedded cancer tissue. Am J Pathol. 2001;158:1623–1631. doi: 10.1016/S0002-9440(10)64118-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheung VG, Nelson SF. Whole genome amplification using a degenerate oligonucleotide primer allows hundreds of genotypes to be performed on less than one nanogram of genomic DNA. Proc Natl Acad Sci USA. 1996;93:14676–14679. doi: 10.1073/pnas.93.25.14676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guillaud-Bataille M, Valent A, Soularue P, Perot C, Inda MM, Receveur A, Smaili S, Roest CH, Benard J, Bernheim A, Gidrol X, Danglot G. Detecting single DNA copy number variations in complex genomes using one nanogram of starting DNA and BAC-array CGH. Nucleic Acids Res. 2004;32:e112. doi: 10.1093/nar/gnh108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mill J, Yazdanpanah S, Guckel E, Ziegler S, Kaminsky Z, Petronis A. Whole genome amplification of sodium bisulfite-treated DNA allows the accurate estimate of methylated cytosine density in limited DNA resources. Biotechniques. 2006;41:603–607. doi: 10.2144/000112266. [DOI] [PubMed] [Google Scholar]
- 8.Pinard R, de Winter A, Sarkis GJ, Gerstein MB, Tartaro KR, Plant RN, Egholm M, Rothberg JM, Leamon JH. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 2006;7:216. doi: 10.1186/1471-2164-7-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lizardi PM, Huang X, Zhu Z, Bray-Ward P, Thomas DC, Ward DC. Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat Genet. 1998;19:225–232. doi: 10.1038/898. [DOI] [PubMed] [Google Scholar]
- 10.Dean FB, Hosono S, Fang L, Wu X, Faruqi AF, Bray-Ward P, Sun Z, Zong Q, Du Y, Du J, Driscoll M, Song W, Kingsmore SF, Egholm M, Lasken RS. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci USA. 2002;99:5261–5266. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blanco L, Bernad A, Lazaro JM, Martin G, Garmendia C, Salas M. Highly efficient DNA synthesis by the phage phi 29 DNA polymerase. Symmetrical mode of DNA replication. J Biol Chem. 1989;264:8935–8940. [PubMed] [Google Scholar]
- 12.Eckert KA, Kunkel TA. DNA polymerase fidelity and the polymerase chain reaction. PCR Methods Appl. 1991;1:17–24. doi: 10.1101/gr.1.1.17. [DOI] [PubMed] [Google Scholar]
- 13.Esteban JA, Salas M, Blanco L. Fidelity of phi 29 DNA polymerase. Comparison between protein-primed initiation and DNA polymerization. J Biol Chem. 1993;268:2719–2726. [PubMed] [Google Scholar]
- 14.Spits C, Le Caignec C, De Rycke M, Van Haute L, Van Steirteghem A, Liebaers I, Sermon K. Optimization and evaluation of single-cell whole-genome multiple displacement amplification. Hum Mutat. 2006;27:496–503. doi: 10.1002/humu.20324. [DOI] [PubMed] [Google Scholar]
- 15.Uda A, Tanabayashi K, Fujita O, Hotta A, Yamamoto Y, Yamada A. Comparison of whole genome amplification methods for detecting pathogenic bacterial genomic DNA using microarray. Jpn J Infect Dis. 2007;60:355–361. [PubMed] [Google Scholar]
- 16.Hosono S, Faruqi AF, Dean FB, Du Y, Sun Z, Wu X, Du J, Kingsmore SF, Egholm M, Lasken RS. Unbiased whole-genome amplification directly from clinical samples. Genome Res. 2003;13:954–964. doi: 10.1101/gr.816903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lage JM, Leamon JH, Pejovic T, Hamann S, Lacey M, Dillon D, Segraves R, Vossbrinck B, Gonzalez A, Pinkel D, Albertson DG, Costa J, Lizardi PM. Whole genome analysis of genetic alterations in small DNA samples using hyperbranched strand displacement amplification and array-CGH. Genome Res. 2003;13:294–307. doi: 10.1101/gr.377203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aviel-Ronen S, Qi Zhu C, Coe BP, Liu N, Watson SK, Lam WL, Tsao MS. Large fragment Bst DNA polymerase for whole genome amplification of DNA from formalin-fixed paraffin-embedded tissues. BMC Genomics. 2006;7:312. doi: 10.1186/1471-2164-7-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Devries S, Nyante S, Korkola J, Segraves R, Nakao K, Moore D, Bae H, Wilhelm M, Hwang S, Waldman F. Array-based comparative genomic hybridization from formalin-fixed, paraffin-embedded breast tumors. J Mol Diagn. 2005;7:65–71. doi: 10.1016/S1525-1578(10)60010-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bredel M, Bredel C, Juric D, Kim Y, Vogel H, Harsh GR, Recht LD, Pollack JR, Sikic BI. Amplification of whole tumor genomes and gene-by-gene mapping of genomic aberrations from limited sources of fresh-frozen and paraffin-embedded DNA. J Mol Diagn. 2005;7:171–182. doi: 10.1016/S1525-1578(10)60543-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehmann U, Kreipe H. Real-time PCR analysis of DNA and RNA extracted from formalin-fixed and paraffin-embedded biopsies. Methods. 2001;25:409–418. doi: 10.1006/meth.2001.1263. [DOI] [PubMed] [Google Scholar]
- 22.Lewis F, Maughan NJ, Smith V, Hillan K, Quirke P. Unlocking the archive—gene expression in paraffin-embedded tissue. J Pathol. 2001;195:66–71. doi: 10.1002/1096-9896(200109)195:1<66::AID-PATH921>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 23.Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, Burkhard C, Schuler D, Probst-Hensch NM, Maiorka PC, Baeza N, Pisani P, Yonekawa Y, Yasargil MG, Lutolf UM, Kleihues P. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64:6892–6899. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 24.Huang J, Grotzer MA, Watanabe T, Hewer E, Pietsch T, Rutkowski S, Ohgaki H. Mutations in the Nijmegen breakage syndrome gene in medulloblastomas. Clin Cancer Res. 2008;14:4053–4058. doi: 10.1158/1078-0432.CCR-08-0098. [DOI] [PubMed] [Google Scholar]
- 25.Nigro JM, Takahashi MA, Ginzinger DG, Law M, Passe S, Jenkins RB, Aldape K. Detection of 1p and 19q loss in oligodendroglioma by quantitative microsatellite analysis, a real-time quantitative polymerase chain reaction assay. Am J Pathol. 2001;158:1253–1262. doi: 10.1016/S0002-9440(10)64076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tohma Y, Gratas C, Biernat W, Peraud A, Fukuda M, Yonekawa Y, Kleihues P, Ohgaki H. PTEN (MMAC1) mutations are frequent in primary glioblastomas (de novo) but not in secondary glioblastomas. J Neuropathol Exp Neurol. 1998;57:684–689. doi: 10.1097/00005072-199807000-00005. [DOI] [PubMed] [Google Scholar]
- 27.Rollbrocker B, Waha A, Louis DN, Wiestler OD, von Deimling A. Amplification of the cyclin-dependent kinase 4 (CDK4) gene is associated with high cdk4 protein levels in glioblastoma multiforme. Acta Neuropathol. 1996;92:70–74. doi: 10.1007/s004010050491. [DOI] [PubMed] [Google Scholar]
- 28.Wang G, Brennan C, Rook M, Wolfe JL, Leo C, Chin L, Pan H, Liu WH, Price B, Makrigiorgos GM. Balanced-PCR amplification allows unbiased identification of genomic copy changes in minute cell and tissue samples. Nucleic Acids Res. 2004;32:e76. doi: 10.1093/nar/gnh070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rook MS, Delach SM, Deyneko G, Worlock A, Wolfe JL. Whole genome amplification of DNA from laser capture-microdissected tissue for high-throughput single nucleotide polymorphism and short tandem repeat genotyping. Am J Pathol. 2004;164:23–33. doi: 10.1016/S0002-9440(10)63092-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar G, Garnova E, Reagin M, Vidali A. Improved multiple displacement amplification with phi29 DNA polymerase for genotyping of single human cells. Biotechniques. 2008;44:879–890. doi: 10.2144/000112755. [DOI] [PubMed] [Google Scholar]