
Fig. 3.
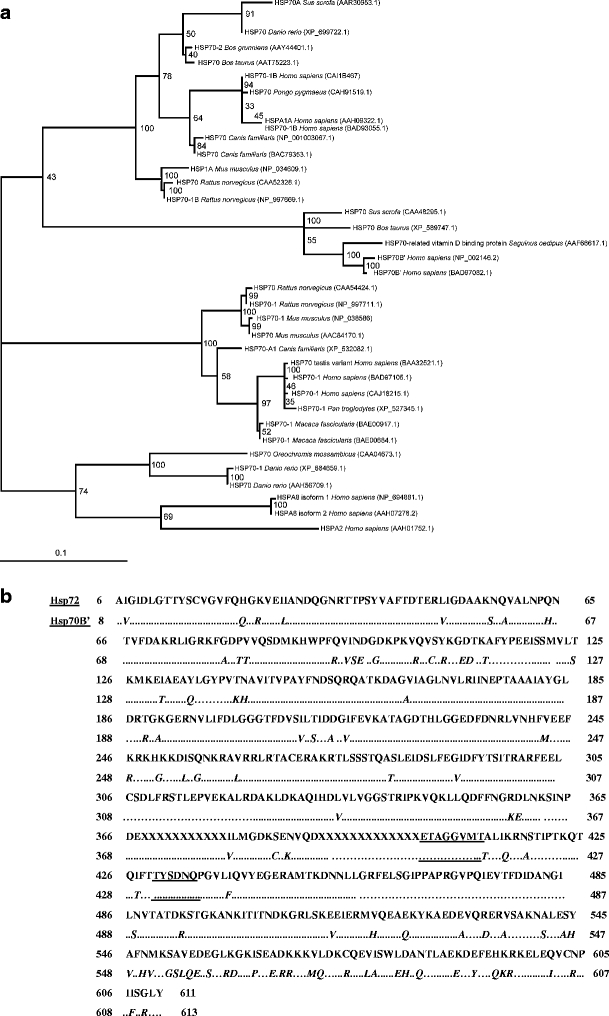
Molecular phylogenetic analysis. To establish the phylogenetic relationship of Hsp70B’ to all other known human Hps70s, protein sequences of interest were collected from the GenBank database at the National Center for Biotechnology Information (NCBI). Additionally, a BlastP search of Hsp70B’ was performed to identify the 50 most similar Hsp70 homologs in vertebrates. As a control, database searches with separated N-terminal and C-terminal regions were performed to check for possible gene fusion events (sequences with close similarity to non-Hsp proteins not identified in the search, with a full Hsp70 sequence). Identical lineage-specific duplicates (isoforms) were removed from this list before alignment. Sequence alignments were then performed using the ClustalW and MUSCLE algorithms (default settings). Phylogenetic analysis of the MUSCLE alignment was performed via bootstrapped maximum likelihood analysis using the PHYML algorithm (Guindon and Gascuel 2003). a Phylogenetic tree of vertebrate Hsp70s, most similar to the human Hsp70B’ sequence. The number at each branch indicates the number of times each node was supported in 100 bootstrap resamplings. Each protein is named based on the sequence definition in NCBI database and its corresponding accession number. Hsp70B’ and Hsp72 subgroups are indicated on the left at the appropriate branch points. The scale bar indicates and evolutionary distance of 0.1 amino acid substitution per position in the sequence. b Alignment of human Hsp72 (top) and human Hsp70B’ (bottom) protein sequences. As much of the sequence information pertaining to the location of the peptide-binding pocket is available for the bacterial Hsp70 homolog, DNAK, BlastP, and Blast2seq (NCBI database) were used to align Hsp70B’ and Hsp72 protein sequences to these proteins and extrapolate the location of the putative binding pocket. Results were checked using secondary and tertiary structure prediction software Geno3D (http://pbil.ibcp.fr/htm/index.php). Hsp70B’ and Hsp72 were then aligned to each other to determine dissimilarities and/or similarities among amino acid residues in regions of interest. Mismatches within the Hsp70B’ sequence are underlined. Sequences corresponding to the putative peptide-binding pocket are underlined