

Abstract
The activator protein 1 (AP-1) transcription factor c-Jun is crucial for neuronal apoptosis. However, c-Jun dimerization partners and the regulation of these proteins in neuronal apoptosis remain unknown. Here we report that c-Jun-mediated neuronal apoptosis requires the concomitant activation of activating transcription factor-2 (ATF2) and downregulation of c-Fos. Furthermore, we have observed that c-Jun predominantly heterodimerizes with ATF2 and that the c-Jun/ATF2 complex promotes apoptosis by triggering ATF activity. Inhibition of c-Jun/ATF2 heterodimerization using dominant negative mutants, small hairpin RNAs, or decoy oligonucleotides was able to rescue neurons from apoptosis, whereas constitutively active ATF2 and c-Jun mutants were found to synergistically stimulate apoptosis. Bimolecular fluorescence complementation analysis confirmed that, in living neurons, c-Fos downregulation facilitates c-Jun/ATF2 heterodimerization. A chromatin immunoprecipitation assay also revealed that c-Fos expression prevents the binding of c-Jun/ATF2 heterodimers to conserved ATF sites. Moreover, the presence of c-Fos is able to suppress the expression of c-Jun/ATF2-mediated target genes and, therefore, apoptosis. Taken together, our findings provide evidence that potassium deprivation-induced neuronal apoptosis is mediated by concurrent upregulation of c-Jun/ATF2 heterodimerization and downregulation of c-Fos expression. This paradigm demonstrates opposing roles for ATF2 and c-Fos in c-Jun-mediated neuronal apoptosis.
The activator protein 1 (AP-1) transcription factor signals by formation of homodimers and heterodimers among the basic-region leucine zipper (bZIP) family proteins Jun, Fos, ATF, and Maf. AP-1 dimers regulate the transcription of many target genes involved in cell differentiation, apoptosis, and stress responses via preferential binding to consensus DNA-regulatory elements (43). For example, c-Jun homodimers and c-Jun/c-Fos heterodimers bind to the consensus 12-O-tetradecanoylphorbol-13-acetate-responsive element (TRE) sequence TGAC/GTCA, whereas ATF2 homodimers and c-Jun/ATF2 heterodimers favor the ATF consensus sequence TG/TACNTCA (46). Accumulating evidence in the literature suggests that the transcriptional regulation and posttranslational regulation of AP-1 proteins are two important mechanisms in regulating the composition and distinct functions of nuclear AP-1 dimers (43).
Neuronal apoptosis contributes to the progression of neuropathological disorders such as stroke and neurodegenerative disease (34). It is well established that c-Jun N-terminal protein kinase (JNK)-dependent transactivation of c-Jun is able to mediate several neuronal death paradigms, including those initiated by β-amyloid toxicity (38), trophic factor withdrawal (7, 24), and ischemia (23). Inhibition of JNKs using either pharmacological agents or c-Jun itself via a dominant negative mutation or small hairpin RNA (shRNA) is able to prevent apoptosis (15, 31, 35, 53), thus indicating the importance of c-Jun signaling in neuronal death (9). Our lab previously indentified death protein 5/harakiri (dp5), a BH3-only Bcl-2 family member, as a critical target gene of c-Jun in neuronal apoptosis (31). However, the dimerization partners of c-Jun and their regulation in neuronal apoptosis remained unknown.
ATF2 is an AP-1 transcription factor constitutively expressed in the brain that dimerizes with c-Jun to exert its function in the cell. Some reports suggest that ATF2 contributes to neuronal survival, and rapid and long-lasting suppression of ATF2 expression has been characterized as a common response to neuronal injury (33). Conversely, ATF2 phosphorylation is observed to increase in neurons upon extreme stimuli (25, 50), suggesting a controversial role for ATF2 in neuronal apoptosis.
c-Fos has been characterized as another important dimerization partner for c-Jun (43). c-Fos has been demonstrated to be essential for stress-evoked death in retinal cells (13, 52). However, in some neurons, including cerebellar granule neurons (CGNs), high levels of c-Fos expression are indispensable for cell differentiation and survival (6, 56). It is therefore unclear how c-Fos signaling participates in neuronal apoptosis.
In this study we investigated the roles of ATF2 and c-Fos in c-Jun-mediated CGN apoptosis induced by potassium withdrawal. We demonstrate that potassium deprivation leads to JNK-dependent transactivation of ATF2 and c-Jun and results in c-Fos downregulation via reduced calmodulin-dependent protein kinase (CaMK) activity. c-Jun/ATF2 heterodimers mediate neuronal apoptosis, and c-Fos downregulation facilitates c-Jun/ATF2 heterodimerization and subsequent apoptosis.
MATERIALS AND METHODS
CGN cultures.
Rat CGNs were prepared from 7- to 8-day-old Sprague-Dawley rat pups (15 to 19 g) as described previously (27, 28, 31). Potassium deprivation and apoptosis assays were performed as described previously (27, 28, 31). More than 800 transfected cells were counted for each treatment. For administration of inhibitors, DIV7 CGNs maintained in 25 mM KCl (25 K) medium plus serum were switched to 25 K or 5 mM KCl (5 K) medium in the presence or absence of the inhibitors SP600125 (Calbiochem), CEP11004 (a kind gift from Cephalon Inc.), nifedipine (Sigma), and KN62 (Calbiochem). Dimethyl sulfoxide was used as a control. The final concentration of dimethyl sulfoxide was less than 0.1%.
Western blotting.
Western blotting analyses were performed as described previously (27, 28, 31). Primary antibodies included phospho-ATF2 (Thr69 Thr71), phospho-c-Jun (Ser73) (Cell Signaling Technology), ATF2, c-Fos (Santa Cruz Biotechnology), c-Jun (BD Bioscience), tubulin, Flag (Sigma), and V5 tag antibody (AbD Serotec).
Reverse transcription (RT)-PCR.
Total RNA was extracted and isolated from CGNs using the TRIzol reagent (Invitrogen) as described previously (31). With the Primer Premier 5.0 software, we designed primers that were specific for c-fos (forward, 5′-CAGCCTTTCCTACTACCAT-3′, and reverse, 5′-CTTATTCCTTTCCCTTCG-3′), β-actin (forward, 5′-CAACTGGGACGATATGGAGAAG-3′, and reverse, 5′-TCTCCTTCTGCATCCTGTCAG-3′), c-jun (forward, 5′-TGGGCACATCACCACTACAC-3′, and reverse, 5′-AGTTGCTGAGGTTGGCGTA-3′), dp5 (forward, 5′-AGACCCAGCCCGGACCGAGCAA-3′, and reverse, 5′-ATAGCACTGAGGTGGCTATC-3′), and atf3 (forward, 5′-TCACCTCCTGGGTCACTG-3′, and reverse, 5′-CCGCCTCCTTTTTCTCTC-3′).
IP and immunodepletion.
Immunoprecipitation (IP) assays were performed as described previously (28). For each trial, neuronal extracts composed of 6.0 × 106 cells were prepared by solubilization in 400 μl cell lysis buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 5 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride, 1 μM aprotinin) for 10 min at 4°C. After brief sonication, the lysates were cleared by centrifugation at 15,000 × g for 10 min at 4°C, and the cell extract was immunoprecipitated with 2 μg of ATF2 (Santa Cruz Biotechnology), c-Jun (BD Biosciences), or c-Fos antibody (Cell Signaling Technology) and incubated with 30 μl protein A plus G agarose hydrazide beads (Calbiochem). For immunodepletion assays, IP with the specified antibody was performed in three rounds. The immunocomplexes were washed three times with cell lysis buffer, and the supernatants were subjected to Western blotting. The percentages of specific proteins in the supernatant were calculated by normalization to those in a tubulin loading control.
For HEK 293 coIP, V5-C2/c-Jun, Flag-C2/ATF2, and control plasmids, with or without c-Fos or A-Fos, were cotransfected into HEK 293 cells using Lipofectamine 2000 as described previously (31). Thirty-six hours after transfection, cells were lysed and immunoprecipitated with 2 μg of V5 or Flag antibody. The precipitated immunocomplexes were subjected to Western blotting.
Gel mobility shift assay.
A gel mobility shift assay was performed using nuclear extracts immunodepleted of c-Jun or ATF2. Nuclear extracts were prepared as described in detail previously (27, 31). For immunodepletion, nuclear extracts (100 μg) were incubated for two rounds at 4°C with 4 μg of c-Jun or ATF2 antibody (Santa Cruz Biotechnology). The atf probes containing a conserved ATF sequence (jun2 site [underlined]) (forward, 5′-AGCTAGCATTACCTCATCCCGATC-3′, and reverse, 5′-GATCGGGATGAGGTAATGCTAGCT-3′) and a synthetic oligonucleotide dp5 probe containing another conserved ATF sequence, TTACATCA, were annealed and labeled with γ-32P (Perkin Elmer Life and Analytical Sciences) using a T4 polynucleotide kinase. 32P-labeled probes were then incubated with 5 μg of immunodepleted nuclear protein in 20 μl DNA binding reaction buffer at 4°C for 1 hour. DNA-protein complexes were resolved by 4% polyacrylamide gel electrophoresis and exposed to photography.
Constructs, transfection, and luciferase assay.
DIV5-6 CGNs were transfected using a calcium phosphate transfection method, and a reporter assay was performed as described previously (28, 31). The plasmid 2×jun2-luc (designated the atf-luciferase [atf-luc] reporter in this paper) was a kind gift from Hans van Dam (48). The plasmids C2/ATF2, C2/c-Jun, and ATF2-DN and a collagenase promoter reporter (coll-luc) were described previously (45); for coIP, C2/c-Jun was subcloned into the pcDNA3.1/V5 vector (Invitrogen). A-ATF2 and A-Fos were also described previously (49). 7×AP-1-luc (designated tre-luc in this paper [an artificial reporter containing seven TRE sites]) was purchased from Stratagene. The dp5-luc and c-Jun-DN constructs were described previously (31). An ATF2-expressing plasmid was constructed by cloning the cDNA encoding ATF2 into pShuttle with a Flag tag at the C terminus, and Flag-tagged c-Fos was cloned into pcDNA 3.1.
RNA interference.
Two 19-nucleotide atf2 shRNA constructs, shatf2a and shatf2b, were designed as described previously (31); they target the sequences GAAGAAGTGGGTTTGTTTA (nucleotides 521 to 540) and GCTATTCCTGCATCAATTA (nucleotides 972 to 990) of atf2 mRNA (NCBI accession number NM_031018), respectively. Two c-fos shRNA constructs, shc-fosa and shc-fosb, were used; they target the sequences GGAGACAGATCAACTTGAA (nucleotides 653 to 671) and GCTGAAGGCTGAACCCTTT (nucleotides 941 to 959) of c-fos mRNA (NCBI accession number NM_022197). Two c-jun shRNAs, shc-juna and shc-junb, were described previously (35).
Immunofluorescence.
Immunocytochemistry and confocal imaging were performed as described previously (35). Antibodies used in these studies include an ATF2 rabbit monoclonal antibody (Cell Signaling Technology), a β-galactosidase (β-Gal) mouse monoclonal antibody (Cell Signaling Technology), a c-Jun mouse monoclonal antibody (BD Biosciences), a β-Gal rabbit polyclonal antibody (5 Prime 3 Prime, Boulder, CO), a Flag mouse monoclonal antibody (Sigma), and Cy3- and fluorescein isothiocyanate-conjugated donkey secondary antibodies (Jackson ImmunoResearch).
Decoy oligonucleotides.
The decoy plasmid was constructed by a previously described method (17). To construct 12 copies of the atf decoy, three oligonucleotides each containing four ATF elements (TTACCTCA) in tandem separated by several unrelated nucleotides were synthesized. The oligonucleotides were 5′-AATTCTTACCTCAGATGATTACCTCATCCCGCTTACCTCACGATCAGTTACCTCAC-3′, 5′-TCGAGTTACCTCAGATGATTACCTCATCCCGCTTACCTCACGATCAGTTACCTCAT-3′, and 5′-GATTACCTCAGATGATTACCTCATCCCGCTTACCTCACGATCAGTTACCTCAA-3′. ATF elements are shown in boldface type, and the restrictive blunt ends are underlined. The decoys were ligated to 12 copies and then subcloned into the pMD19 vector (Takara). To construct a mutant atf decoy, all of the ATF elements (TTACCTCA) involved in the atf decoy were replaced by different nucleotides (underlined in GGACCTCG) (31). All constructs were confirmed by sequencing.
Bimolecular fluorescence complementation (BiFC) assay.
Plasmids encoding c-Jun and ATF2 were fused to N-terminal residues 1 to 172 (JunVN173) or C-terminal residues 155 to 238 (ATF2VC155), respectively, of the Venus fluorescent protein as described previously (30). JunΔL3VN173 served as a negative control because it lacked the capacity to bind to ATF2 (30). CGNs were cotransfected with 2 μg each of JunVN173 and ATF2VC155 and then subjected to a depolarizing (25 K medium) or potassium deprivation (5 K medium) treatment for 1 h at 12 to 16 h posttransfection. To compare the efficiencies of fluorescence complementation, the cells were cotransfected with vectors encoding cyan fluorescent protein (CFP). CFP expression was measured using excitation at 420 nm and emission at 470 nm. Venus fluorescence (green) was measured by excitation at 490 nm and emission at 530 nm. The exposed time for Venus fluorescence was controlled to maximally decrease overlapping signals from CFP. More than 500 cells were counted in each trial.
Construction and infection of Ads.
Recombinant adenoviruses (Ads) containing Flag-tagged c-Fos or A-Fos were generated according to the protocol described for the AdEasy XL Ad vector system (Stratagene). The shuttle vector (Stratagene) delivering either c-Fos or A-Fos was homologously recombined with AdEasy-1 in BJ5183 strains to generate Ad-c-Fos or Ad-A-Fos. Ads were amplified and purified as described previously (29, 44). DIV5 neurons were infected with either Ad-c-Fos or Ad-A-Fos in addition to a control Ad, Ad-green fluorescent protein (GFP), at a multiplicity of infection of 100 for 36 h and then subjected to chromatin IP (ChIP) assays, RT-PCR, and apoptosis assays.
ChIP assays.
ChIP assays were performed using the ChIP assay kit (Upstate Cell Signaling Solutions) as previously described (31). Two micrograms of rabbit c-Jun antibody or rabbit ATF2 antibody (Santa Cruz Biotechnology) were used for IP. Purified DNA was subjected to PCR amplification using the primers spanning the ATF site on the dp5 promoter (forward, 5′-AGGGTTAAAAGTTACCTCTCGGC-3′; reverse, 5′-ACCCCAAGTTTCGCTCTGC-3′, the c-jun promoter (forward, 5′-CTAGACAGCCAAACCAAGAC-3′; reverse, 5′-GCTCACGGGATGAGGTAAT-3′) (31), and the atf3 promoter (forward, 5′-TGTGACCGCCCCTTCTCG-3′; reverse, 5′-CTGAGTGAGACTGTGACTGGGAG-3′) (5).
RESULTS
Potassium deprivation results in JNK-dependent transactivation of c-Jun and ATF2, which coincided with rapid c-Fos downregulation.
To determine whether ATF2 is involved in potassium deprivation-induced neuronal apoptosis, we examined the phosphorylation status of ATF2 using Western blotting. We observed that, in comparison to what occurred with unchallenged CGNs in depolarizing (25 K) medium, potassium deprivation (5 K treatment) led to a dramatic elevation in phosphorylated ATF2 starting at 1 hour postdeprivation and lasting up to 4 hours postdeprivation (Fig. 1A), and this elevation also paralleled increases in levels of c-Jun phosphorylation and expression (Fig. 1A). The ATF2 total protein level remained constant in response to potassium deprivation, and c-Jun expression was induced as previously reported (Fig. 1A) (31, 51). Treatment of CGNs with the JNK inhibitor SP600125 (3, 31) or the mixed-lineage kinase inhibitor CEP11004 (31, 39) completely abrogated ATF2 phosphorylation (Fig. 1B), suggesting that ATF2 phosphorylation is dependent upon JNK activity (12). Thus, c-Jun and ATF2 are activated in the early stages of CGN apoptosis induced by potassium deprivation.
FIG. 1.

Potassium deprivation induces the transactivation of c-Jun and ATF2 and the downregulation of c-Fos. (A) CGNs were cultured in 25 K or 5 K medium for the indicated times, and cell lysates were subjected to Western blotting with antibodies against phospho-c-Jun (p-c-Jun), c-Jun, phospho-ATF2, or ATF2. (B) CGNs in 25 K or 5 K medium were treated with 10 μM SP600125 (SP) or 2 μM CEP11004 (CEP) for 4 h, and cell lysates were subjected to Western blotting (WB) with antibodies against phospho-ATF2 or ATF2. (C) CGNs in 25 K or 5 K medium were lysed at the times indicated and subjected to Western blotting with antibodies against c-Fos. Total mRNA was extracted and subjected to RT-PCR with primers specific to c-fos. (D) CGNs were cultured in 25 K or 5 K medium for 4 h in the presence or absence of 1 μM nifedipine (Nif) or 10 μM KN62 (KN). Lysates were then subjected to Western blotting with antibodies against c-Fos. Total mRNA was extracted and subjected to RT-PCR with primers specific to c-fos. Tubulin was reprobed to verify equal loadings, and β-actin was amplified to verify equal inputs.
We next sought to examine whether c-Fos, another important dimerization partner of c-Jun (43), is involved in CGN apoptosis. Both the mRNA and protein levels of c-Fos were observed to be downregulated sharply at 1 hour after 5 K treatment, and this effect lasted for up to 4 hours (Fig. 1C), in contrast to the increase in ATF2 and c-Jun phosphorylation. These results suggest that potassium deprivation leads to transcriptional downregulation of c-Fos at the onset of apoptosis.
It is well established that depolarization-induced calcium influx via L-type calcium channels sustains elevated c-Fos expression in neurons (37). Blockage of L-type calcium channels by nifedipine consistently lowered depolarization-induced c-Fos expression to levels comparable to those seen in 5 K medium (Fig. 1D). KN62, a specific inhibitor of the Ca2+/CaMKs (8), inhibited depolarization-evoked c-Fos expression (Fig. 1D), suggesting that c-Fos transcriptional downregulation in response to potassium deprivation may result from the impaired activation of CaMKs caused by a loss of calcium influx. In brief, potassium withdrawal results in the JNK-dependent transactivation of c-Jun and ATF2 and concurrent c-Fos downregulation.
The majority of c-Jun and ATF2 proteins heterodimerize with each other, and c-Jun/ATF2 heterodimers predominantly occupy the ATF site in CGNs deprived of potassium.
To test whether potassium deprivation facilitates c-Jun/ATF2 heterodimerization, we performed coIP and immunodepletion assays with cell lysates from CGNs incubated in either 25 K or 5 K medium. As has been reported previously (51), precipitates of c-Jun in CGNs in 25 K medium were below the limits of detection (Fig. 2A). In contrast, under the 5 K condition, significant heterodimerization between c-Jun and ATF2 was detected by coIP (Fig. 2A). Subsequently, we performed immunodepletion of either ATF2 or c-Fos and analyzed the supernatants via Western blotting with a c-Jun antibody. Upon depletion of ATF2, it was determined that approximately 90% of c-Jun was bound to ATF2 when CGNs were deprived of potassium (Fig. 2B, upper panel). In contrast, only a small percentage of c-Jun (∼33%) was associated with ATF2 under the 25 K condition (Fig. 2B, upper panel). The c-Fos depletion experiment revealed that a greater amount of c-Jun (∼55%) interacted with c-Fos than with ATF2 under the 25 K condition but that none of the c-Jun was associated with c-Fos under 5 K conditions (Fig. 2B, lower panel). Thus, potassium deprivation induces substantial heterodimerization between c-Jun and ATF2.
FIG. 2.

The majority of c-Jun heterodimerizes with ATF2, and c-Jun/ATF2 heterodimers predominantly occupy the ATF sites in CGNs deprived of potassium. (A) CGNs in 25 K or 5 K medium for 4 h were lysed and subjected to IP with either ATF2 or c-Jun antibodies. The precipitates were analyzed by Western blotting (WB). (B) Cell lysates from CGNs incubated in either 25 K or 5 K medium for 4 h were subjected to immunodepletion (ID) using ATF2 or c-Fos antibodies. The supernatants were analyzed by Western blotting with a c-Jun antibody. The intensity of the c-Jun band was normalized to tubulin. (C) Gel mobility shift assays were performed using 32P-labeled atf probes or dp5 probes with nuclear extracts depleted of c-Jun or ATF2. Immunoglobulin G (IgG) was used as an antibody control. SB, specific DNA-protein band.
We next performed a gel mobility shift assay to determine whether c-Jun/ATF2 heterodimers bind to conserved ATF sites under conditions of potassium deprivation. To do this, we used end-labeled atf and dp5 probes. The atf probe contains the ATF sequence in the promoter of c-jun (jun2 site, TTACCTCA) (47). The dp5 probe contains another 8-bp ATF element (TTACATCA), which was identified by our group (31) and contains only one base that does not match the jun2 site. Potassium deprivation caused a marked increase in the intensity of the complexes captured by the two probes in comparison to what occurred with 25 K medium (Fig. 2C). Sequential IP of the nuclear extracts collected from CGNs in 5 K medium using either c-Jun or ATF2 antibody resulted in substantial reduction of the specific band (Fig. 2C), thus confirming that both c-Jun and ATF2 are present in the complex. Similar results were obtained using alternative end-labeled probes containing the conserved 8-bp ATF element (TTACGTCA) located in the promoter of the atf3 gene (5, 35; data not shown). In summary, potassium deprivation induces the formation of c-Jun/ATF2 heterodimers, which bind predominantly to the conserved ATF sites.
ATF, but not TRE, activity is increased in CGNs deprived of potassium.
To determine whether potassium deprivation-induced c-Jun/ATF2 heterodimerization contributes to the activation of promoters containing ATF elements, we carried out reporter gene assays using an atf-luc reporter containing 2×jun2 sites (TTACCTCA) to monitor the ATF activity (45, 47). 5 K treatment caused an approximately 2.9-fold increase in the activity of luciferase compared to the level of activity caused by the 25 K treatment (Fig. 3A, left panel). ATF2-DN and c-Jun-DN are dominant negative mutants that lack the transactivation domain. A-ATF2 contains an amphipathic acidic α-helical sequence replacing the basic region, which promotes higher-affinity binding of A-ATF2 to its partner but inhibits binding to ATF sites (49). c-Jun-DN, ATF2-DN, or A-ATF2 abolished the activation of atf-luc stimulated by potassium deprivation (Fig. 3A, left panel), suggesting that c-Jun/ATF2 heterodimers activated atf-luc under potassium deprivation. We also tested the activity of the dp5 and atf3 promoters, both of which contain an ATF sequence, TTACCTCA or TTACGTCA. Comparably, the expression of dp5-luc or atf3-luc was found to be markedly stimulated by potassium deprivation (Fig. 3A, right panel). In accordance with previous reports (31, 35, 36, 51), these studies determined that potassium deprivation promotes ATF activation and upregulation of c-jun, dp5, and atf3 (Fig. 3B).
FIG. 3.

Potassium deprivation increases ATF activity rather than TRE activity. (A) CGNs were transfected with atf-luc plasmids and pCMV-RL in combination with plasmids encoding c-Jun-DN, ATF2-DN, or A-ATF2 (left panel). A dual-reporter analysis of dp5-luc or atf3-luc was performed with CGNs incubated in either 25 K or 5 K medium (right panel). The levels of luciferase activity were normalized to Renilla luciferase activity. (B) The levels of c-jun, dp5, and atf3 mRNA in CGNs in 25 K or 5 K medium for the indicated durations of time were measured, and β-actin mRNA was amplified as an input control. (C) Dual-reporter analysis of tre-luc or coll-luc in CGNs cultured in 25 K or 5 K medium. Data in this figure are presented as means ± standard errors of the means (SE) (three experiments). P < 0.05 (Student's t test).
To monitor whether potassium deprivation alters TRE activity via downregulation of c-Fos or upregulation of c-Jun, coll-luc, containing the TRE site (TGACTCA), and tre-luc were utilized (48). The activities of the two reporter genes were found to be consistent between the 25 K and 5 K treatment groups (Fig. 3C). These results further supported the possibility that the majority of 5 K-induced c-Jun heterodimerized with ATF2. Because of the low levels of c-Jun in the 25 K group and the downregulation of c-Fos in 5 K medium, the absence of c-Fos and c-Jun coexpression may explain the lack of change in TRE activity in 5 K medium. Taken together, these data demonstrate that c-Jun/ATF2 heterodimers promote ATF activity rather than TRE activity in response to potassium deprivation.
c-Jun/ATF2 heterodimerization is necessary and sufficient for induction of apoptosis.
We examined whether interfering c-Jun/ATF2 heterodimers protected CGN from apoptosis. c-Jun-DN can compete with endogenous c-Jun for binding to ATF2 and target genes (31). In agreement with previous reports (14, 51, 53), overexpression of c-Jun-DN significantly protected CGNs from apoptosis (Fig. 4A). This protection upon 5 K-induced CGN apoptosis may be explained by the disruption of c-Jun/ATF2 heterodimers, and as such, blockage of ATF2 should display a similar protection against CGN apoptosis. Indeed, expressing ATF2-DN or A-ATF2 in CGNs attenuated apoptosis in comparison to that in the vector control (Fig. 4A). Since c-Jun-DN and ATF2-DN may affect the activities of other AP-1 transcription factors, we introduced shRNA targeting c-Jun or ATF2. c-jun or atf2 shRNAs can effectively reduce the levels of c-Jun or ATF2 ectopically expressed either in HEK 293 cells or in CGNs (Fig. 4B and C). Knockdown of c-jun was able to suppress 5 K-induced CGN apoptosis (Fig. 4B), which is in agreement with our previous data (35). Comparably, knockdown of atf2 by shatf2a or shatf2b rescued CGNs from apoptosis too (Fig. 4C). In summary, c-Jun/ATF2 heterodimerization and ATF activity were essential for apoptosis induced by potassium deprivation in CGNs.
FIG. 4.

c-Jun/ATF2 heterodimers are necessary and sufficient for CGN apoptosis. (A) CGNs transfected with c-Jun-DN, ATF2-DN, or A-ATF2 together with GFP (to mark the transfected cells) were maintained in 25 K or 5 K medium for 12 h and stained with Hoechst 33258. (B) HEK 293 cells were cotransfected with the constructs BS/U6, shc-juna, or shc-junb together with Flag-c-Jun and subjected to Western blotting with a Flag antibody (left panel). CGNs transfected with BS/U6, shc-juna, or shc-junb plasmids were maintained in 25 K or 5 K medium and then subjected to c-Jun staining, with β-Gal staining as a marker for transfection (middle panel) or with GFP to analyze apoptosis (right panel). Scale bar = 10 μm. (C) Knockdown efficiency and apoptotic analysis for shatf2a and shatf2b were performed as described for panel B. (D) HEK 293 cells were transfected with V5-C2/c-Jun and Flag-C2/ATF2 and then lysed for CoIP with V5 or Flag antibodies. Western blotting (WB) was performed using a Flag or V5 antibody. (E) CGNs were cotransfected with C2/c-Jun and C2/ATF2 in the indicated doses and in the absence or presence of atf-luc plasmids (left panel). A dual-reporter analysis was performed. An apoptotic analysis was performed 48 h posttransfection. All apoptotic rates in this figure were quantified by scoring the percentages of transfected neurons with pyknotic nuclei in total transfected cells. The data are presented as means ± SE of the means (three experiments). *, P < 0.05; **, P < 0.01 (Student's t test).
The proapoptotic effects of the active c-Jun/ATF2 heterodimer were examined using a combinational transfection of C2/ATF2 and C2/c-Jun, two constitutively active mutants. The phosphorylation-dependent activation domain of the two mutants was replaced with the constitutively active transcriptional activation domain of the transcription factor CREB2, and as a result, the mutants were able to potently stimulate ATF site-containing promoters (45). CoIP experiments confirmed that C2/ATF2 and C2/c-Jun formed a complex when overexpressed in HEK 293 cells (Fig. 4D). In CGNs, as shown in Fig. 4E (left panel), C2/ATF2 or C2/c-Jun alone had a subtle effect on atf-luc activity, whereas the combination of the two activated atf-luc, thus indicating that ATF2 and c-Jun activities synergistically upregulated ATF signaling. C2/ATF2 did not influence CGN apoptosis due to the low levels of endogenous c-Jun available to heterodimerize in serum-containing cultures, whereas C2/c-Jun showed a slight activation in CGN apoptosis. Coexpression of C2/ATF2 and C2/c-Jun stimulated more neuronal apoptosis than expression of either construct alone (Fig. 4E, right panel). In summary, c-Jun/ATF2 heterodimers mediate potassium deprivation-induced CGN apoptosis.
Blocking c-Jun/ATF2 heterodimers using decoy oligonucleotides suppresses CGN apoptosis.
Decoy analysis has been described as a promoter competition strategy to capture the functional transcriptional factors or transcriptional complex from targeting the endogenously conserved promoter sites (17, 32). To investigate whether the c-Jun/ATF2 heterodimers play a role in CGN apoptosis, we employed decoy oligonucleotides to prevent the c-Jun/ATF2 heterodimers from binding to the endogenous ATF sites. The same ATF sequence (TTACCTCA) in the atf probe used in gel mobility shift assays was cloned into the pMD vector using 12 copies in tandem to construct an atf decoy. Also, a mutant atf decoy in which the entire ATF sequence was replaced by GGACCTCG (where underlining indicates mutated oligonucleotides) was constructed, resulting in a loss of binding capacity to c-Jun/ATF2 heterodimers. As shown in Fig. 5A, 5 K-evoked activation of atf-luc was impaired by the atf decoy but not the mutant atf decoy, indicating that the atf decoy was able to successfully inhibit the activation of c-Jun/ATF2 heterodimers. Consistently, transfection of the atf decoy was observed to markedly rescue CGNs from apoptosis (Fig. 5B). These results support the idea that c-Jun/ATF2 heterodimers promote CGN apoptosis by binding to promoter ATF sites.
FIG. 5.

Blocking c-Jun/ATF2 heterodimers using decoy oligonucleotides suppresses ATF activity and apoptosis. CGNs transfected with the atf decoy (atf-dec) or mutant atf decoy (m-atf-dec) together with atf-luc were maintained in 25 K or 5 K medium and were subjected to dual-reporter analysis. Transfection of either the atf decoy or the mutant atf decoy alone as well as apoptotic analyses of the cells were performed as described in the legend to Fig. 4A. Data are presented as means ± SE (three experiments). P < 0.05 (Student's t test).
c-Fos downregulation facilitates heterodimerization between c-Jun and ATF2.
The finding that rapid downregulation of c-Fos (Fig. 1C) was accompanied by an increased formation of c-Jun/ATF2 heterodimers (Fig. 2A) raised the possibility that c-Fos downregulation facilitates the formation of c-Jun/ATF2 heterodimers. It has previously been determined in the literature that c-Fos possesses a much higher affinity for c-Jun than ATF2 does (21, 49) (for details, see Discussion). To visualize Jun/ATF2 heterodimerization, a BiFC assay was performed using c-Jun or ATF2 fused to N-terminal residues 1 to 172 or C-terminal residues 155 to 238 of the Venus fluorescent protein (30). To evaluate the efficiency of the BiFC assay, we cotransfected CFP to identify transfected cells, and the percentage of CFP-positive cells exhibiting Venus fluorescence (i.e., BiFC) was assessed. As shown in Fig. 6A, BiFC signal intensities derived from JunVN173 and ATF2VC155 increased by twofold within 1 hour in 5 K medium compared to those in 25 K medium. The constant expression level of JunVN173 or ATF2VC155 between 25 K and 5 K confirmed that the 5 K-increased BiFC signal resulted from enhanced association between c-Jun and ATF2 (Fig. 6A). The combination of ATF2VC155 and JunΔL3VN173, a mutant of c-Jun unable to bind ATF2, did not yield a detectable signal either in 5 K or in 25 K medium (Fig. 6A). Thus, potassium withdrawal facilitated heterodimerization between c-Jun and ATF2.
FIG. 6.

c-Fos downregulation facilitates c-Jun/ATF2 heterodimerization in response to potassium deprivation in CGNs. (A) CGNs transfected with plasmids encoding JunVN173 (JunVN) and ATF2VC155 (ATF2VC), in combination with plasmids encoding CFP, were maintained in 25 K or 5 K medium for 1 h, and photographs were obtained, or cells were lysed for Western blotting to determine the expression levels of JunVN and ATF2VC. JunΔL3VN served as a control. The white arrows indicate BiFC signals (Venus fluorescence). The percentage of CFP-positive cells exhibiting Venus fluorescence was scored. Data are presented as means ± SE (three experiments). P < 0.05 (Student's t test). (B) CGNs cotransfected with plasmids encoding c-Fos or A-Fos together with plasmids encoding JunVN, ATF2VC, and CFP were subjected to BiFC analysis. Data are presented as means ± SE (three experiments). P < 0.05 (Student's t test). (C) HEK 293 cells cotransfected with BS/U6, shc-fosa, or shac-fosb together with plasmids encoding Flag-c-Fos were subjected to Western blotting with a Flag antibody. CGNs were transfected with the BS/U6, shc-fosa, or shc-fosb plasmids together with plasmids encoding JunVN, ATF2VC, and CFP for BiFC analysis. Data are presented as means ± SE (three experiments). P < 0.05 (Student's t test).
If the increased BiFC signal subsequent to potassium deprivation resulted from c-Fos downregulation, then overexpression of c-Fos should prevent the increased signal. Indeed, this was exactly what is shown in Fig. 6B. A-Fos (49), a mutant with higher affinity for heterodimerization with c-Jun than c-Fos, was also able to completely abolished the BiFC signal (Fig. 6B). In addition, the expression of JunVN173 or ATF2VC155 in 25 K or 5 K medium was unaffected by the expression of c-Fos or A-Fos (data not shown). These results suggested that the existence of c-Fos impeded the association between c-Jun and ATF2. Conversely, a specific c-fos knockdown increased the BiFC signal (Fig. 6C) with an effect similar to that of c-Fos downregulation in 5 K-treated cells. As a control, c-fos knockdown failed to affect the expression of either JunVN173 or ATF2VC155 (data not shown). Taken together, these data demonstrate that c-Fos downregulation facilitated the association between c-Jun and ATF2.
Prevention of c-Jun/ATF2 heterodimerization by c-Fos suppresses ATF activity, expression of target genes, and apoptosis.
To address whether c-Fos activity affected the function of c-Jun/ATF2 heterodimers, we tested the effects of c-Fos or A-Fos expression on c-Jun/ATF2 heterodimerization, binding of c-Jun and ATF2 to ATF sites, ATF activity, and potassium deprivation-induced apoptosis. As shown in Fig. 7A, expression of either c-Fos or A-Fos abrogated activation of atf-luc provoked by 5 K. CoIP experiments revealed that the presence of c-Fos or A-Fos predominantly acted to compete with C2/c-Jun for binding to C2/ATF2 (Fig. 7B, left panel). The expression of c-Fos or A-Fos in CGNs consistently repressed the atf-luc activation typically evoked by the combination of C2/c-Jun and C2/ATF2 (Fig. 7B, right panel). These results indicated that the expression of c-Fos suppressed c-Jun/ATF2-mediated ATF activity. A-Fos exhibited enhanced inhibition compared to c-Fos due to its higher affinity for c-Jun and the loss of the capacity of A-Fos/c-Jun complexes to bind TRE or ATF sites (49).
FIG. 7.

Overexpression of c-Fos or A-Fos attenuates c-Jun/ATF2 heterodimer-mediated ATF activity, expression of target genes, and apoptosis. (A) CGNs transfected with plasmids encoding c-Fos or A-Fos together with atf-luc were maintained in 25 K or 5 K medium and subjected to a dual-reporter assay. (B) HEK 293 cells cotransfected with V5-C2/c-Jun and Flag-C2/ATF2 with or without c-Fos or A-Fos were lysed for IP with a V5 antibody. Input and precipitates were analyzed by Western blotting (WB) (left panel). CGNs were transfected with atf-luc plasmids together with plasmids encoding C2/ATF2, C2/c-Jun, c-Fos, and A-Fos and subjected to a dual-reporter assay (right panel). (C) CGNs infected with Ad-c-Fos or Ad-A-Fos were stained with a Flag antibody or not stained with a primary antibody (−1°) (left panel). Western blotting was performed to detect the expression of c-Fos or A-Fos (right panel). (D) Cell lysates were subjected to a ChIP assay with c-Jun or ATF2 antibodies. Chromatin was sonicated into fragments of approximately 500 bp (left panel). Western blotting analysis of the precipitates was performed using c-Jun or ATF2 antibodies (middle panel). Purified chromatin was assessed by PCR amplification of the regions proximal to the c-jun, dp5, and atf3 promoters (right panel). Equal amounts of total genomic DNA (Input) were used for IP under each condition. (E) CGNs infected with Ad-GFP, Ad-c-Fos, or Ad-A-Fos were incubated in 25 K or 5 K medium and subjected to RT-PCR with primers specific to c-jun, dp5, atf3, and bim. β-actin was amplified to verify equal inputs. (F) CGNs infected with Ad-GFP, Ad-c-Fos, or Ad-A-Fos were maintained in 25 K or 5 K medium for apoptotic analysis. Data were presented as means ± SE (three experiments). P < 0.05 (Student's t test).
We then performed a ChIP assay to address whether c-Fos prevents c-Jun and ATF2 from binding to ATF sites in the promoters of c-jun, dp5, and atf3. Due to the low transfection efficiency of CGNs, we constructed recombinant Ads expressing either c-Fos or A-Fos to achieve efficient infection. Ad-mediated c-Fos or A-Fos was expressed in approximately 70 to 80% of CGNs (Fig. 7C, left panel), and equal amounts of c-Fos or A-Fos were expressed between the 25 K and 5 K treatment groups (Fig. 7C, right panel). Chromatin was sonicated into fragments of about 500 bp for ChIP assays (Fig. 7D, left panel). In the captured immunoprecipitates, consistently with a previous report (31), c-Jun protein cross-linked to DNA was readily detectable in CGNs deprived of potassium but not in CGNs in the 25 K group, whereas levels of ATF2 protein cross-linked to DNA were equal in CGNs under the 25 K and 5 K conditions (Fig. 7D, middle panel). ChIP assays using an ATF2 antibody revealed that binding of ATF2 to ATF sites showed no difference between CGNs maintained in 25 K or 5 K medium (Fig. 7D, right panel). This finding is consistent with previous studies demonstrating that binding of ATF2 to ATF sites is unaltered during activation and repression of c-Jun activity (19, 41). In contrast, potassium deprivation led to enhanced binding of c-Jun to the c-jun, dp5, and atf3 promoter regions, and Ad-mediated c-Fos or A-Fos expression significantly attenuated binding of c-Jun to these promoters (Fig. 7D, right panel). Thus, c-Fos expression prevented binding of c-Jun to ATF sites.
Moreover, expression of either c-Fos or A-Fos prevented 5 K-induced upregulation of c-jun, dp5, and atf3 but not upregulation of bim (Fig. 7E), indicating that the prevention of c-Jun/ATF2 heterodimerization by c-Fos was able to suppress the transcription of c-Jun/ATF2 target genes but that bim is not a c-Jun/ATF2 target gene in CGNs deprived of potassium, as demonstrated by our previous reports (31, 44). Furthermore, recombinant Ad expressing either c-Fos or A-Fos was able to significantly prevent CGN apoptosis (Fig. 7F), thus indicating that c-Fos protection against neuronal apoptosis does not involve an active transcriptional mechanism. These results demonstrated that downregulation of c-Fos contributed to c-Jun/ATF2-mediated CGN apoptosis secondary to potassium deprivation.
DISCUSSION
We demonstrated here that in response to potassium deprivation in CGNs, (i) JNK-dependent transactivation of ATF2 and c-Jun is accompanied by c-Fos downregulation via inactivation of CaMKs, (ii) c-Jun predominantly heterodimerizes with ATF2, (iii) c-Jun/ATF2 heterodimers promote apoptosis by triggering ATF activity, (iv) c-Fos downregulation facilitates c-Jun/ATF2 heterodimerization, and (v) c-Fos expression suppresses the transcription of c-Jun/ATF2 target genes and subsequent apoptosis.
Numerous studies have addressed the importance of c-Jun activation in neuronal apoptosis in several systems (2, 9, 20, 31, 51, 53); however, c-Jun binding partners in neuronal apoptosis remain unknown. With CGNs following potassium deprivation, our results appear to be consistent with those of previous reports (20, 24, 36, 51) in that we not only confirmed c-Jun phosphorylation and upregulation in a JNK-dependent manner but also showed that ATF2 acts as a c-Jun partner to promote apoptosis. Although ATF2 is constitutively expressed in the brain, its role in neuronal fate is less clear. In the present study, we showed that (i) potassium deprivation resulted in JNK-dependent transactivation of ATF2, (ii) a dominant negative ATF2 or knockdown of atf2 by shRNAs rescued neurons from apoptosis, and (iii) ectopic coexpression of the constitutively active mutants C2/ATF2 and C2/c-Jun synergistically induced apoptosis. These findings define a role for the requirement of ATF2 in neuronal apoptosis.
Contrary to our findings here, a recent study suggested that neither c-Jun nor ATF2 is involved in potassium deprivation-induced apoptosis in CGNs on the basis of evidence that shRNA knockdown of c-Jun or atf2 does not prevent neuronal apoptosis (4). Although this apparent discrepancy could be due to differences in experimental conditions or the use of different experimental techniques, consistent results were, however, obtained in our own studies when several different experimental approaches were pursued. Thus, our results, as listed below, which are in agreement with those of several other previously published studies (31, 35, 51), collectively demonstrate that c-Jun and ATF2 are vital for CGN apoptosis. First, the results presented in this study have consistently shown that the introduction of an exogenous dominant negative c-Jun construct exerts significant protection against CGN apoptosis. Second, specific knockdown of c-jun by shRNA is able to rescue neurons from apoptosis. Third, either transfection of dominant negative ATF2 mutants or specific knockdown of atf2 by shRNA prevented neuronal apoptosis. Finally, interference of c-Jun/ATF2 binding to ATF sites using a decoy strategy is again able to prevent the apoptotic cascade. Consistent with our findings in this study, the role of c-Jun in neuronal apoptosis has also been addressed under various conditions by several other laboratories (20, 24, 26, 51, 55).
c-Fos has been documented to be an essential player in the differentiation and survival of cultured CGNs (6). However, the mechanism by which c-Fos promotes neuronal survival is elusive. Our results herein suggest that the prosurvival effect of c-Fos may include a nontranscriptional mechanism, since a c-Fos mutant (A-Fos) without transcriptional activity is also able to prevent apoptosis. Moderate Ca2+ influx through L-type calcium channels subsequent to neuronal depolarization in response to potassium-rich medium activates Ca2+/calmodulin-dependent kinases (42), which can elevate c-Fos expression by signaling through MEK/extracellular signal-regulated kinase or CREB (1, 22). Potassium deprivation results in an activation of proapoptotic JNKs concomitantly with decreased activity of the prosurvival CaMK pathways. Similar effects have been observed in nerve growth factor withdrawal-induced PC12 apoptosis, in which activation of JNK and p38 is accompanied by the loss of prosurvival extracellular signal-regulated kinase activity (54). The consequences of JNK activation and CaMK inactivation following potassium deprivation converge to balance the activity of AP-1 factors, in which c-Jun and ATF2 are transactivated to mediate apoptosis via heterodimerization while c-Fos is downregulated to facilitate c-Jun/ATF2 heterodimerization and the apoptotic process (Fig. 8). The dynamic interactions between c-Jun, ATF2, and c-Fos are carefully orchestrated to coordinate CGN apoptosis following potassium deprivation.
FIG. 8.
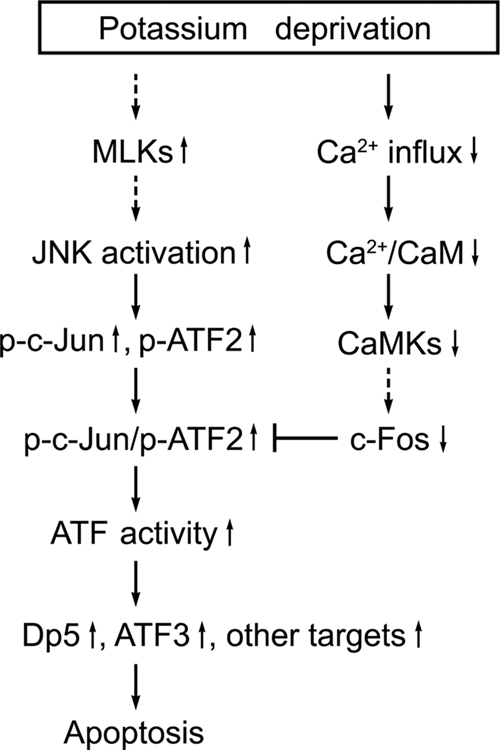
Model for the signaling pathways regulating potassium deprivation-induced CGN apoptosis. Deprivation of the survival agent, potassium, not only results in JNK-dependent transactivation of c-Jun and ATF2 via phosphorylation of their N-terminal regions but also leads to concurrent c-Fos downregulation attributable to the decreased activity of Ca2+/calmodulin (CaM) and CaMKs secondary to a loss of calcium influx. c-Jun primarily heterodimerizes with ATF2, and c-Jun/ATF2 heterodimers are responsible for the upregulation of ATF activity. c-Fos downregulation facilitates the heterodimerization between c-Jun and ATF2. The upregulation of ATF activity triggers target gene expression (dp5, atf3, and other genes) to promote neuronal apoptosis. p-c-Jun, phospho-c-Jun.
The observation that c-Fos downregulation facilitates c-Jun/ATF2 heterodimerization is further supported by the ability of c-Jun to bind to c-Fos with higher affinity than to ATF2. The stability and specificity of dimerization between bZIP members are decided by their hydrophobic residues and the charges of adjacent amino acids (46, 49). For c-Fos, the domain responsible for binding to c-Jun possesses a strong negative charge, whereas for c-Jun, positively charged amino acids predominate. ATF2 contains equal numbers of positive and negative residues. As a result, the coupling energy responsible for c-Jun/c-Fos heterodimerization is far lower than that for c-Jun/ATF2 heterodimerization (49), resulting in a more stable c-Jun/c-Fos heterodimer than the c-Jun/ATF2 heterodimer (11, 40). Thus, the presence of c-Fos impedes ATF2/c-Jun heterodimer formation.
IP assays demonstrated that the majority of c-Jun and ATF2 heterodimerizes with each other during potassium deprivation, and gel mobility shift assays confirmed that c-Jun/ATF2 complexes bind to ATF sites. In order to further substantiate AP-1 dynamics in living cells, we used BiFC analysis (21) to demonstrate that, in living neurons, potassium deprivation induces c-Jun/ATF2 heterodimerization and that coexpression of either c-Fos or A-Fos disassociates c-Jun/ATF2 heterodimers. In addition to performing BiFC analysis, we performed ChIP assays to further confirm that the presence of c-Fos interfered with the binding of c-Jun to ATF2 and ATF sites. Collectively, our data establish that potassium deprivation promotes c-Jun/ATF2 heterodimerization secondary to c-Jun induction and c-Fos downregulation, which is required for the upregulation of target genes containing ATF sites and subsequent neuronal apoptosis.
The function of c-Jun and ATF2 as a complex to regulate target gene expression has been under intense scrutiny for the past several years. An excellent study was published by Hayakawa et al. (16), in which they identified 269 genes via ChIP whose promoters are bound upon phosphorylation of ATF2 and c-Jun, and the binding did not occur in the presence of the JNK inhibitor SP600125 or JNK-specific shRNA (16). A variety of proapoptotic genes, such as tumor necrosis factor alpha (18) and Fas L (10), have been found to serve as direct downstream targets of c-Jun/ATF2 in apoptosis. Dp5, a BH3-only protein, was identified by our group as a target gene product of c-Jun and is required for potassium withdrawal-induced CGN apoptosis, but its induction is only partially mediated by transactivation of the c-Jun/ATF2 complex (31). Therefore, further studies are necessary in order to identify the target genes of c-Jun/ATF2 heterodimers essential for neuronal apoptosis.
In conclusion, our results revealed that increased c-Jun/ATF2 heterodimerization is coupled with the downregulation of c-Fos expression during potassium deprivation-induced CGN apoptosis. c-Jun/ATF2 heterodimers mediate neuronal apoptosis by upregulation of ATF activity, while the concurrent downregulation of c-Fos facilitates c-Jun/ATF2 heterodimerization and subsequent apoptosis (Fig. 8). Our findings suggest a model for selective dimerization of AP-1 proteins coupled with the transcriptional regulation of AP-1 genes in the regulation of neuronal death.
Acknowledgments
We thank Hans van Dam for kindly providing us with the plasmids.
This work was supported by National Natural Science Foundation of China grants U0632006, 30870786, 30629002, and 30831160511.
Footnotes
Published ahead of print on 2 March 2009.
REFERENCES
- 1.Bading, H., D. D. Ginty, and M. E. Greenberg. 1993. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260181-186. [DOI] [PubMed] [Google Scholar]
- 2.Behrens, A., M. Sibilia, and E. F. Wagner. 1999. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet. 21326-329. [DOI] [PubMed] [Google Scholar]
- 3.Bennett, B. L., D. T. Sasaki, B. W. Murray, E. C. O'Leary, S. T. Sakata, W. Xu, J. C. Leisten, A. Motiwala, S. Pierce, Y. Satoh, S. S. Bhagwat, A. M. Manning, and D. W. Anderson. 2001. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 9813681-13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bjorkblom, B., J. C. Vainio, V. Hongisto, T. Herdegen, M. J. Courtney, and E. T. Coffey. 2008. All JNKs can kill, but nuclear localization is critical for neuronal death. J. Biol. Chem. 28319704-19713. [DOI] [PubMed] [Google Scholar]
- 5.Cai, Y., C. Zhang, T. Nawa, T. Aso, M. Tanaka, S. Oshiro, H. Ichijo, and S. Kitajima. 2000. Homocysteine-responsive ATF3 gene expression in human vascular endothelial cells: activation of c-Jun NH(2)-terminal kinase and promoter response element. Blood 962140-2148. [PubMed] [Google Scholar]
- 6.Didier, M., P. Roux, M. Piechaczyk, P. Mangeat, G. Devilliers, J. Bockaert, and J. P. Pin. 1992. Long-term expression of the c-fos protein during the in vitro differentiation of cerebellar granule cells induced by potassium or NMDA. Brain Res. Mol. Brain Res. 12249-258. [DOI] [PubMed] [Google Scholar]
- 7.Eilers, A., J. Whitfield, C. Babij, L. L. Rubin, and J. Ham. 1998. Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J. Neurosci. 181713-1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Enslen, H., and T. R. Soderling. 1994. Roles of calmodulin-dependent protein kinases and phosphatase in calcium-dependent transcription of immediate early genes. J. Biol. Chem. 26920872-20877. [PubMed] [Google Scholar]
- 9.Estus, S., W. J. Zaks, R. S. Freeman, M. Gruda, R. Bravo, and E. M. Johnson, Jr. 1994. Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J. Cell Biol. 1271717-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faris, M., K. M. Latinis, S. J. Kempiak, G. A. Koretzky, and A. Nel. 1998. Stress-induced Fas ligand expression in T cells is mediated through a MEK kinase 1-regulated response element in the Fas ligand promoter. Mol. Cell. Biol. 185414-5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glover, J. N., and S. C. Harrison. 1995. Crystal structure of the heterodimeric bZIP transcription factor c-Fos-c-Jun bound to DNA. Nature 373257-261. [DOI] [PubMed] [Google Scholar]
- 12.Gupta, S., D. Campbell, B. Derijard, and R. J. Davis. 1995. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 267389-393. [DOI] [PubMed] [Google Scholar]
- 13.Hafezi, F., J. P. Steinbach, A. Marti, K. Munz, Z. Q. Wang, E. F. Wagner, A. Aguzzi, and C. E. Reme. 1997. The absence of c-fos prevents light-induced apoptotic cell death of photoreceptors in retinal degeneration in vivo. Nat. Med. 3346-349. [DOI] [PubMed] [Google Scholar]
- 14.Ham, J., C. Babij, J. Whitfield, C. M. Pfarr, D. Lallemand, M. Yaniv, and L. L. Rubin. 1995. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron 14927-939. [DOI] [PubMed] [Google Scholar]
- 15.Harris, C. A., M. Deshmukh, B. Tsui-Pierchala, A. C. Maroney, and E. M. Johnson, Jr. 2002. Inhibition of the c-Jun N-terminal kinase signaling pathway by the mixed lineage kinase inhibitor CEP-1347 (KT7515) preserves metabolism and growth of trophic factor-deprived neurons. J. Neurosci. 22103-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayakawa, J., S. Mittal, Y. Wang, K. S. Korkmaz, E. Adamson, C. English, M. Ohmichi, M. McClelland, and D. Mercola. 2004. Identification of promoters bound by c-Jun/ATF2 during rapid large-scale gene activation following genotoxic stress. Mol. Cell 16521-535. [DOI] [PubMed] [Google Scholar]
- 17.He, S., B. L. Cook, B. E. Deverman, U. Weihe, F. Zhang, V. Prachand, J. Zheng, and S. J. Weintraub. 2000. E2F is required to prevent inappropriate S-phase entry of mammalian cells. Mol. Cell. Biol. 20363-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herr, I., C. Posovszky, L. D. Di Marzio, M. G. Cifone, T. Boehler, and K. M. Debatin. 2000. Autoamplification of apoptosis following ligation of CD95-L, TRAIL and TNF-alpha. Oncogene 194255-4262. [DOI] [PubMed] [Google Scholar]
- 19.Herr, I., H. van Dam, and P. Angel. 1994. Binding of promoter-associated AP-1 is not altered during induction and subsequent repression of the c-jun promoter by TPA and UV irradiation. Carcinogenesis 151105-1113. [DOI] [PubMed] [Google Scholar]
- 20.Hongisto, V., N. Smeds, S. Brecht, T. Herdegen, M. J. Courtney, and E. T. Coffey. 2003. Lithium blocks the c-Jun stress response and protects neurons via its action on glycogen synthase kinase 3. Mol. Cell. Biol. 236027-6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu, C. D., and T. K. Kerppola. 2003. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 21539-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson, C. M., C. S. Hill, S. Chawla, R. Treisman, and H. Bading. 1997. Calcium controls gene expression via three distinct pathways that can function independently of the Ras/mitogen-activated protein kinases (ERKs) signaling cascade. J. Neurosci. 176189-6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuan, C. Y., A. J. Whitmarsh, D. D. Yang, G. Liao, A. J. Schloemer, C. Dong, J. Bao, K. J. Banasiak, G. G. Haddad, R. A. Flavell, R. J. Davis, and P. Rakic. 2003. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc. Natl. Acad. Sci. USA 10015184-15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le-Niculescu, H., E. Bonfoco, Y. Kasuya, F. X. Claret, D. R. Green, and M. Karin. 1999. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol. Cell. Biol. 19751-763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leppa, S., M. Eriksson, R. Saffrich, W. Ansorge, and D. Bohmann. 2001. Complex functions of AP-1 transcription factors in differentiation and survival of PC12 cells. Mol. Cell. Biol. 214369-4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levkovitz, Y., and J. M. Baraban. 2001. A dominant negative inhibitor of the Egr family of transcription regulatory factors suppresses cerebellar granule cell apoptosis by blocking c-Jun activation. J. Neurosci. 215893-5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li, M., D. A. Linseman, M. P. Allen, M. K. Meintzer, X. Wang, T. Laessig, M. E. Wierman, and K. A. Heidenreich. 2001. Myocyte enhancer factor 2A and 2D undergo phosphorylation and caspase-mediated degradation during apoptosis of rat cerebellar granule neurons. J. Neurosci. 216544-6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li, M., X. Wang, M. K. Meintzer, T. Laessig, M. J. Birnbaum, and K. A. Heidenreich. 2000. Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3beta. Mol. Cell. Biol. 209356-9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li, X., B. Monks, Q. Ge, and M. J. Birnbaum. 2007. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature 4471012-1016. [DOI] [PubMed] [Google Scholar]
- 30.Liu, H., X. Deng, Y. J. Shyu, J. J. Li, E. J. Taparowsky, and C. D. Hu. 2006. Mutual regulation of c-Jun and ATF2 by transcriptional activation and subcellular localization. EMBO J. 251058-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma, C., C. Ying, Z. Yuan, B. Song, D. Li, Y. Liu, B. Lai, W. Li, R. Chen, Y. P. Ching, and M. Li. 2007. dp5/HRK is a c-Jun target gene and required for apoptosis induced by potassium deprivation in cerebellar granule neurons. J. Biol. Chem. 28230901-30909. [DOI] [PubMed] [Google Scholar]
- 32.Mann, M. J., and V. J. Dzau. 2000. Therapeutic applications of transcription factor decoy oligonucleotides. J. Clin. Investig. 1061071-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin-Villalba, A., C. Winter, S. Brecht, T. Buschmann, M. Zimmermann, and T. Herdegen. 1998. Rapid and long-lasting suppression of the ATF-2 transcription factor is a common response to neuronal injury. Brain Res. Mol. Brain Res. 62158-166. [DOI] [PubMed] [Google Scholar]
- 34.Mattson, M. P. 2000. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 1120-129. [DOI] [PubMed] [Google Scholar]
- 35.Mei, Y., Z. Yuan, B. Song, D. Li, C. Ma, C. Hu, Y. P. Ching, and M. Li. 2008. Activating transcription factor 3 up-regulated by c-Jun NH(2)-terminal kinase/c-Jun contributes to apoptosis induced by potassium deprivation in cerebellar granule neurons. Neuroscience 151771-779. [DOI] [PubMed] [Google Scholar]
- 36.Miller, T. M., and E. M. Johnson, Jr. 1996. Metabolic and genetic analyses of apoptosis in potassium/serum-deprived rat cerebellar granule cells. J. Neurosci. 167487-7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Misra, R. P., A. Bonni, C. K. Miranti, V. M. Rivera, M. Sheng, and M. E. Greenberg. 1994. L-type voltage-sensitive calcium channel activation stimulates gene expression by a serum response factor-dependent pathway. J. Biol. Chem. 26925483-25493. [PubMed] [Google Scholar]
- 38.Morishima, Y., Y. Gotoh, J. Zieg, T. Barrett, H. Takano, R. Flavell, R. J. Davis, Y. Shirasaki, and M. E. Greenberg. 2001. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J. Neurosci. 217551-7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murakata, C., M. Kaneko, G. Gessner, T. S. Angeles, M. A. Ator, T. M. O'Kane, B. A. McKenna, B. A. Thomas, J. R. Mathiasen, M. S. Saporito, D. Bozyczko-Coyne, and R. L. Hudkins. 2002. Mixed lineage kinase activity of indolocarbazole analogues. Bioorg. Med. Chem. Lett. 12147-150. [DOI] [PubMed] [Google Scholar]
- 40.O'Shea, E. K., R. Rutkowski, and P. S. Kim. 1992. Mechanism of specificity in the Fos-Jun oncoprotein heterodimer. Cell 68699-708. [DOI] [PubMed] [Google Scholar]
- 41.Rozek, D., and G. P. Pfeifer. 1993. In vivo protein-DNA interactions at the c-jun promoter: preformed complexes mediate the UV response. Mol. Cell. Biol. 135490-5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.See, V., A. L. Boutillier, H. Bito, and J. P. Loeffler. 2001. Calcium/calmodulin-dependent protein kinase type IV (CaMKIV) inhibits apoptosis induced by potassium deprivation in cerebellar granule neurons. FASEB J. 15134-144. [DOI] [PubMed] [Google Scholar]
- 43.Shaulian, E., and M. Karin. 2002. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4E131-136. [DOI] [PubMed] [Google Scholar]
- 44.Shi, L., S. Gong, Z. Yuan, C. Ma, Y. Liu, C. Wang, W. Li, R. Pi, S. Huang, R. Chen, Y. Han, Z. Mao, and M. Li. 2005. Activity deprivation-dependent induction of the proapoptotic BH3-only protein Bim is independent of JNK/c-Jun activation during apoptosis in cerebellar granule neurons. Neurosci. Lett. 3757-12. [DOI] [PubMed] [Google Scholar]
- 45.Steinmuller, L., and G. Thiel. 2003. Regulation of gene transcription by a constitutively active mutant of activating transcription factor 2 (ATF2). Biol. Chem. 384667-672. [DOI] [PubMed] [Google Scholar]
- 46.van Dam, H., and M. Castellazzi. 2001. Distinct roles of Jun: Fos and Jun: ATF dimers in oncogenesis. Oncogene 202453-2464. [DOI] [PubMed] [Google Scholar]
- 47.van Dam, H., M. Duyndam, R. Rottier, A. Bosch, L. de Vries-Smits, P. Herrlich, A. Zantema, P. Angel, and A. J. van der Eb. 1993. Heterodimer formation of cJun and ATF-2 is responsible for induction of c-jun by the 243 amino acid adenovirus E1A protein. EMBO J. 12479-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Dam, H., D. Wilhelm, I. Herr, A. Steffen, P. Herrlich, and P. Angel. 1995. ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J. 141798-1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vinson, C., M. Myakishev, A. Acharya, A. A. Mir, J. R. Moll, and M. Bonovich. 2002. Classification of human B-ZIP proteins based on dimerization properties. Mol. Cell. Biol. 226321-6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walton, M., A. M. Woodgate, E. Sirimanne, P. Gluckman, and M. Dragunow. 1998. ATF-2 phosphorylation in apoptotic neuronal death. Brain Res. Mol. Brain Res. 63198-204. [DOI] [PubMed] [Google Scholar]
- 51.Watson, A., A. Eilers, D. Lallemand, J. Kyriakis, L. L. Rubin, and J. Ham. 1998. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J. Neurosci. 18751-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wenzel, A., C. Grimm, A. Marti, N. Kueng-Hitz, F. Hafezi, G. Niemeyer, and C. E. Reme. 2000. c-fos controls the “private pathway” of light-induced apoptosis of retinal photoreceptors. J. Neurosci. 2081-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Whitfield, J., S. J. Neame, L. Paquet, O. Bernard, and J. Ham. 2001. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 29629-643. [DOI] [PubMed] [Google Scholar]
- 54.Xia, Z., M. Dickens, J. Raingeaud, R. J. Davis, and M. E. Greenberg. 1995. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 2701326-1331. [DOI] [PubMed] [Google Scholar]
- 55.Yamagishi, S., M. Yamada, Y. Ishikawa, T. Matsumoto, T. Ikeuchi, and H. Hatanaka. 2001. p38 mitogen-activated protein kinase regulates low potassium-induced c-Jun phosphorylation and apoptosis in cultured cerebellar granule neurons. J. Biol. Chem. 2765129-5133. [DOI] [PubMed] [Google Scholar]
- 56.Zhang, J., D. Zhang, J. S. McQuade, M. Behbehani, J. Z. Tsien, and M. Xu. 2002. c-fos regulates neuronal excitability and survival. Nat. Genet. 30416-420. [DOI] [PubMed] [Google Scholar]