

Abstract
Herpes simplex virus 1 (HSV-1) enters cells either via fusion of the virion envelope and host cell plasma membrane or via endocytosis, depending on the cell type. In the study reported here, we investigated a viral entry pathway dependent on the paired immunoglobulin-like type 2 receptor α (PILRα), a recently identified entry coreceptor for HSV-1 that associates with viral envelope glycoprotein B (gB). Experiments using inhibitors of endocytic pathways and ultrastructural analyses of Chinese hamster ovary (CHO) cells transduced with PILRα showed that HSV-1 entry into these cells was via virus-cell fusion at the cell surface. Together with earlier observations that HSV-1 uptake into normal CHO cells and those transduced with a receptor for HSV-1 envelope gD is mediated by endocytosis, these results indicated that expression of PILRα produced an alternative HSV-1 entry pathway in CHO cells. We also showed that human and murine PILRα were able to mediate entry of pseudorabies virus, a porcine alphaherpesvirus, but not of HSV-2. These results indicated that viral entry via PILRα appears to be conserved but that there is a PILRα preference among alphaherpesviruses.
Herpesviruses (family Herpesviridae) are divided into three subfamilies (alpha-, beta-, and gammaherpesviruses) on the basis of biological characteristics and genomic organization (36). Human herpes simplex virus 1 (HSV-1), HSV-2, and porcine pseudorabies virus (PRV) are representative members of the alphaherpesvirus subfamily. The alphaherpesviruses have a very broad host range in cultured cells and can infect and cause diseases in selected laboratory animals, such as rodents (41). These features indicate that each of these viruses can recognize structural features of receptors conserved among human and animal species. Consistent with this hypothesis, these alphaherpesviruses appear to have common pathways for entry into cells (41). The initial interaction of these viruses with cells is binding of virion envelope glycoprotein C (gC) and gB to cell surface glycosaminoglycans, preferentially heparan sulfate (3-O-S-HS) (15, 16, 41). Subsequent viral penetration requires fusion between the virion envelope and the host cell membrane and depends on gB, the heterodimer gH/gL, gD, and a gD receptor (33, 42, 46). For HSV-1 entry, this fusion occurs either on the cell surface or within an endosome, depending on the cell type (11, 25, 29-31, 40). To date, three HSV-1 entry pathways have been reported: (i) direct fusion between the virion envelope and cell plasma membrane, as seen for Vero and HEp-2 cell entry (10, 11, 40, 50); (ii) low-pH-dependent endocytosis, as seen for cell entry with HeLa cells, Chinese hamster ovary (CHO) cells expressing gD receptors, and human primary epidermal keratinocytes (29, 30); and (iii) low-pH-independent endocytosis, as seen for C10 murine melanoma cell entry (25). A salient question that remains is what determines the viral entry choice between fusion with the cell plasma membrane for some cell types and endocytosis for other cell types.
The gD receptors reported to date fall into three classes (41): (i) HVEM (herpesvirus entry mediator), a member of the tumor necrosis factor receptor family; (ii) nectin-1 and nectin-2, members of the immunoglobulin (Ig) superfamily; and (iii) specific sites on 3-O-S-HS generated by certain 3-O-sulfotransferases (12, 26, 39, 47). Different alphaherpesvirus gD glycoproteins appear to have somewhat different receptor preferences. For example, although both HVEM and nectin-1 are excellent entry receptors for both HSV-1 and HSV-2, nectin-2 is a poor receptor for HSV-1 entry but does have entry activity for HSV-2 (12, 26, 47). In contrast to nectin-2, 3-O-S-HS is a poor receptor for HSV-2 entry but does have entry activity for HSV-1 (39). In the case of PRV, the virus can use human nectin-1 or nectin-2 but not human HVEM or 3-O-S-HS (12, 26, 39, 47).
In addition to the interaction of virion gD with a cell membrane gD receptor, gB binding to a cellular receptor(s) other than 3-O-S-HS has been suggested to mediate HSV-1 entry (4). Consistent with this suggestion, we have recently reported that paired Ig-like type 2 receptor α (PILRα) associates with gB and functions as an entry coreceptor for HSV-1 (37). PILRα is one of the paired receptor families expressed mainly in immune cells, in which one receptor has inhibitory functions and the other mediates activation functions (9, 28, 38). Transduction of PILRα into HSV-1-resistant CHO cells produces cells susceptible to HSV-1 infection, but this HSV-1 infection can be blocked by treating the cells with a soluble form of PILRα or antibodies against PILRα. More importantly, HSV-1 infection of primary human CD14-positive peripheral blood mononuclear cells (PBMCs) expressing both HVEM and PILRα is blocked by either anti-PILRα or anti-HVEM antibody, suggesting that cellular receptors for both gD and gB are required for HSV-1 infection. Since expression of PILRα is restricted mostly to immune cells (9) and is not expressed in cells commonly used for in vitro study of HSV-1, such as Vero cells (H. Arase, unpublished observation), we propose that there is another gB receptor(s) that plays a role in HSV-1 entry into PILRα-negative cells.
Although our previous studies presented the first data that PILRα can serve as an entry receptor for HSV-1 in the cell types that were examined (including CHO cells expressing PILRα and primary human CD14-positive PBMCs), the details of PILRα-dependent viral entry remain to be elucidated. In the studies reported here, we have characterized some of the aspects of PILRα-dependent herpesvirus entry, such as the entry pathway and conservation of this entry in other alphaherpesviruses.
MATERIALS AND METHODS
Cells and viruses.
Vero, CPK, and IC21 cells were described previously (18, 22, 44). CHO-hPILRα, CHO-mPILRα, and CHO-neo cells are transfectants stably expressing human PILRα, mouse PILRα, and empty vector, respectively, and were propagated in Ham's F-12 medium supplemented with 10% fetal calf serum (FCS) and 250 μg G418/ml (17). Cells were subcultured in nonselective medium prior to use in all experiments. HEK293T cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FCS. Human PBMCs were obtained using Ficoll-Paque Plus (GE Healthcare), and CD14-positive cells were separated from PBMCs using a MACS purification system (Miltenyi Biotec). Primary human CD14-positive PBMCs were cultured in RPMI 1640 medium supplemented with 10% FCS. Ham's F-12 medium and Advanced RPMI 1640 (Invitrogen) medium supplemented with 1% FCS were used for virus infection with various types of CHO cells and primary human CD14-positive PBMCs, respectively.
Wild-type HSV-1(F), HSV-2(G), HSV-2 186, and PRV Indiana strain were described previously (18, 27, 44). YK333 is a derivative of HSV-1(F) and carries an enhanced green fluorescent protein (EGFP) expression cassette under the control of the Egr-1 promoter in the unique long 3 (UL3)-UL4 intergenic region (45). YK338 and YK382 are derivatives of HSV-1(F) and HSV-2 186, respectively, carrying the Venus FP cassette under the control of the HSV-1 UL26.5 promoter in the UL50-UL51 intergenic region, as described elsewhere (27). HSV-1 YK338 and YK333 and HSV-2 YK382 have been shown to have phenotypes identical to those of wild-type HSV-1(F) and HSV-2 186, respectively, in cell cultures, and cells infected with YK333 or with YK338 and YK382 efficiently express EGFP or Venus FP, respectively (27, 45).
Monensin treatment.
CHO-hPILRα and CHO-hNectin-1 cells were preincubated in various concentrations of monensin for 1 h at 37°C. The cells in each sample then were mixed with YK333 in the presence of the drug at a multiplicity of infection (MOI) of 1, followed by centrifugation at 32°C at 1,100 × g for 2 h. After the supernatant was removed, infected cells were washed twice with phosphate-buffered saline, refed with the appropriate medium containing monensin, and incubated at 37°C for a further 5 h. The cells then were fixed with 4% paraformaldehyde and analyzed by FACSCalibur with Cell Quest software (Becton Dickinson).
Energy depletion.
YK333 was bound to CHO-hPILRα or CHO-hNectin-1 cells (at an MOI of 1) at 4°C for 1 h. The inoculum was then removed, and the cells were treated with glucose-free medium containing 2% bovine serum albumin, 0.3% 2-deoxy-d-glucose, and 0.05% sodium azide or with control medium and held at 4°C for 15 min and then at 37°C for 30 min. Extracellular virus was acid inactivated by removing the medium and replacing it with acid citrate buffer (40 mM citric acid [pH 4.7], 135 mM NaCl, 10 mM KCl) for 2 min. The cells then were washed, refed with the appropriate medium, and incubated for a further 7 h. The cells were then fixed with 4% paraformaldehyde and analyzed by FACSCalibur.
Electron microscopy (EM) analysis.
CHO-hNectin-1 or CHO-hPILRα cells or primary human CD14-positive PBMCs were mixed with HSV-1(F) (at an MOI of 50), followed by centrifugation at 4°C at 1,100 × g for 2 h or 30 min, respectively, to allow attachment, and then incubated at 37°C for 2 min. Infected cells were fixed with 1% glutaraldehyde and 2% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.4) for 1 h on ice. The cells then were harvested and fixed for 1 h with the same fixative. Small pieces of the fixed pellet were washed with the same phosphate buffer containing 3% sucrose and postfixed with 1% osmium tetroxide in the same phosphate buffer for 1 h on ice. For primary human CD14-positive PBMCs, infected cells were fixed with 2.5% glutaraldehyde in 0.1 M ice-cold sodium cacodylate buffer (pH 7.2) containing 0.5 mg ruthenium red/ml for 1 h, during which time cells were allowed to warm up to room temperature. The cells were then washed with 0.1 M cacodylate buffer (pH 7.2) and postfixed with 2% OsO4 in the same cacodylate buffer containing 0.5 mg ruthenium red/ml for 1 h at room temperature, followed by routine embedding in Epon. The samples were then dehydrated with an ethanol gradient series followed by propylene oxide, embedded in an Epon 812 resin mixture, and polymerized at 70°C for 2 days. Thin sections were cut, stained with uranyl acetate and lead citrate, and examined with a Hitachi H7500 electron microscope.
Virus entry assays.
CHO cells or primary human CD14-positive PBMCs were inoculated with PRV or an HSV at an MOI of 5, followed by centrifugation at 32°C at 1,100 × g for 2 h. The inoculum was then removed, and the cells were refed with the appropriate medium. (i) In experiments in which various types of CHO cells or primary human CD14-positive PBMCs were infected with recombinant HSVs carrying a fluorescent marker, infected cells were fixed with 4% paraformaldehyde at 8 h or 14 h postinfection, respectively, and analyzed by fluorescent microscopy or FACSCalibur. (ii) In experiments in which various types of CHO cells were infected with wild-type HSVs, infected cells were stained with anti-gD antibody at 8 h postinfection and fixed with 4% paraformaldehyde. The stained cells were then incubated with Alexa Fluor 488-conjugated anti-mouse IgG (Invitrogen) and analyzed by FACSCalibur. (iii) In experiments in which primary human CD14-positive PBMCs were infected with wild-type HSVs, infected cells were harvested at 14 h postinfection and analyzed by immunoblotting (19) with anti-ICP27 antibody. (iv) In experiments in which various types of CHO cells were infected with PRV, infected cells were fixed with 4% paraformaldehyde at 8 h postinfection, permeabilized with 0.1% Triton X-100, and stained with anti-EP0 antibody. The stained cells were then incubated with Alexa Fluor 488-conjugated anti-rabbit IgG (Invitrogen) and analyzed by FACSCalibur. (v) In experiments in which primary human CD14-positive PBMCs were infected with PRV, infected cells were harvested at 7 h postinfection and analyzed by immunoblotting with anti-EP0 antibody.
Antibodies.
Mouse monoclonal antibodies (MAb) to gD (DL6), ICP27 (8.F.137B), and α-tubulin (DM1A) were purchased from Santa Cruz Biotechnology, Abcam, and Sigma-Aldrich, respectively. Rabbit polyclonal antibody to PRV EP0 was described previously (48). For blocking experiments, anti-PILRα MAb (M4) (IgG1 isotype) (37) and anti-Flag MAb (M2) (IgG1 isotype), the latter used as a control, were purchased from Sigma-Aldrich.
Binding of soluble PILRα-Ig fusion protein to the surfaces of infected cells.
Soluble PILRα-Ig fusion proteins were produced and used for the detection of gB on the surfaces of HEK293T cells infected with HSV-1(F), HSV-2 186, or PRV at an MOI of 5 for 12 h, as described previously (37).
RESULTS
Effect of ATP synthesis inhibitors and monensin treatment on HSV-1 infection of CHO cells expressing PILRα.
Endocytosis is an active, energy-dependent process and, therefore, is sensitive to ATP synthesis inhibitors (24). It has been reported that treatment with energy depletion medium containing sodium azide and 2-deoxy-d-glucose resulted in inhibition of both pH-dependent and pH-independent endocytic entries of HSV-1 into cells such as HeLa cells, CHO cells expressing nectin-1, and C10 cells, while this treatment did not affect HSV-1 entry into Vero cells (25, 30). pH-dependent endocytic entry of HSV-1 requires that virion particles be delivered to an acidic endosomal compartment (30). Monensin is a carboxylic ionophore that blocks endosomal acidification, and it has been reported that pH-dependent endocytic entry of HSV-1 into cells such as HeLa cells and CHO cells expressing nectin-1 is inhibited by monensin treatment but that monensin has no effect on pH-independent endocytic entry of HSV-1 into C10 cells or on nonendocytic entry into Vero cells (25, 30). To investigate an HSV-1 PILRα-dependent entry pathway, CHO-hPILRα and CHO-hNectin-1 cells treated with or without energy depletion medium containing sodium azide and 2-deoxy-d-glucose were infected with recombinant HSV-1 YK333, which expresses EGFP, at an MOI of 5 for 7 h, and the infected cells were analyzed for EGFP fluorescence by flow cytometry. In addition, cells were infected with HSV-1 YK333 at an MOI of 5 for 7.5 h in the presence or absence of various concentrations of monensin, and HSV-1 infection was analyzed as described above. We note that CHO-hPILRα cells are able to produce low but consistent yields of progeny virus, based on the observation that virus titers in the supernatants of CHO-hPILRα and CHO-hNectin-1 cells infected with wild-type HSV-1(F) at an MOI of 5 for 24 h were 1.3 × 102 and 1.3 × 104, respectively, while that of CHO-neo cells was not detectable. As shown in Fig. 1A, HSV-1 entry into CHO-hNectin-1 cells treated with energy depletion medium was significantly inhibited. Furthermore, there was a dose-dependent inhibition of HSV-1 entry when CHO-hNectin-1 cells were treated with increasing concentrations of monensin (Fig. 1B). These results were consistent with previous reports (30). In contrast, when CHO-hPILRα cells were treated with energy depletion medium or monensin, no inhibitory effect on HSV-1 entry was evident (Fig. 1A and B). These results indicated that the HSV-1 entry pathway into CHO cells expressing PILRα was distinct from that into CHO cells expressing the gD receptor. Since HSV-1 entry by fusion of the virion envelope and cell plasma membrane is not affected by treatment with energy depletion medium or monensin (30), these results supported the conclusion that HSV-1 entry into CHO-hPILRα cells was the result of fusion of the virion envelope and cell plasma membrane.
FIG. 1.

Effects of endocytosis inhibitors on HSV-1 entry into cells. (A) CHO-hPILRα and CHO-hNectin-1 cells were infected with YK333 at an MOI of 1 at 4°C for 1 h. The inoculum was then removed, and cells were treated with glucose-free medium containing 2% bovine serum albumin, 0.3% 2-deoxy-d-glucose, and 0.05% sodium azide and held at 4°C for 15 min and then at 37°C for 30 min. Virus that had not penetrated the cells was inactivated by acid treatment, and infection continued for an additional 7 h, after which infected cells were analyzed by flow cytometry. The relative mean fluorescence intensity (MFI) of infected cells treated with energy depletion medium was calculated as the percentage relative to that of mock-treated infected cells. The mean and standard deviation from three independent experiments is shown for each cell type. (B) CHO-hPILRα or CHO-hNectin-1 cells were pretreated with the indicated concentrations of monensin for 1 h at 37°C. Cells were then infected with YK333 at an MOI of 1 for 7 h in the presence of the drug and analyzed by flow cytometry. The relative MFI of infected cells was calculated as the percentage relative to that in the absence of the drug. The mean and standard deviation from three independent experiments is shown for each data point for each cell type.
We note that treatment of CHO-hPILRα cells with 50 mM monensin increased viral entry a little (Fig. 1B). Although it is unknown how monensin affects viral entry into CHO-hPILRα cells, similar results were also observed with HSV-1-infected Vero cells in the presence of lysosomotropic agents (monensin and ammonium chloride) (30).
Ultrastructural analysis of PILRα-dependent HSV-1 entry.
HSV-1 entry into CHO-hPILRα cells was studied further by EM. CHO-hPILRα and CHO-hNectin-1 cells were incubated with wild-type HSV-1(F) at an MOI of 50 for 2 h at 4°C, and the temperature was raised to 37°C for 2 min. Infected cells were then placed on ice, fixed, and processed for EM. In addition, primary human CD14-positive PBMCs were prepared by the procedure described above except that the cells were incubated with HSV-1 at 4°C for 30 min. Primary human CD14-positive PBMCs endogenously express PILRα, as well as gD receptors for alphaherpesviruses, including HVEM, nectin-2, and poliovirus receptor (34). We previously demonstrated that HSV-1 infects these cells in a PILRα-dependent manner, based on the observation that HSV-1 infection was blocked by anti-PILRα MAb (37). EM of HSV-1-infected CHO-hNectin-1 cells showed enveloped virions surrounded by invaginations of the cell plasma membrane (Fig. 2A) and within vesicles (Fig. 2B and C), both of which are typical features of HSV-1 endocytic entry (30). In contrast, EM analysis of HSV-1-infected CHO-hPILRα cells showed that virions on the cell surface were not in membrane invaginations (Fig. 2D and E) and that naked or unenveloped nucleocapsids were in the cytosol adjacent to the cell plasma membrane (Fig. 2F). These features were consistent with those observed with HSV-infected Vero cells, as reported earlier (11, 40). When approximately 50 virions were counted and categorized, 62% of virions in CHO-hNectin-1 cells exhibited typical features of endocytic HSV-1 entry. In contrast, 83% of virions in CHO-hPILRα cells exhibited typical features of HSV-1 entry by fusion at the plasma membrane. Results similar to those obtained with HSV-1-infected CHO-hPILRα cells were also obtained with HSV-1(F)-infected primary human CD14-positive PBMCs (Fig. 3). These results confirm that PILRα-dependent entry of HSV-1 into cells such as CHO-hPILRα cells and primary human CD14-positive PBMCs is by fusion of the virion envelope and cell plasma membrane.
FIG. 2.

EM of HSV-1 entry into CHO cells expressing PILRα. HSV-1(F) was added to CHO-hNectin-1 (A to C) or CHO-hPILRα (D to F) cells for 2 h at 4°C, followed by a shift to 37°C for 2 min and then preparation of the cells for EM. Bar, 200 nm.
FIG. 3.

EM of HSV-1 entry into CD14-positive PBMCs. HSV-1(F) was added to primary human CD14-positive PBMCs for 30 min at 4°C, followed by a shift to 37°C for 2 min and then preparation of the cells for EM. Bar, 200 nm.
HSV-2 infection of PILRα transfectants.
To test whether PILRα is able to mediate HSV-2 entry, CHO-hNectin-1, CHO-hPILRα, and CHO-neo cells were incubated with recombinant HSV-1 YK338 or recombinant HSV-2 YK382, both of which express Venus FP, at an MOI of 5 for 8 h, and HSV infection was analyzed for Venus FP fluorescence by fluorescence microscopy (data not shown) and flow cytometry (Fig. 4A). As reported previously (12), transduction of CHO cells with human nectin-1 greatly enhanced entry of both HSV-1 YK338 and HSV-2 YK382 (Fig. 4). In contrast, transduction with human PILRα efficiently enhanced entry of HSV-1 YK338, as described previously (37), but failed to enhance entry of HSV-2 YK382 (Fig. 4). Similar results were obtained when CHO-hPILRα cells were infected with HSV-2 YK382 at an MOI of 50 (data not shown). To eliminate the possibility that insertion of the Venus FP expression cassette into the HSV-2 genome affected PILRα-dependent entry of HSV-2 YK382, we used two wild-type HSV-2 strains [HSV-2 186 and HSV-2(G)] for further experiments. CHO-hNectin-1, CHO-hPILRα, and CHO-neo cells were incubated with wild-type HSV-2 186, HSV-2(G), or HSV-1(F) at an MOI of 5 for 8 h, harvested, stained with anti-gD antibody, and analyzed by flow cytometry. As shown in Fig. 4B, the results were consistent with those obtained with fluorescent viruses (Fig. 4A): there was good infection of CHO-hNectin-1 cells by all three viruses, there was good infection of CHO-hPILRα cells by HSV-1(F) but little infection by the two HSV-2 strains, and there was little infection of CHO-neo cells by all three viruses. These observations indicated that PILRα was unable to mediate HSV-2 entry into CHO cells.
FIG. 4.
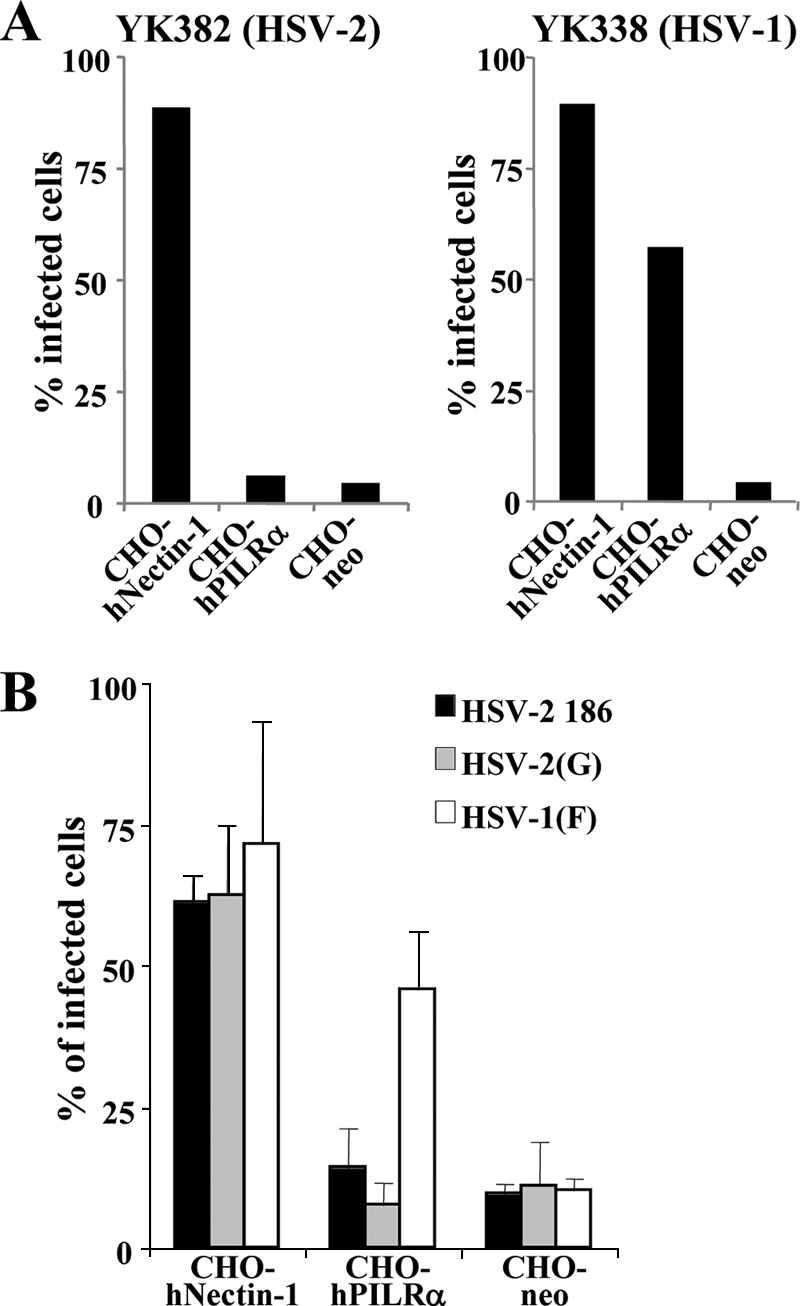
HSV-2 infection of CHO cells expressing PILRα. (A) CHO-hNnectin-1, CHO-hPILRα, and CHO-neo cells were infected with HSV-2 YK382 or HSV-1 YK338 at an MOI of 5. At 8 h postinfection, infected cells were divided into two aliquots. One aliquot was examined by fluorescence microscopy (data not shown), and the other was analyzed by flow cytometry (A) to determine the percentages of infected cells. (B) CHO-hNnectin-1, CHO-hPILRα, and CHO-neo cells were infected with wild-type HSV-2 186, HSV-2(G), or HSV-1(F) at an MOI of 5. At 8 h postinfection, infected cells were stained with anti-gD antibody and analyzed by flow cytometry to determine the percentages of infected cells. The mean and standard deviation from three independent experiments is shown for each cell type.
PRV infection of PILRα transfectants.
To investigate whether both human and murine PILRα are able to serve as receptors for PRV, CHO-hNectin-1, CHO-hPILRα, CHO-mPILRα, and CHO-neo cells were incubated with PRV at an MOI of 5 for 8 h, harvested, stained with anti-PRV EP0 antibody, and analyzed by flow cytometry. As shown in Fig. 5A, transduction with human nectin-1, human PILRα, and murine PILRα resulted in efficient PRV infection. To study the direct involvement of PILRα in PRV entry, PRV entry was analyzed using anti-PILRα MAb (M4), which has been shown to block HSV-1 entry mediated by PILRα (37). As shown in Fig. 5B, when CHO-hPILRα cells were infected with PRV in the presence of anti-PILRα MAb, infection was blocked by the MAb in a dose-dependent manner, while the control MAb did not affect PRV infection. These results indicated that PILRα mediated PRV entry into CHO cells.
FIG. 5.

PRV infection of CHO cells expressing PILRα. (A) CHO-hNectin-1, CHO-hPILRα, CHO-mPILRα, and CHO-neo cells were infected with PRV at an MOI of 5. At 8 h postinfection, infected cells were fixed, permeabilized, stained with anti-EP0 antibody, and analyzed by flow cytometry to determine the percentages of infected cells. The mean and standard deviation from three independent experiments is shown for each cell type. (B) CHO-hPILRα cells were infected with PRV at an MOI of 5 in the presence of various concentrations of anti-PILRα MAb or control MAb. At 8 h postinfection, infected cells were stained with anti-EP0 antibody and analyzed by flow cytometry to determine the percentages of infected cells.
Binding of PILRα to the cell surface molecule(s) expressed in HSV-1-, HSV-2-, and PRV-infected cells.
Envelope glycoproteins of HSV-1, HSV-2, and PRV are known to be expressed on the surfaces of infected cells (6). To test whether cell surface molecules induced by HSV-2 and PRV infection bind to PILRα like HSV-1 gB (37), HEK293T cells were mock infected or infected with HSV-1(F), HSV-2 186, or PRV at an MOI of 5 for 12 h, and binding of human PILRα-Ig fusion protein to cell surface molecules on infected cells was analyzed by flow cytometry. As shown in Fig. 6, HSV-1(F)-, HSV-2 186-, or PRV-infected cells were stained with human PILRα-Ig fusion protein. Consistent with our previous report (37), mock-infected cells were not stained with human PILRα-Ig fusion protein, based on the observation that the staining patterns of mock-infected cells with human PILRα-Ig and control-Ig fusion proteins were identical (data not shown). These results indicated that PILRα was able to bind to the cell surface molecule(s) induced by HSV-2 and PRV infection, although these viruses differed in their abilities to use PILRα for viral entry, as shown above.
FIG. 6.
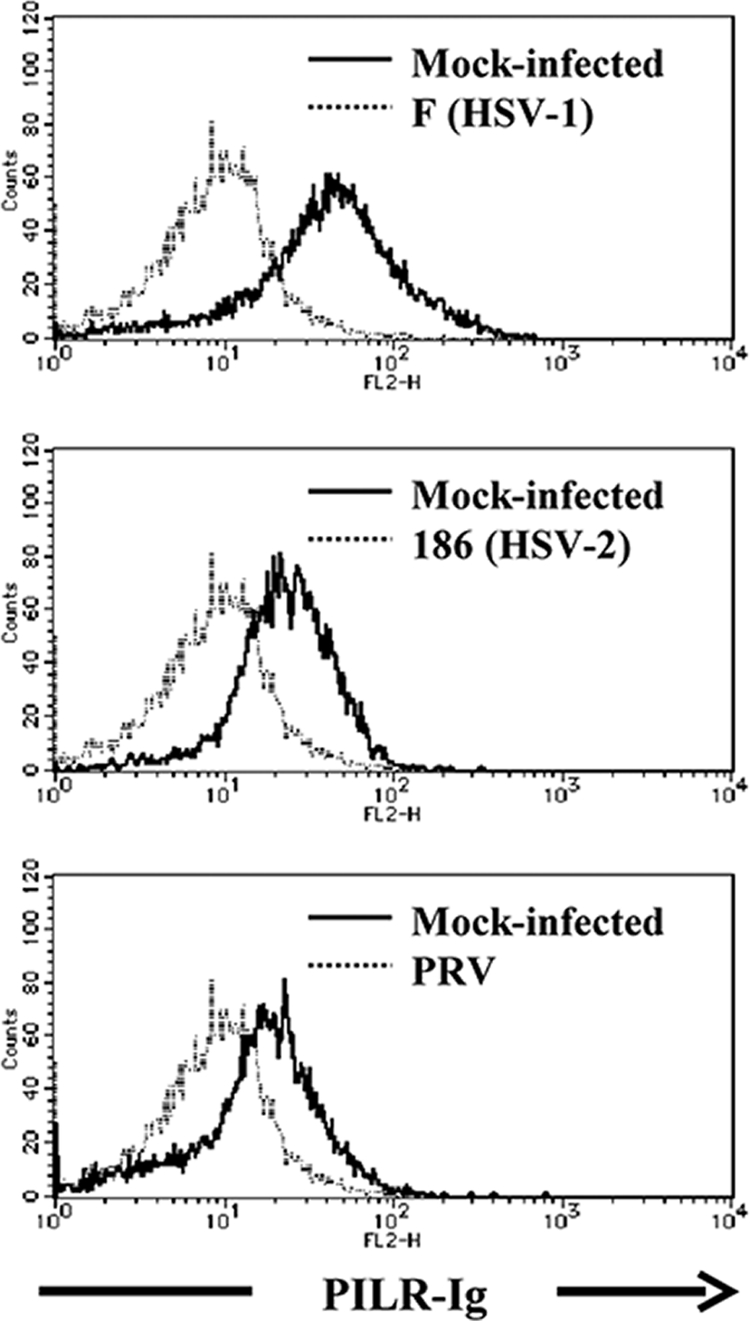
Binding of PILRα to the cell surface molecule(s) in HSV-1-, HSV-2-, or PRV-infected cells. HEK293T cells were mock infected or infected with wild-type HSV-1(F), HSV-2 186, or PRV at an MOI of 5. At 12 h after infection, infected cells were stained with PILRα-Ig fusion protein and analyzed by flow cytometry.
HSV-2 and PRV infection of primary human CD14-positive PBMCs.
We investigated whether the abilities of HSV-1, HSV-2, and PRV to utilize PILRα for viral entry, evidenced by the experiments with CHO cells overexpressing PILRα, were similar in cells expressing PILRα endogenously. In the first series of experiments, primary human CD14-positive PBMCs were incubated with wild-type HSV-1 YK338 or HSV-2 YK382 at an MOI of 5 for 14 h, harvested, and analyzed by flow cytometry. As reported previously (37), primary human CD14-positive PBMCs were efficiently infected with HSV-1 YK338 (Fig. 7A). In contrast, these cells were not infected with HSV-2 YK382 (Fig. 7A). In the second series of experiments, primary human CD14-positive PBMCs were infected with wild-type HSV-1(F), HSV-2 186, or HSV-2(G) at an MOI of 5 for 14 h, harvested, and immunoblotted with mouse MAb to ICP27. Immunoblotting was used instead of flow cytometry in these studies to detect viral infection because immunostained cells showed a very high background in flow cytometry. Consistent with the observations with fluorescent viruses, wild-type HSV-1(F) efficiently infected primary human CD14-positive PBMCs, while the wild-type HSV-2 strains did not (Fig. 7B). The anti-ICP27 antibody reacted with ICP27 proteins of both HSV-1 and HSV-2 in lysates of Vero cells infected with HSV-1(F), HSV-2 186, and HSV-2(G) (Fig. 7B). Taken together, these observations indicated that HSV-2 was unable to infect primary human CD14-positive PBMCs. In the third series of experiments, primary human CD14-positive PBMCs were infected with PRV at an MOI of 5 for 7 h in the absence or presence of anti-PILRα MAb (M4) or control MAb, harvested, and immunoblotted with anti-PRV EP0 antibody. As shown in Fig. 7C, PRV infected primary human CD14-positive PBMCs and this infection was blocked by anti-PILRα MAb. These results indicated that PRV infection of primary human CD14-positive PBMCs was dependent on the presence of cell surface PILRα. Similarly, HSV-2 did not infect IC21 cells, a PILRα-positive murine macrophage line, although these cells could be infected by HSV-1 and PRV in a PILRα-dependent manner (data not shown). Thus, the ability of HSV-1, HSV-2, or PRV to infect CHO cells was a function of whether PILRα was being expressed in these host cells. These observations further support our conclusion that PILRα is able to mediate entry of PRV but not HSV-2.
FIG. 7.
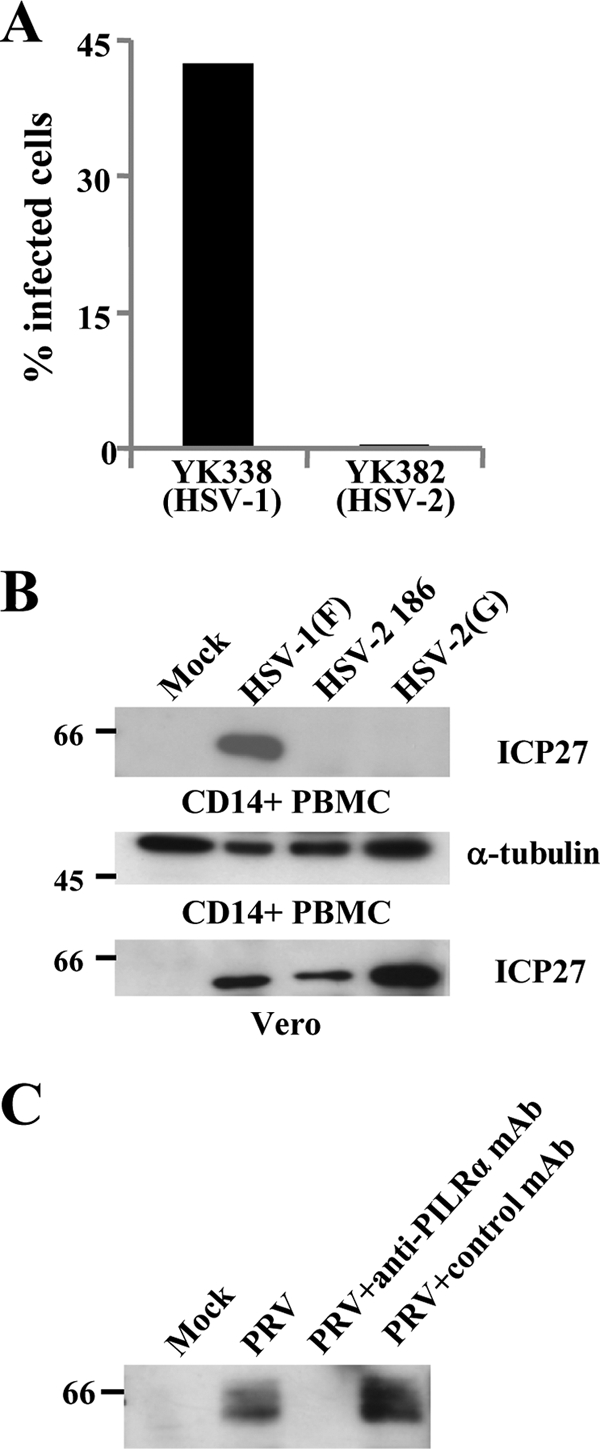
HSV-2 and PRV infections of primary human CD14-positive PBMCs. (A) Primary human CD14-positive PBMCs were infected with HSV-1 YK338 or HSV-2 YK382 at an MOI of 5. At 14 h postinfection, infected cells were analyzed by flow cytometry to determine the percentages of infected cells. (B) Primary human CD14-positive PBMCs were infected with wild-type HSV-1(F), HSV-2 186, or HSV-2(G) at an MOI of 5 for 14 h, harvested, and analyzed by immunoblotting with ICP27 or α-tubulin. Vero cells were analyzed by the same procedure as described above and immunoblotted with ICP27. (C) Primary human CD14-positive PBMCs were infected with PRV at an MOI of 5 in the presence (20 μg/ml) or absence of anti-PILRα MAb or control MAb. At 7 h postinfection, infected cells were analyzed by immunoblotting with anti-EP0 antibody. Values to the left of the blots indicate molecular size markers in kilodaltons.
DISCUSSION
Since the discovery of host cell gD receptors for alphaherpesviruses (12, 26, 39, 47), studies of entry mechanisms for HSV and other alphaherpesviruses have focused mainly on the interaction between virion gD and host cell membrane gD receptors. However, it has long been believed that a molecule(s) other than 3-O-S-HS mediates HSV-1 entry by associating with gB (4), although there has been a lack of data on the gB receptor(s). The recently discovered PILRα is the only HSV-1 entry receptor reported to date that associates with gB, and studies of PILRα-dependent entry of alphaherpesviruses are still in an early stage. In the studies reported here, we investigated aspects of PILRα-dependent entry of alphaherpesviruses. The salient features of this study are summarized below.
(i) HSV-1 entry into CHO cells expressing PILRα was mediated by fusion of the viral envelope and the host cell plasma membrane. It has been reported that HSV-1 entry into normal CHO cells is by endocytosis but that the virus then undergoes lysosomal degradation (30). However, expression of a gD receptor on CHO cells has been reported to enable the virus to enter the cytosol and initiate viral gene expression, although this viral entry is also by endocytosis (30). Consistent with these reports, we have shown here that HSV-1 entered CHO-hNectin-1 cells via endocytosis, indicating that CHO cells typically take up HSV-1 by endocytosis. In contrast, experiments in this study using energy depletion medium and an endosome ionophore and ultrastructural analyses of infected CHO-hPILRα cells showed that PILRα expression on CHO cells altered the HSV-1 entry pathway, with virus entry being via fusion of the virion envelope and the CHO-hPILRα cell plasma membrane. These observations suggested that a gB receptor is one of the factors that determine whether HSV-1 entry is via fusion at the cell surface or endocytosis. Since gB has been reported to play a central role in fusion mediated by HSV-1 glycoproteins (2, 3, 13, 14, 43), high-level expression of gB receptors on a cell surface might bias HSV-1 entry toward fusion at the cell plasma membrane instead of endocytosis. To our knowledge, these are the first data suggesting that a cell factor may be involved in the choice between two alternative entry pathways for HSV-1. The possibility that a gB receptor may be a determinant between two alternative entry pathways for HSV-1 provides a unique opportunity for studying the mechanism by which the choice is made between entry by fusion of the virion envelope and cell plasma membrane or by endocytosis.
(ii) PILRα mediates PRV entry but not HSV-2 entry. gB and PILRα are highly conserved among all Herpesviridae subfamilies and mammals, respectively (36, 49). Therefore, entry of a herpesvirus via PILRα may be a common feature of mammalian herpesviruses. Consistent with this hypothesis, we have demonstrated here that porcine alphaherpesvirus PRV can utilize human and murine PILRα as an entry receptor, based on the observations that transduction of CHO cells, which are relatively resistant to PRV entry (32), with human and murine PILRα made the cells susceptible to PRV infection, and that this infection was blocked by anti-PILRα MAb. The studies reported here also showed that PRV can infect cells expressing PILRα endogenously (primary human CD14-positive PBMCs and IC21 cells) in a PILRα-dependent manner. The use of PILRα as a receptor by both HSV-1 and PRV suggests that PILRα may mediate entry of other mammalian alphaherpesviruses and possibly other mammalian beta- and gammaherpesviruses. In contrast, different alphaherpesviruses differ in their abilities to use PILRα, as previously reported that alphaherpesviruses differ in their abilities to use gD receptors (12, 26, 39, 47). Thus, we showed that expression of PILRα on CHO cells did not make the cells susceptible to HSV-2 infection, unlike the results for HSV-1 and PRV. We also showed that HSV-2 was not able to infect cells expressing PILRα endogenously (primary human CD14-positive PBMCs and IC21 cells), although both HSV-1 and PRV enter these cells in a PILRα-dependent manner. Therefore, PILRα cannot mediate entry of HSV-2, even though the homology between HSV-1 and HSV-2 gB is much higher than that between HSV-1 and PRV gB (7, 23, 35). These observations indicated that there is not only a PILRα preference among alphaherpesviruses but also a specificity for PILRα-dependent entry of HSV-1 and PRV, which reinforces the conclusion that PILRα is an entry receptor for HSV-1 and PRV.
(iii) HSV-2 was not able to infect primary human CD14-positive PBMCs, which predominantly consist of monocytes (37), or murine IC21 macrophages, but HSV-1 efficiently infects both of these cell types. It is possible that the different susceptibilities of PILRα-positive cells to the two HSV serotypes, observed in vitro, reflect HSV serotype-dependent susceptibility and/or infection efficiency of PILRα-positive cells in vivo. It is well established that HSV infections in vivo are controlled by the host immune response (8, 21), including the activities of monocytes, macrophages, and dendritic cells, all of which express PILRα and have been suggested to play roles in host innate resistance to HSV infection (1, 5, 20). The different susceptibilities and/or infection efficiencies of these PILRα-positive immune cells to the two HSV serotypes might affect the immunomodulatory activities of cells in vivo, leading to different immune responses in HSV-1 and HSV-2 infections. This possibility might be significant for understanding the pathological differences between HSV-1 and HSV-2.
Acknowledgments
We thank Shihoko Koyama and Makoto Fukuda for excellent technical assistance.
This study was supported in part by grants for scientific research and grants for scientific research in priority areas from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan and a grant from the Takeda Science Foundation. J.A. was supported by a research fellowship from the Japanese Society for the Promotion of Science (JSPS) for Young Scientists.
Footnotes
Published ahead of print on 25 February 2009.
REFERENCES
- 1.Ahmad, R., J. Ennaciri, P. Cordeiro, S. El Bassam, and J. Menezes. 2007. Herpes simplex virus-1 up-regulates IL-15 gene expression in monocytic cells through the activation of protein tyrosine kinase and PKC zeta/lambda signaling pathways. J. Mol. Biol. 36725-35. [DOI] [PubMed] [Google Scholar]
- 2.Atanasiu, D., J. C. Whitbeck, T. M. Cairns, B. Reilly, G. H. Cohen, and R. J. Eisenberg. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. USA 10418718-18723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avitabile, E., C. Forghieri, and G. Campadelli-Fiume. 2007. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J. Virol. 8111532-11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bender, F. C., J. C. Whitbeck, H. Lou, G. H. Cohen, and R. J. Eisenberg. 2005. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J. Virol. 7911588-11597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bosnjak, L., M. Miranda-Saksena, D. M. Koelle, R. A. Boadle, C. A. Jones, and A. L. Cunningham. 2005. Herpes simplex virus infection of human dendritic cells induces apoptosis and allows cross-presentation via uninfected dendritic cells. J. Immunol. 1742220-2227. [DOI] [PubMed] [Google Scholar]
- 6.Brideau, A. D., L. W. Enquist, and R. S. Tirabassi. 2000. The role of virion membrane protein endocytosis in the herpesvirus life cycle. J. Clin. Virol. 1769-82. [DOI] [PubMed] [Google Scholar]
- 7.Bzik, D. J., B. A. Fox, N. A. DeLuca, and S. Person. 1984. Nucleotide sequence specifying the glycoprotein gene, gB, of herpes simplex virus type 1. Virology 133301-314. [DOI] [PubMed] [Google Scholar]
- 8.Ellermann-Eriksen, S. 2005. Macrophages and cytokines in the early defence against herpes simplex virus. Virol. J. 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fournier, N., L. Chalus, I. Durand, E. Garcia, J. J. Pin, T. Churakova, S. Patel, C. Zlot, D. Gorman, S. Zurawski, J. Abrams, E. E. Bates, and P. Garrone. 2000. FDF03, a novel inhibitory receptor of the immunoglobulin superfamily, is expressed by human dendritic and myeloid cells. J. Immunol. 1651197-1209. [DOI] [PubMed] [Google Scholar]
- 10.Fuller, A. O., R. E. Santos, and P. G. Spear. 1989. Neutralizing antibodies specific for glycoprotein H of herpes simplex virus permit viral attachment to cells but prevent penetration. J. Virol. 633435-3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuller, A. O., and P. G. Spear. 1987. Anti-glycoprotein D antibodies that permit adsorption but block infection by herpes simplex virus 1 prevent virion-cell fusion at the cell surface. Proc. Natl. Acad. Sci. USA 845454-5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geraghty, R. J., C. Krummenacher, G. H. Cohen, R. J. Eisenberg, and P. G. Spear. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 2801618-1620. [DOI] [PubMed] [Google Scholar]
- 13.Gianni, T., C. Forghieri, and G. Campadelli-Fiume. 2007. The herpesvirus glycoproteins B and H.L are sequentially recruited to the receptor-bound gD to effect membrane fusion at virus entry. Proc. Natl. Acad. Sci. USA 1043668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heldwein, E. E., H. Lou, F. C. Bender, G. H. Cohen, R. J. Eisenberg, and S. C. Harrison. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313217-220. [DOI] [PubMed] [Google Scholar]
- 15.Herold, B. C., R. J. Visalli, N. Susmarski, C. R. Brandt, and P. G. Spear. 1994. Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulphate and glycoprotein B. J. Gen. Virol. 751211-1222. [DOI] [PubMed] [Google Scholar]
- 16.Herold, B. C., D. WuDunn, N. Soltys, and P. G. Spear. 1991. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 651090-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato, A., J. Arii, I. Shiratori, H. Akashi, H. Arase, and Y. Kawaguchi. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J. Virol. 83250-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawaguchi, Y., T. Matsumura, B. Roizman, and K. Hirai. 1999. Cellular elongation factor 1δ is modified in cells infected with representative alpha-, beta-, or gammaherpesviruses. J. Virol. 734456-4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawaguchi, Y., C. Van Sant, and B. Roizman. 1998. Eukaryotic elongation factor 1δ is hyperphosphorylated by the protein kinase encoded by the UL13 gene of herpes simplex virus 1. J. Virol. 721731-1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kodukula, P., T. Liu, N. V. Rooijen, M. J. Jager, and R. L. Hendricks. 1999. Macrophage control of herpes simplex virus type 1 replication in the peripheral nervous system. J. Immunol. 1622895-2905. [PubMed] [Google Scholar]
- 21.Lopez, C. 1981. Resistance to herpes simplex virus - type 1 (HSV-1). Curr. Top. Microbiol. Immunol. 9215-24. [DOI] [PubMed] [Google Scholar]
- 22.Mauel, J., and V. Defendi. 1971. Infection and transformation of mouse peritoneal macrophages by simian virus 40. J. Exp. Med. 134335-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGeoch, D. J., C. Cunningham, G. McIntyre, and A. Dolan. 1991. Comparative sequence analysis of the long repeat regions and adjoining parts of the long unique regions in the genomes of herpes simplex viruses types 1 and 2. J. Gen. Virol. 723057-3075. [DOI] [PubMed] [Google Scholar]
- 24.Mellman, I. 1996. Endocytosis and molecular sorting. Annu. Rev. Cell Dev. Biol. 12575-625. [DOI] [PubMed] [Google Scholar]
- 25.Milne, R. S., A. V. Nicola, J. C. Whitbeck, R. J. Eisenberg, and G. H. Cohen. 2005. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 796655-6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montgomery, R. I., M. S. Warner, B. J. Lum, and P. G. Spear. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87427-436. [DOI] [PubMed] [Google Scholar]
- 27.Morimoto, T., J. Arii, H. Akashi, and Y. Kawaguchi. 2009. Identification of multiple sites in herpes simplex virus genomes suitable for insertion of foreign genes. Microbiol. Immunol. doi: 10.1111/j. 1348-0421.2008.00104.x. [DOI] [PubMed]
- 28.Mousseau, D. D., D. Banville, D. L'Abbe, P. Bouchard, and S. H. Shen. 2000. PILRalpha, a novel immunoreceptor tyrosine-based inhibitory motif-bearing protein, recruits SHP-1 upon tyrosine phosphorylation and is paired with the truncated counterpart PILRbeta. J. Biol. Chem. 2754467-4474. [DOI] [PubMed] [Google Scholar]
- 29.Nicola, A. V., J. Hou, E. O. Major, and S. E. Straus. 2005. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 797609-7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicola, A. V., A. M. McEvoy, and S. E. Straus. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 775324-5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicola, A. V., and S. E. Straus. 2004. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J. Virol. 787508-7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nixdorf, R., J. Schmidt, A. Karger, and T. C. Mettenleiter. 1999. Infection of Chinese hamster ovary cells by pseudorabies virus. J. Virol. 738019-8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pertel, P. E., A. Fridberg, M. L. Parish, and P. G. Spear. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279313-324. [DOI] [PubMed] [Google Scholar]
- 34.Reymond, N., A. M. Imbert, E. Devilard, S. Fabre, C. Chabannon, L. Xerri, C. Farnarier, C. Cantoni, C. Bottino, A. Moretta, P. Dubreuil, and M. Lopez. 2004. DNAM-1 and PVR regulate monocyte migration through endothelial junctions. J. Exp. Med. 1991331-1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robbins, A. K., D. J. Dorney, M. W. Wathen, M. E. Whealy, C. Gold, R. J. Watson, L. E. Holland, S. D. Weed, M. Levine, J. C. Glorioso, et al. 1987. The pseudorabies virus gII gene is closely related to the gB glycoprotein gene of herpes simplex virus. J. Virol. 612691-2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roizman, B., D. M. Knipe, and R. J. Whitley. 2007. The family Herpesviridae: a brief introduction, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 37.Satoh, T., J. Arii, T. Suenaga, J. Wang, A. Kogure, J. Uehori, N. Arase, I. Shiratori, S. Tanaka, Y. Kawaguchi, P. G. Spear, L. L. Lanier, and H. Arase. 2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132935-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shiratori, I., K. Ogasawara, T. Saito, L. L. Lanier, and H. Arase. 2004. Activation of natural killer cells and dendritic cells upon recognition of a novel CD99-like ligand by paired immunoglobulin-like type 2 receptor. J. Exp. Med. 199525-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shukla, D., J. Liu, P. Blaiklock, N. W. Shworak, X. Bai, J. D. Esko, G. H. Cohen, R. J. Eisenberg, R. D. Rosenberg, and P. G. Spear. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 9913-22. [DOI] [PubMed] [Google Scholar]
- 40.Sodeik, B., M. W. Ebersold, and A. Helenius. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 1361007-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spear, P. G., R. J. Eisenberg, and G. H. Cohen. 2000. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 2751-8. [DOI] [PubMed] [Google Scholar]
- 42.Spear, P. G., and R. Longnecker. 2003. Herpesvirus entry: an update. J. Virol. 7710179-10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Subramanian, R. P., and R. J. Geraghty. 2007. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc. Natl. Acad. Sci. USA 1042903-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanaka, M., H. Kagawa, Y. Yamanashi, T. Sata, and Y. Kawaguchi. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 771382-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka, M., H. Kodaira, Y. Nishiyama, T. Sata, and Y. Kawaguchi. 2004. Construction of recombinant herpes simplex virus type I expressing green fluorescent protein without loss of any viral genes. Microbes Infect. 6485-493. [DOI] [PubMed] [Google Scholar]
- 46.Turner, A., B. Bruun, T. Minson, and H. Browne. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 72873-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Warner, M. S., R. J. Geraghty, W. M. Martinez, R. I. Montgomery, J. C. Whitbeck, R. Xu, R. J. Eisenberg, G. H. Cohen, and P. G. Spear. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246179-189. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe, S., E. Ono, Y. Shimizu, and H. Kida. 1995. Pseudorabies virus early protein 0 transactivates the viral gene promoters. J. Gen. Virol. 762881-2885. [DOI] [PubMed] [Google Scholar]
- 49.Wilson, M. D., J. Cheung, D. W. Martindale, S. W. Scherer, and B. F. Koop. 2006. Comparative analysis of the paired immunoglobulin-like receptor (PILR) locus in six mammalian genomes: duplication, conversion, and the birth of new genes. Physiol. Genomics 27201-218. [DOI] [PubMed] [Google Scholar]
- 50.Wittels, M., and P. G. Spear. 1991. Penetration of cells by herpes simplex virus does not require a low pH-dependent endocytic pathway. Virus Res. 18271-290. [DOI] [PubMed] [Google Scholar]