

Abstract
Granulocyte-colony stimulating factor (G-CSF) has proved to be a successful therapy for some patients with Crohn's disease. Given the known ability of G-CSF to exert anti-T helper 1 effects and to induce interleukin (IL)-10-secreting regulatory T cells, we studied whether clinical benefit from G-CSF therapy in active Crohn's disease was associated with decreased inflammatory cytokine production and/or increased regulatory responses. Crohn's patients were treated with G-CSF (5 µg/kg/day subcutaneously) for 4 weeks and changes in cell phenotype, cytokine production and dendritic cell subsets were measured in the peripheral blood and colonic mucosal biopsies using flow cytometry, enzyme-linked immunosorbent assay and immunocytochemistry. Crohn's patients who achieved a clinical response or remission based on the decrease in the Crohn's disease activity index differed from non-responding patients in several important ways: at the end of treatment, responding patients had significantly more CD4+ memory T cells producing IL-10 in the peripheral blood; they also had a greatly enhanced CD123+ plasmacytoid dendritic cell infiltration of the lamina propria. Interferon-γ production capacity was not changed significantly except in non-responders, where it increased. These data show that clinical benefit from G-CSF treatment in Crohn's disease is accompanied by significant induction of IL-10 secreting T cells as well as increases in plasmacytoid dendritic cells in the lamina propria of the inflamed gut mucosa.
Keywords: cytokines, FoxP3, plasmacytoid dendritic cells, regulatory T cells, Th1 inflammation
Introduction
Novel therapies for Crohn's disease can exploit our deepening understanding of gut inflammatory conditions [1]. Granulocyte-colony stimulating factor (G-CSF) may be a particularly useful treatment for the T helper type 1 (Th1)/Th17-driven Crohn's inflammation because of its demonstrated immunomodulatory properties. G-CSF treatment results in decreased interferon (IFN)-γ and increased interleukin (IL)-4 expression, decreased IL-12Rβ2 chain expression and up-regulation of GATA-3 (important for Th2 responses) in mouse and human peripheral blood cells [2–4]. G-CSF treatment can also promote the generation and/or mobilization of IL-10-producing regulatory T cells in the peripheral blood [5,6], as well as preferentially mobilize peripheral plasmacytoid dendritic cells (PDCs) [7–9], a dendritic cell (DC) subset that has been linked to development of IL-10-producing Tr1-type regulatory cells [10]. Moreover, G-CSF can exert direct anti-inflammatory effects via inhibition of Toll-like receptor ligand-stimulated release of tumour necrosis factor (TNF)-α, IFN-γ, IL-12 and IL-1β from human monocytes and whole blood cells [2,11]. Currently, G-CSF therapy for Crohn's disease treatment is supported by several case reports and open-label studies that show a benefit for some patients [12–15].
The present study was undertaken to address the mechanisms for the therapeutic effects of G-CSF in patients with active Crohn's disease by measuring the changes in immune cell function that occur with G-CSF treatment and associating these changes with clinical response. A special focus of this study was to address the effect of G-CSF on regulatory cells in Crohn's inflammation in order to clarify how G-CSF might complement existing and emerging therapies for this disease.
Materials and methods
Patients and treatment
Patients were enrolled at the NIH Clinical Center between December 2001 and July 2003. Eligible adult patients had a verifiable diagnosis of Crohn's disease and a Crohn's disease activity index (CDAI) score of ≥ 225 and ≤ 450 respectively. Acceptable concomitant medications included only stable doses of antibiotics, mesalamine drugs, prednisone (≤ 25 mg/day) and probiotics. Other exclusion criteria included short-bowel syndrome, bowel obstruction, splenomegaly, probable requirement for imminent intestinal surgery, active enteric infection, abnormal chest X-ray or electrocardiogram, active hepatitis B or C infection, HIV seropositivity or a history of cancer. Patients were excluded if they reported any reactions to G-CSF (filgrastim) or any proteins derived from Escherichia coli.
After giving informed consent and a 14-day screening period, eligible patients received G-CSF 5 µg/kg/day subcutaneously for 28 days (dose was adjusted to keep the absolute neutrophil count ≤ 50 000 cells/µl). Clinical status was assessed on days 8, 15, 22 and 28, and biochemical and haematological parameters were assessed on days 4, 8, 11, 15, 22 and 28. Patients were assessed as clinical responders if the CDAI score fell at least 100 points from baseline and experienced remission if the absolute CDAI score was ≤ 150 points. Patients were followed for up to 24 weeks after the final injection of G-CSF, assessed at weeks 4, 8, 16 and 24. Colonoscopy was performed just prior to the first injection and within 48 h of the final injection. Biopsy specimens were restricted to areas of endoscopically visible active inflammation.
Cytokine measurement
Peripheral blood mononuclear cells (PBMCs) were prepared from 50 ml whole blood using Ficoll-sodium diatrizoate gradient centrifugation. CD4+CD45RO+ cells were enriched using CD4 and CD45RO magnetic affinity cell sorting (MACS) microbeads (Miltenyi Biotec, Auburn, CA, USA). Cells were stimulated for 48 h with antibodies (1 µg/ml) against CD3 and CD28. All culture supernatant cytokine concentrations were determined using multiplex bead Luminex100 instrumentation (Luminex Corporation, Austin, TX, USA) with Lincoplex assay kits (LINCO Research, Inc., St Charles, MO, USA). The assays were performed and analysed using a Logistic-5PL regression method with the Bio-Plex manager 3·0 software (Bio-Rad Laboratories, Hercules, CA, USA). Percentage coefficient of variation for both intra- and interassay variability was 10–15%.
Peripheral blood DC phenotyping
Isolated PBMCs stained for four-colour flow cytometry and analysed on a fluorescence activated cell sorter (FACSCalibur) using Cellquest software (Becton Dickinson Biosciences, San Jose, CA, USA). List mode parameters were collected for 106 mononuclear cells. DCs were identified by gating on the lineage (CD3, CD14, CD16, CD19, CD20 and CD56)-negative and human leucocyte antigen D-related-positive population. Subpopulations of DCs were then determined by using CD11c+ to identify myeloid and BDCA-2+ to identify plasmacytoid cells. Monoclonal antibodies were obtained from Becton Dickinson Biosciences, except for BDCA-2 (Miltenyi Biotech). All intra-assay variability was < 3%. The interassay variability for B cells and monocytes was 13%, for T cells < 6% and for natural killer cells 18·5%.
Immunohistochemistry
Sections from formalin-fixed mucosal biopsies were deparaffinized and rehydrated prior to heat-induced antigen retrieval (10 mM Tris-HCl, 1 mM ethylenediamine tetraacetic acid, pH 9·0 for 20 min by microwave). Slides were washed with Tris-buffered saline, 0·05% Tween-20 (TBST). Endogenous peroxidase activity was blocked [10 min with 0·3% hydrogen peroxide containing sodium azide from the EnVision+®Dual Lynk System-HRP (DAB+)] (DakoCytomation, Carpinteria, CA, USA). Mouse anti-human CD-123 antibody (BD Biosciences Pharmingen, San Diego, CA, USA) was added at 1:50 dilution for 1 h at 37°C. After washing with TBST, the slides were incubated with a horseradish peroxidase (HRP)-labelled polymer (DakoCytomation), and peroxidase activity was visualized with the substrate 3′-3′ diaminobenzidine. The slides were counterstained with Mayer's haematoxylin, dehydrated and mounted.
For forkhead box P3 (FoxP3) and CD25 double-staining, after blocking the endogenous peroxidase activity, slides were incubated with the mouse monoclonal anti-FoxP3 (Abcam, Cambridge, MA, USA) at 1:50 overnight at 4°C and processed as above. For the second staining, after heat-induced antigen retrieval, the slides were incubated in serum free-protein block for 30 min and then with mouse anti-human CD25 antibody (Novocastra, Bannockburn, IL, USA) at 1:50 for 60 min at 37°C. After washing with TBST, the slides were incubated with goat anti-mouse immunoglobulin (Ig)G-alkaline phosphatase conjugate (Sigma, St Louis, MO, USA) at 1:100 for 45 min and washed with TBST. Alkaline phosphatase activity was visualized using the Vector Red alkaline phosphatase subtrate kit I (Vector Laboratories, Burlingame, CA, USA) containing 0·2 mM of levamisole (DakoCytomation). The slides were washed with distilled water, counterstained and mounted using an aqueous mounting media. Negative double-staining control experiments were performed with mouse IgG2a isotype (AbD Serotec, Raleigh, NC, USA) using similar Ig concentrations. Biopsy slides were counted for stained lamina propria cells included in 8–12 high power fields (hpf)/slide by two pathologists masked to the clinical response.
Statistical analysis
The sample size for this open-label pilot study was designed for the primary outcome of safety (10 treated subjects gave > 80% probability of observing adverse events that had actual rates of 10% or greater). Secondary outcomes such as changes in cytokine production and cell populations were analysed in an exploratory fashion (no adjustments were made for multiple comparisons). Differences between means within groups (baseline and post-treatment results in responders, for instance) were tested using a paired t-test and differences between groups (post-treatment results for responders and non-responders, for instance) were tested using a non-paired t-test (InStat; GraphPad Software, San Diego, CA, USA). In some instances, because of the small sample size, significance testing was repeated using non-parametric Mann–Whitney testing to assess the robustness of the results.
Results
Patient demographics and response to G-CSF treatment
Ten patients were screened and nine were enrolled (one patient had no active Crohn's disease). Of the nine patients, there were six females and three males [34·7 ± 7·8 years old, mean ± standard deviation (s.d.), range 22–42]. The mean duration of disease was 13·8 ± 9·5 years. Eight patients had ileocolonic disease and one had only colonic involvement. One patient had fistulizing disease and five had had previous bowel resections. At the time of enrolment patients were taking no Crohn's medications (two patients) or were taking one or two Crohn's medications: four were taking corticosteroids, five were taking mesalamine, and two had stopped azathioprine 4 weeks prior to enrollment. Patients began the study with mean CDAI of 313 ± 60 points.
During the 28-day G-CSF treatment period six patients reported improvement in their CDAI score, achieving a clinical response (four patients) or remission (two patients). Improvement in three of these patients occurred after 2 weeks and the other three by the end of G-CSF treatment (Fig. 1). One remission was durable for over 60 days post-treatment, and in two patients clinical responses were maintained for 110 days post-treatment. The three non-responders terminated participation early in the treatment (one patient) or early in the follow-up period (two patients) because of lack of efficacy. Five other patients terminated participation coincident with a recrudescence of symptoms during the follow-up period.
Fig. 1.
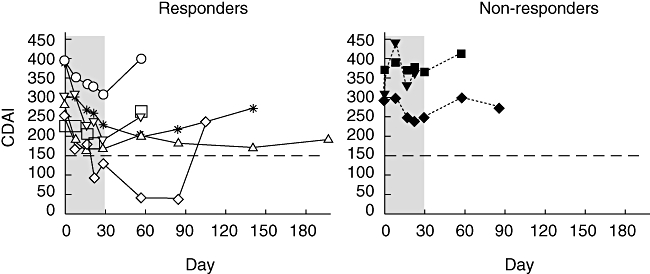
Clinical response to granulocyte-colony stimulating factor (G-CSF) treatment in active Crohn's disease. Patients’ clinical response measured by the Crohn's disease activity index (CDAI) is plotted over the course of treatment (shaded area) and follow-up. Dotted line denotes CDAI = 150, the level at or below defining remission.
The three non-responding patients had significantly longer duration of disease compared with the responding patients (22·3 ± 5·5 years versus 9·5 ± 8·2, P < 0·05) and included the only patient with active fistulae. Additionally, all three non-responding patients reported prior bowel resections compared with only two of six of the responding patients.
Changes in PBMC phenotype following G-CSF treatment
In order to detect differences in immune parameters that might be associated with the clinical response to G-CSF, we measured peripheral blood cell phenotypes before and after G-CSF treatment. Overall, there were no significant differences in numbers of baseline peripheral blood cell phenotypes in responding patients compared with non-responders, except for higher pretreatment numbers of CD3+/DR+ cells in non-responders (343 ± 156 versus 171 ± 51 cells/ul, mean ± s.d., P = 0·04, reference range 0–291 cells/µl). Similarly, there were no differences in cell numbers at the end of treatment between the two groups. However, while within each responder and non-responder group significant increases in some phenotypes could be measured after treatment (Table 1), nearly all the changes, including the non-significant ones, were in a conjugate upward direction. These data did not point to any meaningful differences in the PBMC phenotypes before or after G-CSF treatment that was associated with the presence of a clinical response.
Table 1.
Peripheral blood mononuclear cell phenotyping before and after granulocyte-colony stimulating factor (G-CSF) treatment (cell/µl blood): intragroup changes for responders and non-responders, paired t-test results.
| Responders (n = 6) | Non-responders (n = 3) | |||||
|---|---|---|---|---|---|---|
| Pre- | Post- | P-value | Pre- | Post- | P-value | |
| CD3 | 1356 ± 579 | 2093 ± 400 | 0·04 | 1004 ± 511 | 1577 ± 246 | n.s. |
| CD5 | 1337 ± 586 | 2071 ± 400 | 0·04 | 995 ± 512 | 1592 ± 254 | n.s. |
| CD4 | 877 ± 483 | 1279 ± 263 | 0·05 | 550 ± 291 | 862 ± 143 | n.s. |
| CD8 | 505 ± 122 | 931 ± 337 | 0·02 | 497 ± 242 | 741 ± 37 | n.s. |
| CD4/CD3 | 877 ± 489 | 1272 ± 269 | n.s. | 547 ± 290 | 864 ± 177 | n.s. |
| CD8/CD3 | 430 ± 101 | 747 ± 276 | n.s. | 425 ± 228 | 660 ± 86 | n.s. |
| T4/T8 ratio | 1·9 ± 0·9 | 1·8 ± 0·8 | n.s. | 1·1 ± 0·1 | 1·2 ± 0·2 | n.s. |
| DNT | 29 ± 22 | 42 ± 32 | 0·05 | 19 ± 2 | 37 ± 27 | n.s. |
| CD45RO/CD4 | 395 ± 279 | 540 ± 293 | n.s. | 317 ± 218 | 539 ± 284 | 0·05 |
| CD45RA/CD4 | 276 ± 170 | 429 ± 136 | n.s. | 106 ± 70 | 197 ± 97 | n.s. |
| CD45RO/CD8 | 110 ± 46 | 194 ± 129 | n.s. | 120 ± 71 | 122 ± 6 | n.s. |
| CD45RA/CD8 | 208 ± 88 | 357 ± 138 | n.s. | 159 ± 83 | 287 ± 15 | n.s. |
| CD3/DR | 171 ± 51* | 375 ± 184 | 0·02 | 343 ± 156 | 273 ± 22 | n.s. |
| CD4/DR | 58 ± 15 | 117 ± 49 | 0·04 | 69 ± 51 | 91 ± 18 | n.s. |
| CD8/DR | 113 ± 54 | 277 ± 187 | 0·04 | 179 ± 154 | 175 ± 3 | n.s. |
| CD3/CD25 | 648 ± 527 | 949 ± 441 | 0·05 | 382 ± 226 | 649 ± 310 | 0·03 |
| CD3/CD161 | 200 ± 100 | 336 ± 132 | 0·02 | 206 ± 147 | 390 ± 326 | n.s. |
| CD20 | 156 ± 75 | 214 ± 81 | 0·04 | 75 ± 51 | 155 ± 61 | 0·01 |
| CD20/CD5 | 53 ± 33 | 75 ± 40 | 0·02 | 29 ± 23 | 55 ± 30 | n.s. |
| CD19 | 161 ± 74 | 225 ± 83 | 0·04 | 82 ± 54 | 165 ± 72 | n.s. |
| NK | 141 ± 85 | 344 ± 230 | n.s. | 157 ± 131 | 201 ± 100 | n.s. |
| NK T | 71 ± 56 | 143 ± 105 | 0·02 | 133 ± 49 | 258 ± 6 | n.s. |
P = 0·04 for intergroup pretreatment values responders versus non-responders. NK, natural killer; NK T, natural killer T cells; n.s., not significant.
Granulocyte-colony stimulating factor treatment is associated with changes in memory cell cytokine production
The capacity of circulating CD4+CD45RO+ memory T cells to produce cytokines before and after G-CSF treatment was assessed, and the most striking effect was seen in relation to IL-10 production. In particular, peripheral blood CD4+CD45RO+ cells isolated from responding patients exhibited significant increases in IL-10 production following G-CSF treatment [from 3973 ± 1147 pg/ml to 13 162 ± 2885, P < 0·02, mean ± standard error of the mean (s.e.m.)]; one patient had very high baseline and end-of-treatment IL-10 production (Fig. 2). In contrast, CD4+CD45RO+ cells from non-responding patients did not have a significant change in their IL-10 production following G-CSF treatment (3250 ± 1948 pg/ml to 5517 ± 1188 pg/ml, P > 0·3).
Fig. 2.
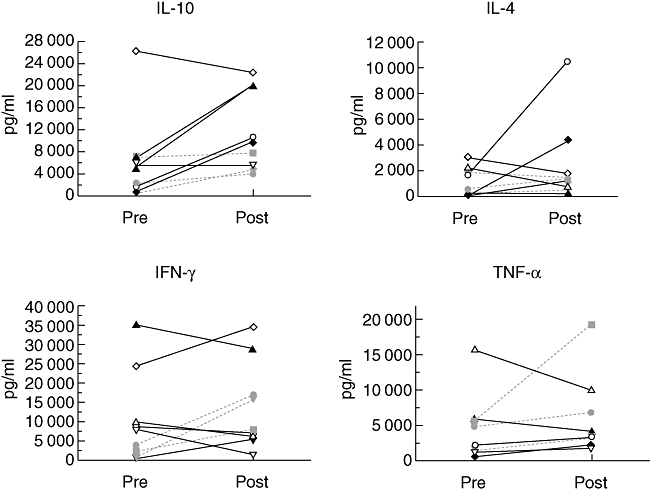
Cytokine production by peripheral CD4+CD45RO+ cells before and at the end of granulocyte-colony stimulating factor (G-CSF) treatment. Isolated cells were cultured and stimulated with anti-CD3 and anti-CD28 antibodies. Supernatant cytokines were measured by enzyme-linked immunosorbent assay. Interleukin-10 levels for responders pre- versus post-treatment were 3973 ± 1147 pg/ml and 13 162 ± 2885 (mean ± standard error of the mean), P < 0·02. Responders, solid lines, non-responders, dashed lines.
No significant differences in peripheral CD4+CD45RO+ cell IL-4 production were observed before or after treatment within or between groups: 1288 ± 524 pg/ml pretreatment to 3215 ± 1579 pg/ml after treatment for responding patients and 879 ± 526 pg/ml pretreatment to 1176 ± 312 pg/ml after treatment for non-responding patients. However, two responding patients, who had shown an increase in IL-10 production, also exhibited a notable increase in CD4+CD45RO+ cell production of IL-4 after G-CSF treatment.
Interferon-γ production by peripheral CD4+CD45RO+ cells from G-CSF-responsive patients did not change at the end of treatment (14 483 ± 5174 pg/ml versus 13 893 ± 5720). In non-responders IFN-γ production, while lower at the start of treatment, increased significantly at the end of treatment (2253 ± 968 pg/ml versus 13 600 ± 2814, P < 0·02). Finally, with respect to TNF-α production by peripheral CD4+CD45RO+ T cells, there were no significant differences before or after treatment in either group.
Granulocyte-colony stimulating factor treatment affects levels of circulating DC subsets
Peripheral blood myeloid (CD11c+) and plasmacytoid (BDCA-2+) DC populations were quantitated at the initiation and termination of G-CSF treatment as well as after the 4-week follow-up period. All patients showed an increase in the number of peripheral blood myeloid and PDCs as well as a decrease in the myeloid dendritic cell(MDC)/PDC ratio at the end of G-CSF treatment (Fig. 3a). Responding patients showed a significant decrease in the MDC/PDC ratio at the end of treatment compared with baseline (2·1 ± 0·2 versus 1·1 ± 0·2, mean ± s.e.m., P < 0·01) that was sustained to follow-up (2·1 ± 0·2 versus 1·1 ± 0·4, P < 0·05). Non-responding patients showed a trend towards a decrease in MDC/PDC ratio at the end of treatment (3·5 ± 1·7 versus 0·9 ± 0·2, P > 0·2) and a return towards baseline at the end of the 4-week follow-up (3·5 ± 1·7 versus 2·3 ± 0·4, P > 0·6).
Fig. 3.
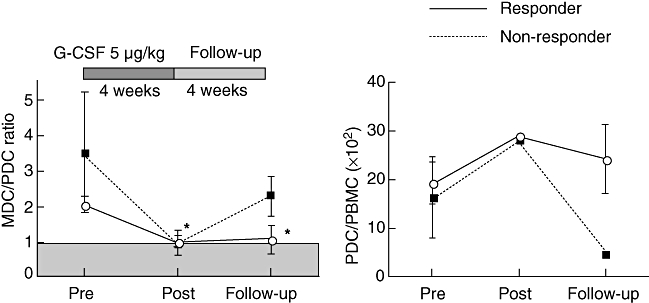
Peripheral blood dendritic cell responses to granulocyte-colony stimulating factor (G-CSF) treatment. Myeloid dendritic cells (MDC), lin–DR+CD11c+ and plasmacytoid dendritic cells (PDC), lin–DR+BDCA2+, were measured by flow cytometry before (Pre), at the end (Post), and 4 weeks after (follow-up) G-CSF treatment. The MDC/PDC ratio and PDCs as percentage of total peripheral blood mononuclear cells are shown (mean ± standard error of the mean). *P < 0·01 post-treatment, P < 0·05 at follow-up Responders compared with pretreatment. Responders, solid lines, non-responders, dashed lines.
The above results were reflected in the PDC/total PBMC ratios of the responding and non-responding patients (Fig. 3b). Thus, responding patients showed an increase from 19 ± 4% PDC/PBMC × 102(mean ± s.e.m.) prior to treatment to 29 ± 8 at end of treatment and 24 ± 7 at end of follow-up (4 weeks after the final dose), whereas non-responders showed an increase from 17 ± 8% PDC/PBMC × 102prior to treatment to 28 ± 9 at end of treatment, dropping to 5% at the end of follow-up. Despite changes within the G-CSF responder group, statistical testing for differences between responders and non-responders found no significance at any time-point.
Lamina propria cell CD123 expression following G-CSF treatment
To determine if any changes in peripheral blood PDCs were reflected in the lamina propria, biopsies of endoscopically inflamed colonic mucosa were stained for cells expressing CD123. Of all pretreatment biopsies, only one patient in the responder group showed CD123-positive cells in the lamina propria (≤ 1 cell/hpf) or in any lymphoid follicles included in the biopsy. However, at the end of treatment, four patients in the responder group showed the notable appearance of CD123-positive cells in the lamina propria (a mean of five CD123+ cells/hpf, range 2–14) which were neither endothelial cells associated with mucosal blood vessels nor basophils (Table 2 and Fig. 4). No non-responder patients accumulated any CD123-positive cells in the lamina propria after G-CSF treatment.
Table 2.
Lamina propria mononuclear cell CD123 expression in colonic biopsies before and after granulocyte-colony stimulating factor (G-CSF) treatment.
| Responder | Non-responder | |||
|---|---|---|---|---|
| Pre-* | Post- | Pre- | Post- | |
| CD123+ | 0·1 ± 0·1 | 4·8 ± 2·3 | 0 | 0 |
Number of cells/high power fields (mean ± standard error of the mean).
Fig. 4.
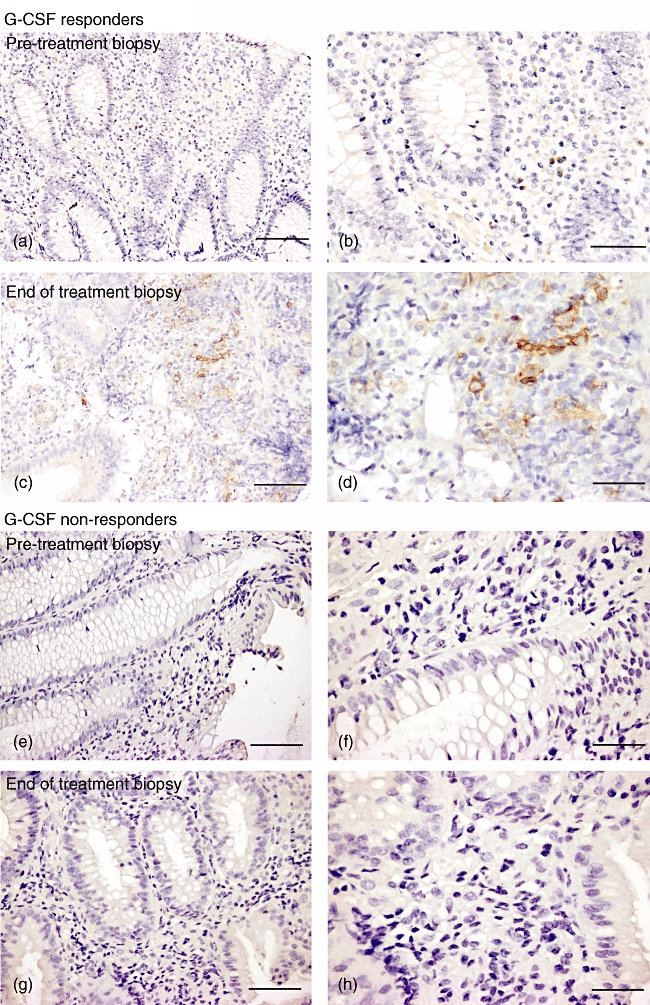
CD123 staining in colonic biopsies from Crohn's patients treated with granulocyte-colony stimulating factor (G-CSF). Colonoscopic biopsies were taken before and within 1 week of completing G-CSF treatment from a responder (a–d) and a non-responder patient (e–h). CD123+ cells have brown cytoplasmic staining. Scale bar: 100 µm for (a, c, e, g); 50 µm for (b, d, f, h).
Lamina propria cell CD25 and FoxP3 expression following G-CSF treatment
To assess the effect of G-CSF treatment on FoxP3-expressing T cell populations, biopsies of endoscopically inflamed colonic mucosa were examined for expression of CD25 and FoxP3 among lamina propria mononuclear cells by immunohistochemical staining. CD25-stained cells were counted as CD25high (intensely stained cytoplasm) or CD25low (intermediate or very low staining). While numbers of CD25high/FoxP3– cells/hpf were similar between responder and non-responder patients (1·6 ± 0·5 versus 2·2 ± 1·4, mean ± s.e.m.), the non-responding patients showed significantly higher numbers of FoxP3+ cells, both CD25high(3·4 ± 0·3 versus 0·8 ± 0·3, P < 0·001) and CD25low (22 ± 3·2 versus 6·3 ± 1·8, P < 0·005), in the lamina propria prior to treatment (Table 3, Fig. 5). However, at the end of treatment, the numbers of CD25high/FoxP3–, CD25low/FoxP3+ and CD25high/FoxP3+ cells/hpf were not changed significantly within the groups of patients. The CD25high/FoxP3+ cells decreased significantly as a percentage of total CD25+ cells in non-responders at the end of treatment (69·3 ± 17% to 47·3 ± 20·9%, P < 0·05) (Table 4). However, a non-significant increase in CD25high/FoxP3+ cells in the lamina propria of responding patients at the end of G-CSF treatment (0·8 ± 0·3 to 1·8 ± 0·8) was also seen.
Table 3.
Lamina propria mononuclear cell forkhead box P3 (FoxP3) and CD25 expression in colonic biopsies before and after granulocyte-colony stimulating factor (G-CSF) treatment.
| Responder | Non-responder | |||
|---|---|---|---|---|
| Pre-† | Post- | Pre- | Post- | |
| FoxP3+/CD25high | 0·8 ± 0·3* | 1·8 ± 0·8 | 3·4 ± 0·3 | 3·1 ± 1·2 |
| FoxP3+/CD25low | 6·3 ± 1·8** | 8·9 ± 4·0*** | 22 ± 3·2 | 32·9 ± 13·6 |
| FoxP3−/CD25high | 1·6 ± 0·5 | 1·5 ± 0·5 | 2·2 ± 1·4 | 3·7 ± 1·5 |
P < 0·001;
P < 0·005;
P = 0·059. Responder versus non-responder at that time-point.
Number of cells/high power fields (mean ± standard error of the mean).
Fig. 5.
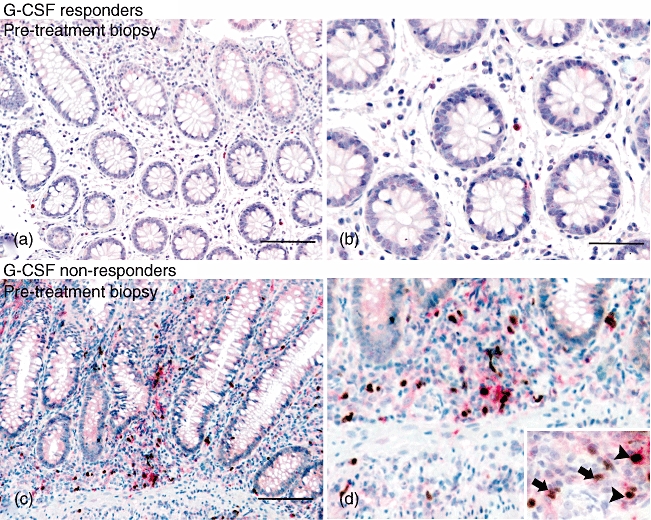
Forkhead box P3 (FoxP3) and CD25 staining in colonic biopsies from Crohn's patients treated with granulocyte-colony stimulating factor (G-CSF). Colonoscopic biopsies taken before treatment with G-CSF, representative biopsies from responder (a, b) and non-responder (c, d) patients. FoxP3-positive cells are seen as brown nuclear staining and CD25-positive cells are seen as red cytoplasmic staining. Scale bar: 100 µm for (a) and (c), 50 µm for (b) and (d). Detail shows FoxP3+/CD25high cells (black arrowheads) and FoxP3+/CD25low cells (black arrows); 60× magnification.
Table 4.
Lamina propria mononuclear cell forkhead box P3 (FoxP3)/CD25 expression as a percentage of total FoxP3 or CD25 expression in colonic biopsies before and after granulocyte-colony stimulating factor (G-CSF) treatment.
| Responder | Non-responder | |||
|---|---|---|---|---|
| Pre- | Post- | Pre- | Post- | |
| DP†/total FoxP3 | 10 ± 2·9% | 13·5 ± 5·0% | 13·7 ± 0·8% | 5·9 ± 3·0% |
| DP/total CD25 | 38·8 ± 13·5% | 32·7 ± 11·5% | 69·3 ± 17% | 47·3 ± 20·9%* |
P ≤ 0·05. Non-responder pre- versus post-;
double-positive FoxP3/CD25high cells.
Discussion
Granulocyte-colony stimulating factor-induced clinical responses in Crohn's disease were accompanied by increased capacity of circulating memory T cells to produce IL-10 and the appearance of plasmacytoid DCs in the lamina propria, a DC phenotype associated with regulatory responses. While non-responders showed none of these findings, this group may have disease activity less amenable to treatment with G-CSF, given that this group reported a significantly longer duration of disease and a higher rate of previous bowel surgery and that the data showed higher expression of FoxP3 in lamina propria cells, a finding reported to be linked to colitis activity [16–18].
Clinical responsiveness to G-CSF was not associated with significant differences in baseline or post-treatment changes in PBMC phenotype. G-CSF effects on PBMC phenotype in all treated Crohn's patients largely recapitulated the expected effects seen in normal volunteer peripheral blood stem cell donors (who received G-CSF 10 µg/kg/day for 4–5 days) [19]. Similar to healthy stem cell donors, all G-CSF-treated Crohn's patients exhibited increased peripheral CD3+ T cell expression of CD25. Importantly, neutrophil numbers were also increased by G-CSF therapy to the level seen in normal blood stem cell donors, but this increase was equivalent in both responders and non-responders. This is of interest, because an alternative mechanism for G-CSF efficacy in Crohn's disease proposes that G-CSF may improve an innate immunodeficiency [14] that predisposes individuals to gut inflammation, as mild neutrophil defects have been reported in Crohn's patients [20] and a Crohn's-like colitis may be seen in states of significant neutrophil defects such as chronic granulomatous disease and glycogen-1b storage disease [21,22]. This study does not offer support for the thesis that Crohn's disease is due to immunodeficiency consequent to a neutrophil defect.
Naive or memory CD4 cells from healthy humans have been shown to produce increased amounts of IL-10 after G-CSF treatment in vivo[23,24] or after in vitro exposure to PDCs [10,25], particularly when the PDCs have been exposed to G-CSF [6]. A striking immunological result of treatment of Crohn's patients with G-CSF was the significant increase in IL-10 production capacity by peripheral CD4+CD45RO+ (memory) T cells, particularly as it was restricted to patients with a clinical response. While these data do not prove that the induced IL-10-producing cells are related causally to the clinical improvement in this group of patients, the complete absence of this effect in the three non-responding patients supports the idea that induction of regulatory effects by G-CSF may be one mechanism for its efficacy. These G-CSF-induced IL-10-producing cells may represent the development of IL-10-producing regulatory T cells known as Tr1 cells, but the exact T cell subset that is the source of the IL-10 cannot be deduced from these data.
Finally, while the generation of Tr1 cells producing IL-10 was observed by previous studies of the effects of G-CSF on IL-10 production, and G-CSF has IL-10-linked beneficial clinical effects in an animal model of graft-versus-host disease [5], the present report suggests that G-CSF treatment and induction of IL-10 might also operate in a human disease.
It is not likely that G-CSF exerts its main therapeutic effect in Crohn's disease by down-modulating the Th1 response. Although two G-CSF responders developed new IL-4 production, there was no significant decrease in responder IFN-γ production. Interestingly, non-responding patient T cells showed a significant increase in mean IFN-γ production after G-CSF treatment. The neutral or enhancing effect of G-CSF on Th1 responses in patients with Crohn's disease stands in contrast to the effect of G-CSF on stem cell donors where IFN-γ production is inhibited significantly by G-CSF exposure [19]. This difference may be related to the effects of G-CSF in inflammatory versus healthy states and the duration of G-CSF treatment in the two studies (29 days versus 5 days).
Previous studies showed that circulating DC numbers increase after G-CSF treatment, an increase favouring plasmacytoid over MDC subsets [7,8,26–28]. We could not show that significant changes in peripheral blood PDCs or the MDC/PDC ratio accounted for significant changes in responders versus non-responder patients. However, in corresponding in situ studies of DCs in the colonic tissue of our patients we found that all patients exhibited almost no CD123+ plasmacytoid DCs prior to G-CSF treatment, consistent with a previous report noting few plasmacytoid DCs present in the tissues of either Crohn's disease or non-inflamed control patients [29]. In contrast, while colonic tissue from a majority of patients responding to G-CSF treatment exhibited an increase in CD123+ plasmacytoid DCs at the end of treatment, tissue from the non-responding patients continued to be devoid of these cells. Thus, studies of CD123+plasmacytoid DCs in the colonic tissues showed changes in the numbers of these cells that were restricted to patients who manifested clinical responses to G-CSF therapy. The changes in IL-10 production and plasmacytoid DCs might be linked, as it has been shown that plasmacytoid cells exert an immunoregulatory effect through induction of IL-10-secreting Tr1 cells [25], possibly through the secretion of IFN-α/β[30,31]. Future studies will need to test whether the failure of G-CSF to ameliorate disease in non-responders in this view, is related to inadequately induced plasmacytoid DCs in these patients by differences in the expression of gut-homing molecules or the ability to induce regulatory T cells.
Although G-CSF may exert beneficial effects in Crohn's disease by inducing T regulatory cells, the FoxP3+‘natural’ regulatory T cell (nTreg) subset does not appear to be a major mechanism, but its involvement cannot be ruled out based on our data. Previous studies have shown that nTregs play an important role in animal models of colitis and in humans with immunodysregulation polyendocrinopathy enteropathy X-linked syndrome who develop severe colitis in their absence [18,32]. However, the relation of such cells to inflammatory bowel disease is unclear because patients with Crohn's disease and ulcerative colitis have significantly higher numbers of lamina propria nTregs compared with healthy controls (or even non-involved or disease-remission mucosa) [16,17,33–35]. Our data show that G-CSF therapy increased the number of CD25+FoxP3+ cells in responders, but to a small and non-significant degree. In addition, while Crohn's patients who were unresponsive to G-CSF therapy had significantly higher numbers of lamina propria CD25+FoxP3+ cells compared with patients who responded to G-CSF, it is not clear whether these higher numbers of putative regulatory T cells identify non-responding Crohn's patients as actually having a more refractory inflammation, or indicate the presence of conditions that attenuate the induction of regulatory mechanisms. Future studies measuring enhanced suppressive function of these CD25+FoxP3+ cells after G-CSF treatment may establish a role for nTregs in the therapeutic effect. Finally, in contrast to previous studies, we identified a sizable population of CD25lowFoxP3+ cells that were present in greater numbers than the CD25highFoxP3+cells; the regulatory capacity of this population, if any, is currently unknown. Taken together, these data suggest strongly that ‘conventional’ CD25+FoxP3+ cells are probably not responsible for the therapeutic effects of G-CSF in this disease, but further study is required.
In summary, our data suggest that G-CSF holds promise for the treatment of Crohn's disease through its capacity to induce IL-10-mediated regulatory effects, associated possibly with increased plasmacytoid DC numbers in the inflamed gut. Future studies are needed to define more clearly the mechanisms of the observed immune responses to G-CSF and to establish more clearly the causal link between these responses and the improvement of symptoms.
Acknowledgments
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. This project has been funded by the Division of Intramural Research, NIAID, the NIH Clinical Center and the National Cancer Institute under contract N01-CO-12400. The authors thank C. Y. Huang for helpful discussions about the manuscript.
References
- 1.Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–21. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartung T, Doecke W-D, Gantner F, et al. Effect of granulocyte colony-stimulating factor treatment on ex vivo blood cytokine response in human volunteers. Blood. 1995;85:2482–9. [PubMed] [Google Scholar]
- 3.Pan L, Delmonte J, Jalonen C, Ferrara J. Pretreatment of donor mice with granulocyte colony-stimulating factor polarizes donor T lymphocytes toward type-2 cytokine production and reduces the severity of experimental graft-versus-host disease. Blood. 1995;86:4422–9. [PubMed] [Google Scholar]
- 4.Zeng D, Dejbakhsh-Jones S, Strober S. Granulocyte colony-stimulating factor reduces the capacity of blood mononuclear cells to induce graft-versus-host disease: impact on blood progenitor cell transplantation. Blood. 1997;90:453–63. [PubMed] [Google Scholar]
- 5.Morris E, MacDonald K, Rowe V, et al. Donor treatment with pegylated G-CSF augments the generation of IL-10-producing regulatory T cells and promotes transplantation tolerance. Blood. 2004;103:3573–81. doi: 10.1182/blood-2003-08-2864. [DOI] [PubMed] [Google Scholar]
- 6.Rutella S, Bonnano G, Pierelli L, et al. Granulocyte colony-stimulating factor promotes the generation of regulatory DC through induction of IL-10 and IFN-alpha. Eur J Immunol. 2004;34:1291–302. doi: 10.1002/eji.200324651. [DOI] [PubMed] [Google Scholar]
- 7.Arpinati M, Green C, Heimfeld S, Heuser J, Anasetti C. Granulocyte colony-stimulating factor mobilizes T helper 2-inducing dendritic cells. Blood. 2000;95:2484–90. [PubMed] [Google Scholar]
- 8.Klangsinsirikul P, Russell H. Peripheral blood stem cell harvests from G-CSF-stimulated donors contains a skewed Th2 CD4 phenotype and a predominance of type 2 dendritic cells. Exp Hematol. 2002;30:495–501. doi: 10.1016/s0301-472x(02)00785-3. [DOI] [PubMed] [Google Scholar]
- 9.Vela-Ojeda J, Esparza M, Reyes-Maldonaldo E, et al. Peripheral blood mobilization of different lymphocyte and dendritic cell subsets with the use of intermediate doses of G-CSF in patients with non-Hodgkin's lymphoma and multiple myeloma. Ann Hematol. 2006;85:308–14. doi: 10.1007/s00277-006-0090-8. [DOI] [PubMed] [Google Scholar]
- 10.Ito T, Yang M, Wang Y-H, et al. Plasmacytoid dendritic cells prime IL-10-producing T regulatory cells by inducible costimulator ligand. J Exp Med. 2007;204:105–15. doi: 10.1084/jem.20061660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boneberg E, Hareng L, Gantner F, Wendel A, Hartung T. Human monocytes express functional receptors for granulocyte colony-stimulating factor that mediate suppression of monokines and interferon-gamma. Blood. 2000;95:270–6. [PubMed] [Google Scholar]
- 12.Dejaco C, Lichtenberger C, Miehsler W, et al. An open-label pilot study of granulocyte colony-stimulating factor for the treatment of severe endoscopic postoperative recurrence in Crohn's disease. Digestion. 2003;68:63–70. doi: 10.1159/000074517. [DOI] [PubMed] [Google Scholar]
- 13.Fata F, Myers P, Addeo J, Grinberg M, Nawabi I, Cappell M. Cyclic neutropenia in Crohn's ileocolitis: efficacy of granulocyte colony-stimulating factor. J Clin Gastroenterol. 1997;24:253–6. doi: 10.1097/00004836-199706000-00015. [DOI] [PubMed] [Google Scholar]
- 14.Korzenik J, Dieckgraefe B. An open-labelled study of granulocyte colony-stimulating factor in the treatment of active Crohn's disease. Aliment Pharmacol Ther. 2005;21:391–400. doi: 10.1111/j.1365-2036.2005.02287.x. [DOI] [PubMed] [Google Scholar]
- 15.Vaughan D, Drumm B. Treatment of fistulas with granulocyte colony-stimulating factor in a patient with Crohn's disease. N Engl J Med. 1999;340:239–40. doi: 10.1056/NEJM199901213400317. [DOI] [PubMed] [Google Scholar]
- 16.Holmen N, Lundgren A, Lundin S, et al. CD4+CC25high regulatory T cells are enriched in the colonic mucosa of patients with active ulcerative colitis and increase with disease activity. Inflamm Bowel Dis. 2006;12:447–56. doi: 10.1097/00054725-200606000-00003. [DOI] [PubMed] [Google Scholar]
- 17.Maul J, Loddenkemper C, Mundt P, et al. Peripheral and intestinal regulatory CD4+CD25high T cells in inflammatory bowel disease. Gastroenterology. 2005;128:1868–78. doi: 10.1053/j.gastro.2005.03.043. [DOI] [PubMed] [Google Scholar]
- 18.Uhlig H, Coombes J, Mottet C, et al. Characterization of foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J Immunol. 2006;177:5852–60. doi: 10.4049/jimmunol.177.9.5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tayebi H, Kuttler F, Saas P, et al. Effect of granulocyte colony-stimulating factor mobilization on phenotypical and functional properties of immune cells. Exp Hematol. 2001;29:458–70. doi: 10.1016/s0301-472x(01)00613-0. [DOI] [PubMed] [Google Scholar]
- 20.Versaget H, Pena A, Weterman I, Lamers C. Diminished neutrophil function in Crohn's disease and ulcerative colitis identified by decreased oxidative metabolism and low superoxide dismutase content. Gut. 1988;29:223–8. doi: 10.1136/gut.29.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Issacs D, Wright V, Shaw D, Raafat F, Walker-Smith J. Chronic granulomatous disease mimicking Crohn's disease. J Pediatr Gastroenterol Nutr. 1985;4:498–501. doi: 10.1097/00005176-198506000-00030. [DOI] [PubMed] [Google Scholar]
- 22.Roe T, Thomas D, Gilsanz V, Isaacs H, Atkinson J. Inflammmatory bowel disease in glycogen storage disease type 1b. J Pediatr. 1986;109:55–9. doi: 10.1016/s0022-3476(86)80572-8. [DOI] [PubMed] [Google Scholar]
- 23.Meilcarek M, Graf L, Johnson G, Torok-Storb B. Production of interleukin-10 by granulocyte colony-stimulating factor-mobilized blood products: a mechanism for monocyte-mediated suppression of T-cell proliferation. Blood. 1998;92:215–22. [PubMed] [Google Scholar]
- 24.Rutella S, Pierelli L, Bonnano G, et al. Role for granulocyte colony-stimulating factor in the generation of human T regulatory type 1 cells. Blood. 2002;100:2562–71. doi: 10.1182/blood-2001-12-0291. [DOI] [PubMed] [Google Scholar]
- 25.Kvale E, Floisand Y, Lund-Johansen F, et al. Plasmacytoid DCs regulate recall responses by rapid induction of IL-10 in memory T cells. Blood. 2007;109:3369–76. doi: 10.1182/blood-2006-06-031484. [DOI] [PubMed] [Google Scholar]
- 26.Hock B, Haring L, Ebbett A, Patton W, McKenzie J. Differential effects of G-CSF mobilisation on dendritic cell subsets in normal allogeneic donors and patients undergoing autologous transplantation. Bone Marrow Transplant. 2002;30:733–40. doi: 10.1038/sj.bmt.1703734. [DOI] [PubMed] [Google Scholar]
- 27.Lonial S, Hicks M, Rosenthal H, et al. A randomized trial comparing the combination of granulocyte-macrophage colony-stimulating factor plus granulocyte colony-stimulating factor versus granulocyte colony-stimulating factor for mobilization of dendritic cell subsets in hematopoietic progenitor cell products. Biol Blood Marrow Transplant. 2004;10:848–57. doi: 10.1016/j.bbmt.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 28.Shaughnessy P, Bachier C, LeMaistre C, Akay C, Pollack B, Gazitt Y. Granulocyte colony-stimulating factor mobilizes more dendritic cell subsets than granulocyte-macrophage colony-stimulating factor with no polarization of dendritic cell subsets in normal donors. Stem Cells. 2006;24:1789–97. doi: 10.1634/stemcells.2005-0492. [DOI] [PubMed] [Google Scholar]
- 29.Middel P, Raddatz D, Gunawan B, Haller F, Radzun H-J. Increased number of mature dendritic cells in Crohn's disease: evidence for a chemokine mediated retention mechanism. Gut. 2006;55:220–7. doi: 10.1136/gut.2004.063008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aman M, Tretter T, Eisenbeis I, et al. Interferon-alpha stimulates production of interleukin-10 in activated CD4+ T cells and monocytes. Blood. 1996;87:4731–6. [PubMed] [Google Scholar]
- 31.Levings M, Sangregorio R, Galbiati F, Squadrone S, de Waal Malefyt R, Roncarolo M-G. IFN-α and IL-10 induce the differentiation of human type 1 T regulatory cells. J Immunol. 2001;166:5530–9. doi: 10.4049/jimmunol.166.9.5530. [DOI] [PubMed] [Google Scholar]
- 32.Fantini M, Becker C, Tibbe I, et al. F-beta induced foxp3+ regulatory T cells suppress Th1-mediated experimental colitis. Gut. 2005;55:671–80. doi: 10.1136/gut.2005.072801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kelsen J, Agnholt J, Hoffmann H, Romer J, Hvas C, Dahlerup H. Foxp3+CD4+CD25+ T cells with regulatory properties can be cultured from colonic mucosa of patients with Crohn's disease. Clin Exp Immunol. 2005;141:549–57. doi: 10.1111/j.1365-2249.2005.02876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saruta M, Yu Q, Fleshner P, et al. Characterization of Foxp3+CD4+ regulatory T cells in Crohn's disease. Clin Immunol. 2007;125:281–90. doi: 10.1016/j.clim.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 35.Yu Q, Saruta M, Avanesyan A, Fleshner P, Banham A, Papadakis K. Expression and functional characterization of Foxp3+CD4+ regulatory T cells in ulcerative colitis. Inflamm Bowel Dis. 2007;13:191–9. doi: 10.1002/ibd.20053. [DOI] [PubMed] [Google Scholar]