

Abstract
Cytochrome cbb3 is the most distant member of the heme-copper oxidase family still retaining the following major feature typical of these enzymes: reduction of molecular oxygen to water coupled to proton translocation across the membrane. The thermodynamic properties of the six redox centers, five hemes and a copper ion, in cytochrome cbb3 from Rhodobacter sphaeroides were studied using optical and EPR spectroscopy. The low spin heme b in the catalytic subunit was shown to have the highest midpoint redox potential (Em,7 +418 mV), whereas the three hemes c in the two other subunits titrated with apparent midpoint redox potentials of +351, +320, and +234 mV. The active site high spin heme b3 has a very low potential (Em,7 -59 mV) as opposed to the copper center (CuB), which has a high potential (Em,7 +330 mV). The EPR spectrum of the ferric heme b3 has rhombic symmetry. To explain the origins of the rhombicity, the Glu-383 residue located on the proximal side of heme b3 was mutated to aspartate and to glutamine. The latter mutation caused a 10 nm blue shift in the optical reduced minus oxidized heme b3 spectrum, and a dramatic change of the EPR signal toward more axial symmetry, whereas mutation to aspartate had far less severe consequences. These results strongly suggest that Glu-383 is involved in hydrogen bonding to the proximal His-405 ligand of heme b3, a unique interaction among heme-copper oxidases.
The heme-copper oxidases form a family of enzymes that have structural homology of the catalytic subunit in common (1). This family of proteins, characterized by six conserved histidine ligands of the redox cofactors, ranges from classical, mitochondrial terminal oxidases to nitric-oxide reductases, and the members have been classified according to evolutionary relationships of their sequences (2–4). The bacterial cbb3-type cytochrome c oxidases form a distinct, divergent subfamily within the heme-copper oxidases (5). Terminal oxidases share the catalytic activity of four-electron reduction of molecular oxygen to water coupled to translocation of protons across the membrane (6, 7). Cytochrome cbb3, expressed in some bacteria as a sole terminal oxidase, is characterized by its ability to maintain catalytic activity under low oxygen tension (8), and it has also been shown to have the capacity to translocate protons (9).
The Rhodobacter sphaeroides cytochrome cbb3 is encoded by the ccoNOQP operon composed of four genes (10). The catalytic subunit CcoN homes a binuclear active site composed of a high spin heme b3 and a nearby copper ion (CuB). There are altogether four low spin hemes in the enzyme. In addition to a protoheme (heme b) residing in the vicinity of the active site in subunit CcoN, there are three hemes c present in the soluble domains of the two other transmembrane subunits, a monoheme subunit CcoO and a diheme subunit CcoP (11). There is yet one more membrane-spanning subunit, CcoQ, without bound cofactors (12). Although the catalytic subunit shows homology to the other heme-copper oxidases (13), the other three subunits bear no resemblance to subunits of other types of terminal oxidases. However, subunit CcoO has been shown to have sequence homology with the nitric-oxide reductase subunit NorC (14).
The crystal structures of a few heme-copper oxidases have been resolved (15–19), but only structural homology models are currently available for cytochromes cbb3 (20–23). Apart from the signatures common to all heme-copper oxidases, the sequence alignments have revealed only very few other conserved residues when terminal oxidases are compared. Even though some amino acids, absent from cytochrome cbb3, have been shown to be of critical importance to the function of the classical heme-copper oxidases, the major functions still remain the same in all of these enzymes.
The thermodynamic properties of the cbb3-type oxidases have been investigated sparsely. Apart from work yielding partial information about the properties of the hemes (11, 24, 25), two more complete studies have been carried out (5, 26). All the hemes in cytochrome cbb3 were proposed to have high redox potentials, both in the Pseudomonas stutzeri and Bradyrhizobium japonicum enzymes (5, 26). This is also the case in all other studies, except for the enzyme from Rhodothermus marinus, where two low potential redox centers were reported (25). However, little is known about the copper center in the active site (CuB). Early Fourier transform infrared (FTIR)2 spectroscopic measurements identified the presence of a heme/copper binuclear center in R. sphaeroides cytochrome cbb3 (11), and more recent resonance Raman and FTIR studies have given additional information about the structure of the active site (27–29).
In the absence of deconvoluted spectral components and thereby clear assignments of the redox centers in the cbb3-type oxidases, and the lack of consensus about their thermodynamic properties, a complete study was required. In this work we have set out to investigate the thermodynamic properties of all the redox centers in cytochrome cbb3 from R. sphaeroides using a combination of optical and EPR redox titrations with the main focus on the details of the catalytic site. This effort will form a basis for further mechanistic studies.
EXPERIMENTAL PROCEDURES
Chemicals—The detergent n-dodecyl β-d-maltoside (DDM) was purchased from Anatrace (Maumee, OH), and other chemicals were obtained from Sigma.
Bacterial Strain, Mutagenesis, and Enzyme Preparation—Mutagenesis was achieved using the M13 template of the ccoNOQP operon as described earlier (22). The production and purification of the protein were described in Ref. 30. The enzyme was purified to homogeneity, and the protein samples were exchanged into the buffers needed in each experiment using Amicon ultracentrifugal filter (Millipore) concentrators.
Optical Redox Titrations—Anaerobic spectroelectrochemical redox equilibrium titrations were performed using an optically transparent, thin layer electrode cell described in detail previously (31). The measurements were performed in 100 mm potassium phosphate or in 100 mm MES buffers at varying pH values, supplemented with 100 mm K2SO4 and 0.05% DDM. The mediator mixture contained 100 μm ferrocene dimethanol, 100 μm ferrocene ethanol, 100 μm ferrocene methanol, 100 μm pentaaminepyridineruthenium perchlorate, 100 μm hexaamineruthenium chloride, and 1 mm methyl viologen. The redox titrations were performed in both oxidative and reductive directions with the same sample, in the spectral range 350–750 nm at 21 °C. The potential range used was from -300 to +520 mV, and data were collected at 20-mV intervals. The precision of the experiments was ±5 mV. The equilibrium state at each redox potential was determined by optical changes at 425 nm. All potentials were expressed versus normal hydrogen electrode. The data were analyzed by using MATLAB (The Mathworks, South Natick, MA). The redox titrations profiles at selected wavelengths were least squares fitted to a sum of independent 1-electron (n = 1) redox transitions described by a modified Nernst equation (Equation 1),
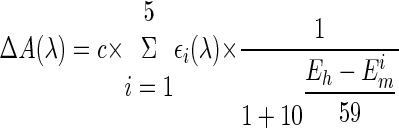 |
(Eq. 1) |
where c is the concentration of the sample;
εi(λ) is the extinction coefficient for the
specific redox center at the selected wavelength; Eh is
the redox potential of the solution; and 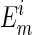 is
the apparent redox midpoint potential of the transition.
is
the apparent redox midpoint potential of the transition.
EPR Redox Titrations—The electrochemical cell used for redox equilibrium titrations by EPR spectroscopy was described in detail previously (32). The redox mediators used were 1 mm benzyl viologen, 200 μm hexaamineruthenium chloride, 100 μm pentaaminepyridineruthenium perchlorate, and 100 μm ferrocene ethanol. The cell was incubated with the mediator mixture in buffer (100 mm MES, pH 6.0, 100 mm K2SO4 supplemented with 0.05% DDM) overnight at low potential (-350 mV) to consume oxygen present within the cell. The sample (300 μl of ∼100 μm enzyme) was loaded and fully reduced at -300 mV to consume traces of oxygen. Titrations were conducted by poising the sample at desired potentials at 21 °C with continuous mixing until the current reached a minimum constant level, after which the sample was frozen in liquid nitrogen, and data were collected. X-band EPR spectra were recorded using a Bruker EMS EPR spectrometer equipped with an Oxford Instruments ESR900 helium flow cryostat together with an ITC4 temperature controller. Spectra at 9.4 GHz were collected at 10 K using a field modulation frequency of 100 kHz, a modulation amplitude of 1.27 millitesla, and a microwave power of 2 milliwatt. The spectra shown are normalized for gain and protein concentration.
RESULTS
Optical Redox Titrations—Cytochrome cbb3 contains altogether six redox centers, five hemes and a copper center. According to a hemochrome spectrum, the B- and C-type hemes are present in the ratio 2:3, and the presence of the copper center has been verified using FTIR spectroscopy (11). Anaerobic optical redox titrations were undertaken to determine the thermodynamic parameters of the redox cofactors. The data obtained from a representative redox titration are presented as the optical changes at different redox potentials versus wavelength using the fully oxidized spectrum as a reference (Fig. 1A). The spectrum at the lowest redox potential (-300 mV) minus the spectrum at the highest potential (+520 mV) was found to equal the reduced minus oxidized spectrum obtained by oxidizing the enzyme with ferricyanide and reducing it anaerobically by dithionite, i.e. full reduction and oxidation were reached in the titrations. The sharp features of the low spin hemes c and b are clearly observed in the α region of the spectra, and the Soret region shows the overlapping bands of all hemes (Fig. 1A).
FIGURE 1.

Optical redox titrations of cytochrome cbb3. A, redox difference data of absorbance changes from an optical titration of a sample in 100 mm potassium phosphate, pH 7, 100 mm K2SO4, and 0.05% DDM supplemented with redox mediators. The α region spectra are multiplied by 2.75. B, first derivative of the optical changes as a function of redox potential at 550 nm (○), 560 nm (224), and 440 nm (□). The experimental data are fitted with a sum of five theoretical Nernstian n = 1 curves at each wavelength (solid lines). Three theoretical Nernstian n = 1 curves (dashed lines) representing the redox transitions of the individual hemes c are shown for comparison. C, equivalent redox titration curves with the fitted lines presented as absorbance changes versus redox potential. In addition, a profile of a slow reductive titration at 550 nm (×) is shown, which required an additional Nernstian component to fit the data well. D, reduced minus oxidized spectral components from a global fit with five theoretical Nernstian curves. The dotted line represents components correlating with redox changes at 550 (only one of the three similar components is presented for clarity), and the solid line shows the redox spectrum reflecting changes at 560 nm, and the dashed line presents a redox spectrum with changes at 440 nm. The α region of the spectra is multiplied by 2.75.
The titrations of the redox cofactors can be followed as the derivative of the optical changes versus the redox potential at each wavelength. In the high potential region (>+100 mV) multiple maxima corresponding to the apparent midpoint potentials of the hemes were observed. The redox changes of the low spin hemes c and b could best be followed in the α region at 550 and 560 nm, respectively. The same redox changes are present also in the Soret region, but here the overlapping bands of the hemes make identification of their origins more demanding. Titration profiles at the earlier mentioned wavelengths are presented in Fig. 1B. The curve at 560 nm shows two well separated components, a single peak both at a high and a low potential. These data can be fitted by two independent theoretical Nernstian n = 1 curves with Em,7 values of +418 ± 3 and -59 ± 2 mV. The extinctions for the α band maxima of the low spin hemes c and b are comparable. This may be used to evaluate the results at different wavelengths because the number of hemes within the enzyme is known. The profile at 550 nm is a sum of several components at high potentials, and at low potentials a single redox transition with Em,7 =-59 mV was observed. The high potential redox transitions at 550 nm were fitted with three independent theoretical Nernstian n = 1 curves with the same extinction as used for fitting the high potential titration at 560 nm. The individual theoretical curves with redox potentials +351 ± 8, +320 ± 7, and +234 ± 5 mV, and their sum, are presented in Fig. 1B; the sum of the three theoretical curves fit the experimental data satisfactorily. A component titrating at a low potential (Em,7 =-59 mV) was observed in both profiles of redox changes. This component could best be followed in the Soret region at 440 nm. The curve can be fitted well with one theoretical Nernstian n = 1 curve in the low potential region, whereas the changes observed in the high potential region originate from the overlapping absorbance bands of low spin hemes. The same titration curves are presented as absorbance changes as a function of redox potential together with the fitted theoretical curves in Fig. 1C. Altogether, five n = 1 Nernstian curves were enough to fit the data well.
To identify the redox components from which the optical transitions arise, the data surface was deconvoluted by global fitting using a set of five independent theoretical Nernstian curves with the determined midpoint redox potential values (see above). The redox difference spectra of the individual components are shown in Fig. 1D. The spectrum with sharp peaks at 430 and 560 nm is characteristic of a low spin heme b, whereas the spectrum having peaks at 418 and 550 nm is typical of low spin heme c (for clarity, only the spectrum of one heme c is shown). The spectrum with a Soret maximum at 440 nm and a very broad band at ∼565 nm is typical of a reduced minus oxidized spectrum of a high spin heme, which is why we tentatively assign it to heme b3. We conclude that three types of heme components have been found in the optical titrations, three hemes c, a low spin heme b, and a high spin heme b3.
An unusual hysteretic behavior of a small fraction of a heme c was also observed, where the results from the oxidative titration differed from those of the reductive titration. The redox midpoint potential of a fraction of heme c in the latter case was lowered considerably (Em,7 ∼-120 ± 10 mV) compared with that of the former, as shown for 550 nm traces in Fig. 1C. This shift of the midpoint potential, pronounced particularly at high pH (not shown), can be due to a partial change in the heme ligand conformation or other changes in the protein upon the long duration of a titration.
Having established the thermodynamic parameters of the hemes, we may ask whether indications of the CuB site are present. As for the aa3-type enzymes, we found no distinct optical signature for this center with sufficiently intense changes of extinction upon oxidoreduction. However, one may attempt to follow the redox changes of CuB indirectly by taking advantage of the very close proximity of the two metal centers in the active site. In the mitochondrial oxidase a ligand to metal charge-transfer (LMCT) band of the ferric high spin heme has been shown to be sensitive to the changes of the reduction state of the copper through the interaction of the ligands of the oxidized redox centers (33). The optical changes in the LMCT band area of the spectrum upon redox titration are presented in Fig. 2A as a redox difference data surface, where the fully reduced spectrum has been used as a reference to enhance the visibility of all the spectral changes. The LMCT band from the high spin ferric heme can be observed in the domain between 600 and 680 nm, whereas the LMCT band centered near 695 nm arises from the cytochromes c with a methionine residue as an axial ligand (34). The fully oxidized heme b3 in cytochrome cbb3 has an LMCT band at 622 nm. At a redox potential of ∼+100 mV, the maximum of this band shifts to 645 nm (Fig. 2A). Therefore, the titrations were followed at these two wavelengths (Fig. 2B). The high potential transition at 622 nm is composed of not only changes of the LMCT band of the high spin heme but also those of the α bands of low spin hemes contributing to the changes of absorbance in this spectral region. However, as the majority of the absorbance change at this wavelength originated from the LMCT band of interest, a theoretical n = 1 Nernstian curve with an Em,7 value of +330 mV was plotted to the experimental data yielding an estimate of the redox midpoint potential. Interestingly, the LMCT band maximum red shifts during this transition to the new position at ∼645 nm (Fig. 2A), the titration of which corresponds to that previously assigned to reduction of heme b3 (Fig. 2B), i.e. with an Em,7 of -59 mV (see above). Because of the broad LMCT band, the two positions overlap, and the low potential transition can indeed also be observed at 622 nm (Fig. 2B). Together, these data strongly suggest that the LMCT band of ferric heme b3 is at ∼622 nm when both the heme and the CuB are oxidized, that it shifts to ∼645 nm upon reduction of CuB, and that the latter band vanishes when heme b3 is also reduced. EPR titrations give further support to this assignment (see below).
FIGURE 2.

Optical changes in the LMCT band area. A, absorbance changes in the LMCT domain upon a redox titration at pH 7 using the fully reduced spectrum as a reference. B, redox titration profiles at 622 nm minus (610 nm + 660 nm)/2 (224) and at 645 nm minus (610 nm + 700 nm)/2 (○). Theoretical Nernstian n = 1 curves fitted to the experimental data are represented by solid lines below +100 mV and above a theoretical Nernstian curve with an Em,7 value of +330 mV is plotted to the experimental data.
These results demonstrate that the thermodynamic properties of the binuclear site in cytochrome cbb3 are fundamentally different from those of the aa3-type enzymes. Although the redox potential of CuB is relatively high in both instances, the potential of the high spin heme of the active site is very low in the cbb3-type enzyme. Redox titrations were further carried out at different pH values, and the full data on all redox centers are summarized in Table 1.
TABLE 1.
Apparent midpoint redox potentials of the redox cofactors measured at different pH values Data were from optical titrations. The errors were determined from the 95% confidence bounds of the fitting results.
| Redox cofactor | Em,6 | Em,7 | Em,8 |
|---|---|---|---|
| mV | mV | mV | |
| Heme b | 428 ± 2 | 418 ± 3 | 410 ± 3 |
| Heme c | 356 ± 9 | 351 ± 8 | 341 ± 4 |
| Heme c | 330 ± 9 | 320 ± 7 | 293 ± 4 |
| Heme c | 258 ± 5 | 234 ± 5 | 210 ± 5 |
| Heme b3 | –38 ± 2 | –59 ± 2 | –77 ± 2 |
| CuBa | ∼350 | ∼330 | ∼290 |
The values are approximations from the optical data
EPR Redox Titrations—All ferric hemes exhibit EPR signals, except those with strong magnetic coupling to another paramagnet, which may broaden the signal beyond detection. The heme in the binuclear center of the heme-copper oxidases is an example of the latter case, because of its magnetic coupling to the copper ion. Fig. 3A shows the EPR spectra of cytochrome cbb3 fully oxidized by ferricyanide (lower trace) and in the presence of redox mediators at a redox potential +520 mV (middle trace). Two different kinds of EPR spectra arising from low spin ferric hemes are present. The gz ∼ 3.5 peak is broad, and most likely the other components of this signal are too broad to be detected. This spectrum is suggested to arise from a single cytochrome c in the monoheme subunit (35), whereas the three remaining low spin hemes are presented in the spectra by overlapping spectral component centered at gzyx ∼ 2.96, 2.25, and 1.51. A signal at g ∼ 6 can also be observed in the fully oxidized preparation, which may arise from a small fraction of high spin heme with broken magnetic coupling to copper, or from a low spin heme that has lost its axial ligand.
FIGURE 3.

X band EPR spectra of the WT enzyme at 10 K. A, spectrum of ferricyanide-oxidized enzyme (bottom) and a sample poised at +520 mV in the presence of redox mediators (middle) both in 100 mm MES, pH 6.0, 100 mm K2SO4, and 0.05% DDM are shown. The top spectrum is obtained at a redox potential of ∼+100 mV. The g values of the main signals are marked. B, changes of a signal intensity at g = 5.20 (•) upon a redox titration are shown. The experimental data are fitted by two theoretical Nernstian n = 1 curves with the Em values -60 and 351 mV.
In strongly magnetically coupled systems, an EPR signal appears only in the case of reduction of one of the partners during a redox titration. Therefore, to establish the midpoint redox potentials of the components of the binuclear center, a redox titration of the EPR sample was performed. According to the optical data, all low spin hemes, along with CuB, should be reduced at a redox potential of ∼+100 mV. The EPR spectrum of cytochrome cbb3 at that redox state is presented in Fig. 3A (top trace). According to the prediction from the optical titrations, this is in the area of redox potential where the ferric heme b3 may exhibit maximal EPR signals, because CuB should be reduced and heme b3 should remain oxidized. As clearly shown in Fig. 3A (top trace), all bands in the range below g ∼ 4 disappear, which confirms that the low spin hemes are fully reduced at this state. Instead, two overlapping bands appear at this redox potential, at g ∼ 5.20 and g ∼ 4.95, and an additional band at g ∼ 6.60 becomes more pronounced.
Redox titrations followed at the g values 6.60, 5.20, and 4.95 yielded similar titration profiles. Fig. 3B shows the redox potential dependence of the peak at g = 5.20. The experimental data were fitted with two Nernstian n = 1 curves, in which the g = 5.2 feature appears with a midpoint potential of +351 ± 7 mV, and disappears with Em,6 =-60 ± 7 mV. This behavior strongly supports the assignments made on the basis of the optical redox titrations. The rise of the g = 5.2 signal with Em,6 = 351 mV thus results from a break of the magnetic coupling between high spin ferric heme b3 and cupric CuB due to reduction of the latter, and it subsequently vanishes with Em,6 = -60 mV due to reduction of the heme.
The feature at g = 5.99, present already in the fully oxidized enzyme, exhibited a different microwave power dependence compared with the other signals in the g = 6 region (not shown). The g = 5.99 signal vanishes completely with an Em,6 of ∼+100 mV (not shown). Therefore, it has no obvious counterpart in the optical redox titrations, which corroborates the conclusion that it represents a small part of the enzyme in which either the magnetic heme b3-CuB interaction is broken, or a small fraction of low spin heme may have become high spin because of a loss of an axial ligand.
Properties of the Active Site—The degree of saddling of the high spin hemes can be compared by calculating a percentage of rhombicity from the extent of splitting of the components in the g ∼ 6 region of the EPR spectra as described in Ref. 36. According to this classification, the EPR signature of ferric heme b3 in cytochrome cbb3 exhibits ∼10% rhombicity, much higher than in the aa3-type enzymes, and its midpoint redox potential is much lower than that of heme a3. The polypeptide sequence in the vicinity of the active site was searched to pin-point a possible structural reason for these differences. According to sequence alignments of the cbb3-type oxidases, there is a well conserved glutamate residue in helix IX, predicted to lie relatively close to the proximal histidine ligand of heme b3. This glutamate is the only well conserved acidic amino acid residue predicted to lie within the membrane domain (21, 22). When this glutamate residue is mutated to an aspartate (E383D), the catalytic activity drops to 1/3 compared with WT, yet retaining full proton translocation capacity, whereas mutation to glutamine (E383Q) leads to virtually complete loss of catalytic activity (22).
Optical redox titrations were performed with both these variants at pH 6. Although all the other spectral properties remained similar to those of the WT enzyme, the reduced minus oxidized heme b3 spectrum was changed in both mutants (Fig. 4A), and the midpoint redox potential was raised to ∼0 mV in both cases (not shown). The less severe mutation to aspartate led to a very small red shift in the Soret region maximum, and the broad α band was not significantly shifted. Elimination of the carboxylic group by introduction of the glutamine residue had a far stronger effect. The Soret peak maximum was blue-shifted by 10 nm; the α band was clearly blue-shifted, and the charge-transfer band also changed shape.
FIGURE 4.

Effect of the glutamate 383 mutations. A, redox difference spectra of heme b3 from WT (solid), E383D (dotted), and E383Q (dashed) mutants obtained from redox titrations are presented. B, ferric high spin heme EPR signal of the E383D (dotted) and E383Q (dashed) variants and the WT (solid) enzyme obtained at ∼+100 mV are presented. The guidelines are located at g = 6.60, 5.98, 5.20, and 4.95.
The mutant enzymes were also studied by EPR spectroscopy. The E383D variant showed only a slight deviation from the WT signal with a small decrease and broadening of the bands arising from the high spin heme, relative to the band at g = 5.99 (Fig. 4B). The mutation to glutamine again had a much stronger effect. The bands near g ∼ 5 were completely abolished, and the g ∼ 6 band increased considerably in size. The shape of the g ∼ 6 band of this variant is still not like that of the classical axial g = 6 signal of the aa3-type cytochromes but is broader with an additional feature at g ∼ 6.6. The effect of the E383Q mutation may nevertheless be described as a shift from rhombic toward more axial symmetry.
DISCUSSION
Thermodynamic Balance in Cytochrome cbb3—The apparent midpoint potentials of the six redox centers in cytochrome cbb3 from R. sphaeroides were determined using a combination of optical and EPR redox titrations. The four low spin hemes located in three different subunits were easily distinguished by optical spectroscopy. The spectra of the low spin hemes obtained by global fitting closely resemble the spectra of low spin hemes c and b from other heme proteins. The heme b in the catalytic subunit has the highest potential of all redox centers, and all hemes c have potentials above +200 mV in the pH range studied (Table 1). These results agree well with the observations for cytochrome cbb3 from other organisms (5, 11, 26), and the pH dependence of the redox potentials of the low spin hemes are similar to those observed in the enzyme from B. japonicum (26).
The observation of hysteresis in the redox titration of a fraction of heme c, particularly at high pH, can have several explanations. One heme c center may be fragile and changes its ligand sphere during the titrations, as observed for other cytochromes c (37, 38). The observation that full reduction leads to partial recovery implies that the heme has perhaps encountered a change in the proximity of its axial ligand at a high redox potential, but it is able to regain its original conformation when incubated in the fully reduced state. Although this observation is likely to be the result of an experimental artifact, we cannot at this time rule out that it may have biological significance.
This is the first time the thermodynamic properties of the entire active site of cytochrome cbb3 were studied. The titration of heme b3 revealed a very low midpoint redox potential and an optical spectrum typical of a high spin heme. Although some previous studies have identified a high spin heme with a high midpoint potential, its optical spectrum was not shown (5, 26), and we ascribe the apparent discrepancy to complications in resolving the individual titration of a high spin heme in the α region in the presence of four additional overlapping spectral components. In earlier work, absolute spectra of the reduced enzyme showed a clear shoulder at 440 nm (26), implying that a heme with properties ascribed here to heme b3 is present also in cytochrome cbb3 from other organisms. However, the redox changes of this heme were presumably not detected previously because of a too narrow potential range in the titrations.
Cytochrome cbb3 is the most distant member of the heme-copper oxidase family, and it has been suggested to have closer evolutionary connections to the nitric-oxide reductases (4, 39). Both enzymes are capable of reducing oxygen as well as nitric oxide (40). The study of the cofactors in Paracoccus denitrificans nitric-oxide reductase revealed that the active site heme b3 also has a low midpoint potential (Em,7.6 =+60 mV) (41), similar though not identical to that observed here for cytochrome cbb3. Although this suggests another close similarity between cytochromes cbb3 and the nitric-oxide reductases, the reasons for the low potential are difficult to assess in the absence of crystal structures of either enzyme.
EPR Studies of the Catalytic Site—The low spin heme signals in the EPR spectrum of the fully oxidized WT cytochrome cbb3 are similar to previously published spectra (11, 24, 35). However, the changes in the EPR spectrum on oxidoreduction of cytochrome cbb3 have not been published previously, and thus the spectrum of the high spin ferric heme b3 magnetically uncoupled from the CuB site by reduction of the latter is reported here for the first time. This signal was found to be broad with a high degree of rhombicity, thereby implying a different catalytic site heme structure in comparison with the classical oxidases. A question about the intensity of the signal arises immediately. The wide rhombic signal is difficult to simulate because of its shape. Two very wide overlapping derivative shaped signals partially cancel each other, and the presence of the fairly broad signal at g ∼ 6 further complicates the situation. The spectrum was obtained in the presence of redox mediators, and although they do not contribute to the spectrum above g ∼ 3, below this value the contribution is significant making it challenging to estimate the size of the possible gx = 2 component of the high spin heme signal. Even though the signal in the g ∼ 6 region appears small in comparison with the low spin heme signals, its width may account for the lack in intensity. Several enzyme preparations were tested yielding similar ratios between the low spin heme intensities and that arising from the high spin ferric heme.
The EPR redox titrations clearly confirmed what the optical titrations implied about the apparent midpoint potentials of the redox cofactors in the active center. The redox midpoint potentials obtained agreed well despite the large temperature difference between the measuring conditions used in the two experiments. The 20-mV difference in the redox potentials obtained for the high spin heme in optical and EPR titrations is easily explained by experimental error or by a temperature dependence of the Em value of the heme. The redox potential of CuB was not accurately determined from the optical data, but the EPR experiments yielded a result that corresponds closely to the estimate made from the absorbance changes of the LMCT band.
Role of Glutamate in the Active Site of Cytochrome cbb3—The position of glutamate 383 has been studied by molecular modeling (21, 22), and this residue was suggested to lie as close as 4 Å from the proximal histidine ligand of the high spin heme (21). According to sequence alignments, this glutamate sometimes appears to be replaced by a glutamine (21), but this concerns few very distant members of the cbb3 subfamily of enzymes. At any rate, all cytochromes cbb3 studied on the molecular level up to now belong to subfamilies with this glutamate residue fully conserved, including the enzyme from R. sphaeroides studied here.
The reduced minus oxidized spectrum of protoheme b3 in cytochrome cbb3 was found to be typical of a high spin heme, although with a Soret maximum at a surprisingly high wavelength. The mutation of glutamate 383 to glutamine shifted this peak maximum to a position more typical of protohemes, implying that the glutamate residue resides close to the heme and that it contributes to the unusual red-shifted Soret band. To account for this result, we suggest that glutamate 383 is hydrogen-bonded to the proximal histidine of heme b3 (Fig. 5), which is also supported by earlier data (see below). The rise of the redox potential of heme b3 by only 40 mV in the mutant enzymes suggests that although the glutamate residues has an effect on this parameter, alone it is not sufficient to explain the low redox potential of the active site heme.
FIGURE 5.

A model of the active site of cytochrome cbb3. The hemes and the copper center of the catalytic subunit are shown together with the axial histidine ligands of the hemes originating from helix X. The glutamate 383 residue suggested to form a hydrogen bond (dotted red line) with histidine 405 is located in helix IX. The figure was prepared using the Visual Molecular Dynamics software (45).
The mutation to glutamine (as opposed to aspartate) abolishes the hydrogen bond from the free nitrogen atom of the proximal histidine 405 to the carboxylate side chain of glutamate 383. Therefore, this mutation may lead to a new configuration of the heme, where the histidine ligand is no longer pulling the iron proximally out of the heme plane, thus resulting in a more planar, axial geometry as observed in our EPR experiments. During a long incubation of the enzyme at alkaline pH, the high spin heme of the WT enzyme also seems to become likely to lose this hydrogen bond, because in a fraction of the sample a shift of the Soret band maximum to 430 nm is observed (not shown). Comparison of the EPR spectra of the WT and mutated enzymes supports the proposed structural changes in the catalytic site, and it suggests that the glutamate residue is located at a hydrogen bonding distance to the proximal histidine ligand, even closer than proposed by molecular modeling of the cbb3 structure (21).
Comparison of the Active Site Heme Structure with Other Hemoproteins—We may now compare our view of the heme in the active site of cytochrome cbb3 with other hemoproteins. The rhombic high spin heme is a unique feature among the heme-copper oxidases. However, the work in Ref. 36 shows that the rhombicity is by no means unique among heme proteins. Some members of the globin family, such as hemoglobin and myoglobin, exhibit even wider rhombic splitting of the hemes. The ruffled shape of the heme b3 might allow the active site of cytochrome cbb3 to be more spacious by widening the gap between the heme and the CuB site. This conclusion is strongly supported by resonance Raman experiments on cytochrome cbb3 as well as by FTIR spectroscopy (11, 27–29). Wider bandwidths arising from the Fe-CO vibrational mode in both spectra implied a larger number of possible conformations allowed for the CO molecule bound to the high spin heme of cytochrome cbb3 than is observed in other terminal oxidases, suggesting that the copper ion is no longer restraining the position of the CO ligand.
The proposed hydrogen bonding between glutamate 383 and the proximal histidine 405 of the high spin heme is likely to be conserved among the cbb3-type oxidases. However, such bonding is not present in other terminal oxidases nor in nitric-oxide reductases, because no carboxylic residue in the vicinity of the high spin heme is found in any of the known aa3 structures nor predicted in the NO reductases. However, a similarity in this respect between cytochrome cbb3 and cytochrome c peroxidase (CcP) is of interest. The catalytic triad in CcP involves a hydrogen bond from the proximal histidine ligand of the protoheme to an aspartate residue (42), and a direct effect of such hydrogen bonding upon the length of the Fe-His bond has been studied in detail (43). EPR experiments of CcP also yielded rhombic heme signals, and mutations of the aspartate led to results very similar to those presented here (42). In this case a mutation to glutamate did not compromise the bonding, but more radical mutations to asparagine and alanine resulted in loss of catalytic activity and a shift of the rhombic EPR signal toward axial symmetry. Although the important interactions formed by the other components of the catalytic triad in CcP cannot be compared directly to those in cytochrome cbb3 because of the absence of the crystal structure of the latter enzyme, these similarities support the conclusions made in this work.
Previous resonance Raman studies on cytochrome cbb3 from R. sphaeroides (28) also provide strong support to our notion of the heme b3 structure. The stretching vibration between high spin heme iron and its proximal histidine ligand was found to be unusually strong among the heme-copper oxidases. This was attributed to strong hydrogen bonding of the histidine ligand, giving it considerable anionic character, in full agreement with the present findings.
Cytochrome cbb3 has to date shown two special structural features of the catalytic site as follows: a cross-link between a histidine ligand of CuB and the active site tyrosine with a different spatial location of the latter in comparison with the aa3 enzymes (30, 44), and unique hydrogen bonding of the proximal histidine ligand of the active site heme to a carboxylate. Both of these features emphasize the fact that the active site structure can be very different in the subfamilies of the heme-copper oxidases, and well conserved within individual subfamilies, even though the overall function is shared. The diversity may reflect the optimization of different terminal oxidase for specific environmental requirements.
This work was supported by the National Graduate School in Informational and Structural Biology (to V. R.), by the Sigrid Jusélius Foundation, Magnus Ehrnrooth Foundation, Biocentrum Helsinki, and the Academy of Finland.
Footnotes
The abbreviations used are: FTIR, Fourier transform infrared; CcP, cytochrome c peroxidase; DDM, n-dodecyl β-d-maltoside; Em,x, apparent redox midpoint potential at pH x; EPR, electron paramagnetic resonance; LMCT, ligand to metal charge-transfer; MES, 2-(N-morpholino)ethanesulfonic acid; WT, wild type.
References
- 1.Ferguson-Miller, S., and Babcock, G. T. (1996) Chem. Rev. 96, 2889-2907 [DOI] [PubMed] [Google Scholar]
- 2.Pereira, M. M., Santana, M., and Teixeira, M. (2001) Biochim. Biophys. Acta 1505, 185-208 [DOI] [PubMed] [Google Scholar]
- 3.Hemp, J., and Gennis, R. B. (2008) Results Probl. Cell Differ. 45, 1-31 [DOI] [PubMed] [Google Scholar]
- 4.Ducluzeau, A. L., Ouchane, S., and Nitschke, W. (2008) Mol. Biol. Evol. 25, 1158-1166 [DOI] [PubMed] [Google Scholar]
- 5.Pitcher, R. S., and Watmough, N. J. (2004) Biochim. Biophys. Acta 1655, 388-399 [DOI] [PubMed] [Google Scholar]
- 6.Wikström, M. K. (1977) Nature 266, 271-273 [DOI] [PubMed] [Google Scholar]
- 7.Wikström, M. (2004) Biochim. Biophys. Acta 1655, 241-247 [DOI] [PubMed] [Google Scholar]
- 8.Preisig, O., Zufferey, R., Thöny-Meyer, L., Appleby, C. A., and Hennecke, H. (1996) J. Bacteriol. 178, 1532-1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raitio, M., and Wikström, M. (1994) Biochim. Biophys. Acta 1186, 100-106 [DOI] [PubMed] [Google Scholar]
- 10.Toledo-Cuevas, M., Barquera, B., Gennis, R. B., Wikström, M., and Garcia-Horsman, J. A. (1998) Biochim. Biophys. Acta 1365, 421-434 [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Horsman, J. A., Berry, E., Shapleigh, J. P., Alben, J. O., and Gennis, R. B. (1994) Biochemistry 33, 3113-3119 [DOI] [PubMed] [Google Scholar]
- 12.Oh, J. I., and Kaplan, S. (2002) J. Biol. Chem. 277, 16220-16228 [DOI] [PubMed] [Google Scholar]
- 13.Zufferey, R., Arslan, E., Thöny-Meyer, L., and Hennecke, H. (1998) J. Biol. Chem. 273, 6452-6459 [DOI] [PubMed] [Google Scholar]
- 14.van der Oost, J., de Boer, A. P., de Gier, J. W., Zumft, W. G., Stouthamer, A. H., and van Spanning, R. J. (1994) FEMS Microbiol. Lett. 21, 1-9 [DOI] [PubMed] [Google Scholar]
- 15.Iwata, S., Ostermeier, C., Ludwig, B., and Michel, H. (1995) Nature 76, 660-669 [DOI] [PubMed] [Google Scholar]
- 16.Tsukihara, T., Aoyama, H., Yamashita, E., Tomizaki, T., Yamaguchi, H., Shinzawa-Itoh, K., Nakashima, R., Yaono, R., and Yoshikawa, S. (1996) Science 72, 1136-1144 [DOI] [PubMed] [Google Scholar]
- 17.Soulimane, T., Buse, G., Bourenkov, G. P., Bartunik, H. D., Huber, R., and Than, M. E. (2000) EMBO J. 19, 1766-1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Svensson-Ek, M., Abramson, J., Larsson, G., Törnroth, S., Brzezinski, P., and Iwata, S. (2002) J. Mol. Biol. 321, 329-339 [DOI] [PubMed] [Google Scholar]
- 19.Qin, L., Hiser, C., Mulichak, A., Garavito, R. M., and Ferguson-Miller, S. (2006) Proc. Natl. Acad. Sci. U. S. A. 103, 16117-16122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hemp, J., Christian, C., Barquera, B., Gennis, R. B., and Martinez, T. J. (2005) Biochemistry 44, 10766-10775 [DOI] [PubMed] [Google Scholar]
- 21.Hemp, J., Han, H., Roh, J. H., Kaplan, S., Martinez, T. J., and Gennis, R. B. (2007) Biochemistry 46, 9963-9972 [DOI] [PubMed] [Google Scholar]
- 22.Sharma, V., Puustinen, A., Wikström, M., and Laakkonen, L. (2006) Biochemistry 45, 5754-5765 [DOI] [PubMed] [Google Scholar]
- 23.Sharma, V., Wikström, M., and Laakkonen, L. (2008) Biochemistry 47, 4221-4227 [DOI] [PubMed] [Google Scholar]
- 24.Gray, K. A., Grooms, M., Myllykallio, H., Moomaw, C., Slaughter, C., and Daldal, F. (1994) Biochemistry 33, 3120-3127 [DOI] [PubMed] [Google Scholar]
- 25.Pereira, M. M., Carita, J. N., Anglin, R., Saraste, M., and Teixeira, M. (2000) J. Bioenerg. Biomembr. 32, 143-152 [DOI] [PubMed] [Google Scholar]
- 26.Verissimo, A. F., Sousa, F. L., Baptista, A. M., Teixeira, M., and Pereira, M. M. (2007) Biochemistry 46, 13245-13253 [DOI] [PubMed] [Google Scholar]
- 27.Wang, J. L., Gray, K. A., Daldal, F., and Rousseau, D. L. (1995) J. Am. Chem. Soc. 117, 9363-9364 [Google Scholar]
- 28.Varotsis, C., Babcock, G. T., Garcia-Horsman, J. A., and Gennis, R. B. (1995) J. Phys. Chem. 99, 16817-16820 [Google Scholar]
- 29.Stavrakis, S., Koutsoupakis, K., Pinakoulaki, E., Urbani, A., Saraste, M., and Varotsis, C. (2002) J. Am. Chem. Soc. 24, 3814-3815 [DOI] [PubMed] [Google Scholar]
- 30.Rauhamäki, V., Baumann, M., Soliymani, R., Puustinen, A., and Wikström, M. (2006) Proc. Natl. Acad. Sci. U. S. A. 103, 16135-16140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bogachev, A. V., Bertsova, Y. V., Bloch, D. A., and Verkhovsky, M. I. (2006) Biochemistry 45, 3421-3428 [DOI] [PubMed] [Google Scholar]
- 32.Euro, L., Bloch, D. A., Wikström, M., Verkhovsky, M. I., and Verkhovskaya, M. (2008) Biochemistry 7, 3185-3193 [DOI] [PubMed] [Google Scholar]
- 33.Mitchell, R., Mitchell, P., and Rich, P. R. (1991) FEBS Lett. 280, 321-324 [DOI] [PubMed] [Google Scholar]
- 34.Pettigrew, G. V., and Moore, G. R. (1987) Cytochromes c. Biological Aspects (Rich, A., ed) p. 25, Springer-Verlag, Cambridge, MA
- 35.Pitcher, R. S., Cheesman, M. R., and Watmough, N. J. (2002) J. Biol. Chem. 277, 31474-31483 [DOI] [PubMed] [Google Scholar]
- 36.Peisach, J., Blumberg, W. E., Ogawa, S., Rachmilewitz, E. A., and Oltzik, R. (1971) J. Biol. Chem. 246, 3342-3355 [PubMed] [Google Scholar]
- 37.Murgida, D. H., and Hildebrandt, P. (2005) Phys. Chem. Chem. Phys., 3773-3784 [DOI] [PubMed]
- 38.Ye, T., Kaur, R., Senguen, F. T., Michel, L. V., Bren, K. L., and Elliott, S. J. (2008) J. Am. Chem. Soc. 130, 6682-6683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Castresana, J., and Saraste, M. (1995) Trends Biochem. Sci. 20, 443-448 [DOI] [PubMed] [Google Scholar]
- 40.Forte, E., Urbani, A., Saraste, M., Sarti, P., Brunori, M., and Giuffre, A. (2001) Eur. J. Biochem. 268, 6486-6491 [DOI] [PubMed] [Google Scholar]
- 41.Grönberg, K. L., Roldán, M. D., Prior, L., Butland, G., Cheesman, M. R., Richardson, D. J., Spiro, S., Thomson, A. J., and Watmough, N. J. (1999) Biochemistry 38, 13780-13786 [DOI] [PubMed] [Google Scholar]
- 42.Goodin, D. B., and McRee, D. E. (1993) Biochemistry 32, 3313-3324 [PubMed] [Google Scholar]
- 43.Sinclair, R., Hallam, S., Chen, M., Chance, B., and Powers, L. (1996) Biochemistry 35, 15120-15128 [DOI] [PubMed] [Google Scholar]
- 44.Hemp, J., Robinson, D. E., Ganesan, K. B., Martinez, T. J., Kelleher, N. L., and Gennis, R. B. (2006) Biochemistry 45, 15405-15410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Humphrey, W., Dalke, A., and Schulten, K. (1996) J. Mol. Graphics 14, 33-38 [DOI] [PubMed] [Google Scholar]