

Abstract
To test the hypothesis that somatic mitochondrial (mt)DNA mutation accumulation predisposes mice to β-cell loss and diabetes development, transgenic mice expressing a proofreading-deficient mtDNA polymerase-γ under the control of the rat insulin-1 promoter were generated. At 6 wk of age, mtDNA mutations reached 0.01% (1.05 mutations/10,000 bp) in islets isolated from transgenic mice. This mutational burden is associated with impaired glucose tolerance and a diabetes prevalence of 52% in male transgenic mice. Female transgenic mice maintain slightly elevated fasting glucose levels, mild glucose intolerance, and a diabetes prevalence of 14%. Diabetes in transgenic animals is associated with insulin insufficiency that results from a significant reduction in β-cell mass. Importantly, apoptosis of β-cells is increased 7-fold in female and 11-fold in male transgenic mice compared with littermate controls. These results are consistent with a causative role of somatic mtDNA mutation accumulation in the loss of β-cell mass and diabetes development.
Keywords: mitochondrial deoxyribonucleic acid, insulin, islet
the obesity epidemic (14) has accelerated the incidence of type 2 diabetes mellitus, yet not all obese or even insulin-resistant patients develop overt diabetes (7, 19). In individuals who progress to diabetes, the failure of β-cells is progressive; however, the mechanisms responsible for this loss of β-cells have yet to be fully characterized. Free radical-mediated damage associated with elevated levels of metabolic substrates, such as glucose and lipid, appear to participate in β-cell damage during the progression to type 2 diabetes (18). Mitochondria are the primary sites of free radical generation (26), and mitochondrial (mt) DNA is exquisitely sensitive to free radical mediated damage. This sensitivity is associated with the proximity of mtDNA to the source of free radical generation. The resulting DNA damage and mutations due to this damage are likely to affect mitochondrial-expressed genes because mtDNA is primarily composed of coding sequence (26). Indeed, the Mitochondrial Theory of Aging proposes that the vicious cycle of increased mtDNA mutation accumulation results in defects in the components of the oxidative phosphorylation machinery, causing further increases in reactive oxygen species generation and mtDNA mutations (4, 5). It is this accumulation of mtDNA mutations that may be responsible for the development of age-related pathologies (13, 26).
The development of mitochondrial disease is associated with a large proportion of mtDNA molecules (>30%) carrying a single pathogenic mutation. While these maternally inherited diseases often include diabetes within their phenotypic spectra, only 0.2–2% of diabetes cases have been associated with the accumulation of individual mtDNA mutation at high levels (11). During aging, the frequency of mtDNA genomes containing mutations does not generally exceed 1% in normal individuals and these mutations occur randomly (2). The contributions of mtDNA mutations that accumulate at low levels during aging (0.1–1%) to the development of diabetes are unknown. To examine the pathogenic nature of low-level somatic mtDNA mutation accumulation on diabetes development, a mutant mtDNA polymerase-γ [PolγExo(−)] that lacks 3′-5′-exonuclease proofreading activity was expressed in mice under the control of the rat insulin promoter (RIP). With the use of a similar construct expressed under the control of a cardiac-specific promoter, the lack of proofreading activity is associated with the accumulation of mtDNA mutations to levels of 0.01% and these mutations correlate with the development of four-chamber dilated cardiomyopathy (27). Also, knockin expression of a PolγExo(−) leads to several age-related phenotypes that include shortened lifespan in homozygous mice (9, 24). We now show that mice expressing this mutant Polγ in β-cells accumulate mutations in islet mtDNA to levels of 0.01% by 6 wk of age and that mtDNA mutation accumulation is associated with the development of overt diabetes in male mice and impaired glucose tolerance in female mice. Impaired glucose tolerance and diabetes in these mice are due to insulin insufficiency that is a consequence of reduced β-cell mass due to increased neonatal β-cell apoptosis.
MATERIALS AND METHODS
Transgene construction.
Two transgene constructs were developed for the generation of founders. Both constructs were derived from the mutant murine mtDNA polymerase-γ (Polγ) gene containing an aspartate to alanine mutation at residue 181 (D181A) resulting in the inhibition of exonuclease proofreading (27). The rat insulin-1 promoter (RIP1) was a generous gift of Colin Nichols (Washington University School of Medicine). All constructs were confirmed by sequencing. The untagged transgene was generated by ligating RIP1 (SacI-BamHI) to the mutant PolγExo(−) cDNA partially digested at the 5′-end with EcoRI, followed by gel purification. The in vitro ligated construct (linear) was gel purified and used for injections. Oocyte injection of the first transgene construct was performed at Emory University and resulted in a single founder on an FVB background. This line is referred to as RIP1-PolγExo(−). A second construct containing a C-terminal myc tag was prepared using a pCS2 + MT plasmid containing the 6 myc epitope tag provided by Dr. Joseph Baldassarre (St. Louis University). Restriction digest sites were added to the 3′-UTR of the original mutant polymerase construct through high-fidelity PCR (Platinum Pfx DNA polymerase; Invitrogen, Carlsbad, CA) to allow the myc tag to be inserted in frame with Polγ. The entire construct was confirmed by sequencing. Oocyte injection was performed at the Washington University Mouse Genetics Core and resulted in four potential founder lines on an FVB background, three of which transmitted the transgene to subsequent generations. Mice from these lines have been studied to confirm observations made on the original founder line and are reported as RIP1-PolγExo(−)-myc B-D.
Transgene incorporation and expression analysis.
Genotyping and Southern blot analysis were performed as previously described (27), and the copy number was determined from the Southern blot by PhosphorImage quantification of the summed signal from the transgenic bands relative to the endogenous Polγ band. RT-PCR analysis was performed on total mRNA purified using TRI-Reagent (Sigma-Aldrich) from individual tissues, including brain, heart, kidney, liver, pancreas, spleen, and isolated islets, harvested from wild-type and transgenic mice. cDNA was generated using Moloney murine leukemia virus reverse transcriptase (Invitrogen) and amplified by PCR using primers flanking the point mutation, as previously described (27). PCR products were digested with either Taq I or Btg I and analyzed by acrylamide gel electrophoresis.
mtDNA mutation analysis.
All mutation analyses were performed on mtDNA purified from islets isolated from 6-wk-old female wild-type and transgenic mice. mtDNA deletion mutations were evaluated using nested-primer PCR, and mtDNA point mutations were determined by sequencing mtDNA clones isolated as previously described (27).
Glucose homeostasis.
Four-hour-fasted blood glucose was measured from tail vein blood of mice aged 4–12 wk on a MediSense Precision PcX glucometer (Abbott, Abbott Park, IL). Blood glucose measurements of 1-wk-old mice were performed in the fed state. Oral glucose tolerance was determined at 6 wk of age after a 4 h fast. Mice were given 2 g dextrose/kg body wt by oral gavage. Blood glucose was determined before glucose administration and 15, 30, 60, and 120 min after gavage. Intraperitoneal insulin tolerance tests were performed on 4-h-fasted mice given an intraperitoneal injection of insulin (0.5 U/kg body wt). Blood glucose was determined before injection and 30 min after dosing. In vivo β-cell function was determined as change in plasma insulin levels (collected from tail vein) before and 15 min after mice received a 2 g/kg oral dose of dextrose. Blood samples were kept on ice and centrifuged (15 min at 2,400 rpm Eppendorf 5810R centrifuge), and plasma was stored at −70°C until it was assayed for insulin by ELISA (Crystal Chem, Downers Grove, IL).
Insulin secretion assays.
Islets were isolated from wild-type and transgenic mice by collagenase digestion (8), and insulin secretion was measured as previously described (6). Briefly, 15 islets per condition were incubated at 37°C in Krebs-Ringer phosphate buffer. Islets were stimulated with 3, 10, or 20 mM glucose, 30 mM KCl with 3 mM glucose, or 10 mM leucine with 10 mM glucose. Insulin levels were determined by RIA at the Washington University Diabetes Research and Training Center Core Laboratory for Clinical Studies and Immunoassay.
Pancreas insulin content.
Pancreata were harvested from 1- and 8-wk-old male and female fed wild-type and transgenic mice, rinsed in PBS, blotted dry, and weighed. To extract insulin, pancreata were placed in acid ethanol and stored at −20°C, as described previously (15). Tissues were homogenized using a Polytron PT10/35 (Kinematica, Bohemia, NY) and centrifuged in an Eppendorf 5810R centrifuge at 2,400 rpm, and insulin content was determined on the supernatant.
Histology.
Pancreata were harvested from mice, rinsed in PBS, weighed, and arranged head to tail for paraffin embedding. Several serial sections (5 μm) were taken every ∼50 μm, and one from each depth was stained with hematoxylin and eosin or trichrome by the St. Louis University histopathology core. Unstained sections were deparaffinized in xylenes and rehydrated through steps of ethanol to PBS, and antigens were retrieved under pressure using Borg Decloaker (Biocare Medical, Concord, CA). Sections were blocked using Background Sniper (Biocare Medical). Primary antibodies used were guinea pig anti-insulin (1:100; Dako, Carpinteria, CA), rabbit anti-glucagon (1:200; Linco Millipore, St. Charles, MO), rabbit anti-cleaved caspase-3 (Asp175; 1:200; Cell Signaling, Danvers, MA), mouse anti-2′-deoxyuridine:5-fluoro-2′-deoxyuridine (BrdU; 1:100; Amersham, Piscataway, NJ), or rabbit anti-Myc Tag (1:200; Cell Signaling). Fluorescent-labeled secondary antisera were Alexa Fluor-488 labeled donkey anti-guinea pig (1:200; Molecular Probes, Invitrogen), CY3-conjugated donkey anti-rabbit, or CY3-conjugated donkey anti-mouse (1:200; Jackson ImmunoResearch, West Grove, PA). DAPI was used to stain for nucleic acids. Islet architecture of endocrine cells was evaluated on sections costained for insulin, glucagon, and DAPI. β-Cell proliferation was examined in mice injected with BrdU (50 μg/g body wt) 2 h before pancreas harvest. Proliferation was quantified on sections costained for insulin, BrdU, and DAPI. Quantification of proliferating β-cells was performed using random-field counting of a 48-point grid on three sections per pancreas at different depths into the tissue for three mice of each type and gender. Data are expressed as percentage of BrdU-positive, insulin-positive cells. β-Cell apoptosis was determined on sections costained for insulin, cleaved caspase-3, and DAPI. Data are expressed as percentage of cleaved caspase-3-positive/insulin-positive cells.
RESULTS
β-Cell-specific accumulation of mtDNA mutations.
For these studies, mice expressing a proofreading-deficient mtDNA Polγ under the control of the RIP1 were generated [RIP1-PolγExo(−); Fig. 1A]. The transgene encodes a mutant Polγ that lacks the proofreading function due to the inactivation of its 3′-exonuclease activity that results from the substitution of an alanine for an aspartate residue at amino acid 181 of mouse Polγ (Fig. 1A). At the DNA level this substitution changes a restriction site from Taq I to Btg I, thereby allowing quantification of the ratio of transgene to endogenous gene expression by restriction analysis of RT-PCR products. After the first round of DNA injections, one founder line RIP1-PolγExo(−) was generated. As described in the Supplemental Data, three additional founder lines expressing a myc epitope-tagged mutant Polγ gene [RIP1-PolγExo(−)-myc] have been generated (see Supplemental Fig. S1; supplemental data for this article are available online at the Am J Physiol Endocrinol Metab website). The transgene copy number in the original line was 12-fold greater than the endogenous Polγ gene, as determined by Southern blot analysis (Fig. 1B), and transgene expression was limited to the pancreas (Fig. 1C). The ratio of transgene to endogenous Polγ mRNA was significantly greater in islets than in the whole pancreas, consistent with expression restricted to the endocrine pancreas (Fig. 1C).
Fig. 1.

Generation of RIP1-PolγExo(−) transgenic mice. A: schematic representation of the construct used to generate RIP1-PolγExo(−) mice. The transgene, under the control of the rat insulin-1 promoter (RIP1), is comprised of murine polymerase-γ (Polγ)-containing an inactivating D181A mutation in the proofreading region. B: transgene copy number is 12-fold higher than endogenous Polγ as determined by Southern blot analysis. C: RT-PCR followed by restriction digest using Taq I (T) for endogenous and Btg I (B) for transgene demonstrates selective transgene expression in the pancreas and islets isolated from transgenic (Tg) mice. Note that expression of transgenic and endogenous Polγ is observed in the total pancreas, while the transgenic polymerase is only detected in isolated islets that are predominantly composed of β-cells. WT, wild type.
Unfortunately, immunohistochemical analysis for transgene expression is not possible, since commercial anti-Polγ antibodies are only poorly immunoreactive (data not shown; unpublished observations, H. P. Zassenhaus). However, using the epitope-tagged transgene, we were able to confirm expression in insulin-positive cells in the pancreatic islet but not in the exocrine pancreas (Supplemental Fig. S1D). Furthermore, myc staining was punctate and excluded from the nucleus (Supplemental Fig. S1D). This is consistent with a mitochondrial distribution and suggests that the mutant PolγExo(−) protein is not ectopically localized to the nucleus (22).
The accumulation of mutations was determined using mtDNA isolated from islets harvested from 6-wk-old female wild-type and transgenic mice. In addition to single base mutations, the proofreading-deficient PolγExo(−) is also known to generate large-scale deletion mutations (24, 25, 27). With the use of a nested-primer PCR strategy, a deletion mutant of ∼7.5 kbp in length was observed in mtDNA of transgenic but not wild-type mice (Supplemental Fig. S2). Point mutation accumulation was determined by sequencing clones of mtDNA isolated from islets of 6-wk-old wild-type and transgenic mice. This protocol identified 1.05 mutations per 10,000 bp of mtDNA, while mutations were not present in mtDNA harvested from islets isolated from littermate controls. This level of mutations (0.01%) is similar to the burden of mutations identified in the hearts of mice expressing mutator PolγExo(−) under the control of a cardiac-specific promoter (27).
Impaired glucose homeostasis in RIP1-PolγExo(−) transgenic mice.
There are no differences in the total body weight of wild-type or transgenic mice of the same gender (Fig. 2A); however, the average fasting blood glucose levels are dramatically elevated in male transgenic mice compared with littermate controls (4–12 wk of age; Fig. 2B). Female transgenic mice also have significantly higher fasting blood glucose than wild-type female mice. With the use of a fasting blood glucose level of >200 mg/dl to define diabetes, 52% of male transgenic mice are diabetic by 6 wk of age. The fasting blood glucose levels of female transgenic mice are also elevated compared with littermate controls, reflecting the disease prevalence in female transgenic mice of 14% (Fig. 2C).
Fig. 2.

Impaired glucose homeostasis in Tg RIP1-PolγExo(−) mice. Body weights (A) and 4-h-fasted blood glucose levels (B; n = 18–26) were measured on 4- and 12-wk-old wild-type and transgenic male and female mice. Data for A and B are means ± SE. C: incidence of diabetes (defined as 2 consecutive fasting glucose levels >200 mg/dl) is shown for 6-wk-old Tg mice. Note that elevated average fasting glucose levels observed in female Tg mice (B) are likely due to the limited number of mice that develop diabetes (incidence shown in C).
Since fasting glucose levels were elevated in transgenic mice, the response to an oral glucose challenge was examined. At 6 wk of age, glucose tolerance was impaired in both male and female transgenic mice compared with littermate controls (Fig. 3A). Consistent with elevated fasting glucose levels (Fig. 2B), transgenic male mice were significantly more glucose intolerant than female transgenic mice. The oral glucose tolerance results, quantified as area under the curve ± SE of the mean, show statistically significant (Student's t-test between wild-type and transgenic of each gender) impairment with wild-type females area under the curve of 2.0 × 104 ± 8.7 × 102 (n = 16) compared with transgenic females at 2.9 × 104 ± 2.4 × 103 (n = 17; P < 0.005) and wild-type males 2.8 × 104 ± 2.0 × 103 (n = 17) compared with transgenic males at 4.9 × 104 ± 3.7 × 103 (n = 20; P < 0.0001). Similar results were obtained in male and female transgenic mice at 4, 8, and 12 wk of age (data not shown).
Fig. 3.
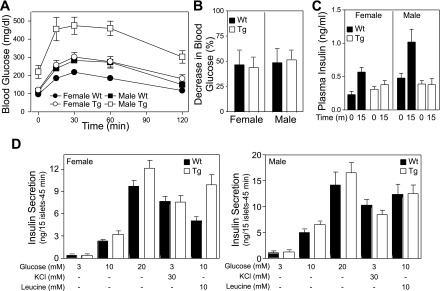
β-Cell defect in Tg RIP1-PolγExo(−) mice. A: male, and to a lesser extent female, RIP1-PolγExo(−) mice are glucose intolerant as determined on 6-wk-old mice fasted 4 h before the tolerance test (n = 16–20). B: intraperitoneal insulin tolerance tests (0.5 U insulin/kg body wt; n = 5–8) show that 11-wk-old PolγExo(−) male and female WT and Tg mice are insulin responsive. Data are presented as percent decrease from baseline (means ± SE) and show ∼50% reductions in blood glucose 30 min after insulin administration in all groups. Similar results were been obtained from mice 4–13 wk of age. C: in vivo assessment of β-cell function was determined by measuring plasma insulin level before and 15 min postoral glucose challenge (2 g/kg body wt; n = 6–15; WT females, P < 0.005; WT males, P < 0.05). D: in contrast to the attenuated in vivo β-cell response in Tg male and female mice, insulin secretion by islets isolated from Tg mice in response to glucose (3, 10, or 20 mM), KCl, and leucine is either comparable or greater than the response of islets isolated from littermate controls.
β-Cell defect in RIP1-PolγExo(−) transgenic mice.
The glucose intolerance observed in transgenic mice expressing a mutator PolγExo(−) under the control of RIP1 is not caused by impaired insulin action. The percent reduction in blood glucose in response to 30-min intraperitoneal insulin challenge is similar in wild-type and transgenic, male and female mice (Fig. 3B; data shown are from 11-wk-old mice, and similar results have been obtained at 4, 6, 8, and 13 wk of age). Glucose intolerance observed in transgenic mice is associated with defects in the β-cell response to an oral glucose challenge (Fig. 3C). Plasma insulin concentrations increase over twofold in 6-wk-old male and female wild-type mice 15 min postadministration of an oral glucose challenge. In contrast, this increase in plasma insulin in response to an oral glucose challenge is significantly attenuated in female transgenic mice and absent in male transgenic mice (Fig. 3C). To determine if the defect in the β-cell response is due to an impairment of β-cell function, insulin secretion assays were performed using isolated islets. In a concentration-related manner, glucose stimulates insulin secretion from isolated transgenic mouse islets to levels that are comparable with the response observed from islets isolated from male and female wild-type mice. Further, insulin secretion in response to KCl, which stimulates secretion by direct plasma membrane depolarization, and leucine, which requires mitochondrial metabolism to stimulate secretion, is similar or greater in islets isolated from transgenic mice compared with wild-type littermates (Fig. 3D). The exaggerated insulin secretory response of transgenic female islets to 10 mM leucine was not consistently observed using islets isolated from mice at other ages. Insulin secretion studies have also been normalized to total insulin content of islets to control for differences in islet size, with results identical to those shown in Fig. 3D. Similar results have been obtained using islets isolated from wild-type and transgenic mice at 4, 8, and 11 wk of age (data not shown). These findings suggest that the impairment in the β-cell response to an oral glucose challenge observed in PolγExo(−) expressing mice is likely a consequence of reduced β-cell mass.
Reduced β-cell mass in RIP1-PolγExo(−) transgenic mice.
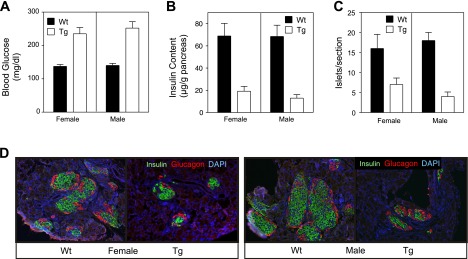
There is an approximate threefold decrease in both the number of islets/section and in the insulin content of pancreata isolated from male transgenic mice compared with littermate controls (6 wk of age; Fig. 4). In female mice, insulin content is reduced by 50% at 6 wk of age (Fig. 4C), while the number of islets/section is similar to littermate control mice (Fig. 4B). The disconnect between islet number and insulin content in female transgenic mice appears to be due to the reduction in islet size (∼50%) compared with wild-type controls (Fig. 4A). Male transgenic mice have significantly fewer islets that are much smaller in size with irregular borders when compared with their wild-type counterparts (Fig. 4A). Consistent with the irregular borders, endocrine hormone staining of pancreatic sections shows a disruption in the normal morphology of islets in transgenic pancreas (Fig. 5). The islets in sections prepared from 6-wk-old male and female wild-type mice contain normal architecture with a dense core of insulin containing β- cells (green) and a peripheral rim of glucagon containing α-cells (red). In contrast, the architecture of nearly all of the islets found in male transgenic mice was disorganized with α-cells present in the islet core, decreased density of β-cells in the core, and a loss of regular islet borders (Fig. 5). Most of the islets observed in male transgenic mice were small clusters of <100 endocrine cells. In pancreatic sections prepared from female transgenic mice, an altered architecture was observed in some (∼50%) but not all of the islets. Taken together, these results indicate that impaired glucose tolerance in transgenic mice is not due to impaired β-cell function but is associated with a loss of β-cell mass accompanied by abnormal islet architecture.
Fig. 4.

Reduced β-cell mass and altered islet morphology in Tg RIP1-PolγExo(−) mice. A: islet morphology is disordered in male and female RIP1-PolγExo(−) as determined by hematoxylin and eosin staining on pancreatic sections harvested from 6-wk-old mice. There is no evidence of islet inflammation, but islets identified in Tg female mice are ∼50% smaller than littermate control mice. Islets identified in pancreatic sections prepared from male Tg mice contain disorganized borders and are smaller than controls. B and C: disorganized structure is consistent with (B) reduced islet numbers/section and (C) insulin content in male Tg mice. Female Tg mice have similar number of islets/section; however, total insulin content is reduced by 50%, consistent with the smaller size of the islets. All data were obtained with 6-wk-old wild-type and RIP1-PolγExo(−) mice. Images are represent 6 mice/group, and quantification of islet number is presented as means ± SE of 6 sections/pancreas from 6 mice/group. Insulin content was determined from 9–12 pancreata per group and is expressed as means ± SE (females, P < 0.0001; males, P < 0.0001).
Fig. 5.

Disordered endocrine hormone localization in Tg RIP1-PolγExo(−) mice. Pancreatic sections prepared from 6-wk-old Wt and Tg mice were stained for the endocrine hormones insulin (green) and glucagon (red). Representative sections demonstrate reduced size of islets in Tg male and female mice as well as an altered islet architecture, which is evident by glucagon expressing cells in the core of ∼50% of islets observed in female mice and nearly all of the islets in male Tg mice. Data represent pancreatic sections prepared from at least 6 mice/group.
β-Cell defects in 1-wk-old RIP1-PolγExo(−) transgenic mice.
Because mice exhibited altered glucose handling as early as 4 wk of age and because rodent islets undergo significant remodeling during the first 2 wk of life (20), serum glucose levels, islet number, and pancreatic insulin content were examined at early time points, 1-wk-old age, in wild-type and mutator PolγExo(−) expressing mice. The body weights of 1-wk-old wild-type and transgenic mice did not differ (data not shown). As early as 1 wk of age, both male and female transgenic mice have elevated blood glucose levels compared with wild-type littermate controls (Fig. 6A). Because the mice are still nursing at this age, these are fed blood glucose values; thus a value >200 mg/dl is not diagnostic of diabetes. At this age, mice exhibited a greater than threefold reduction in pancreas insulin content as well as a decreased number of islets/section (Fig. 6, B and C). Immunohistochemical analysis of endocrine hormones in sections prepared from 1-wk-old wild-type and transgenic mice indicated that, similar to 6-wk-old mice, the architecture of the islets in transgenic mice is altered with α-cells found in the core of the islets and reductions in the size of most of the islets (Fig. 6D).
Fig. 6.
Elevated blood glucose and decreased β-cell content in 1-wk-old Tg RIP1-PolγExo(−) mice. A: blood glucose was measured on fed 1-wk-old WT and Tg mice and was elevated in both female and male Tg mice (n = 14–24; P < 0.0001). B and C: β-cell content was estimated by determining whole pancreas insulin content and analyzing sections for islet numbers. As in older mice, 1-wk-old Tg mice had reduced insulin content (B: females, P < 0.005; males, P < 0.0001; n = 6–10) and fewer islets per histological section (C: females, P < 0.05; males, P < 0.0001; n = 4–7). D: immunohistochemical staining for insulin (green) and glucagon (red) in 1-wk-old mice shows smaller Tg islets and altered Tg islet architecture as observed in older mice.
Reduction in β-cell mass in RIP1-PolγExo(−) transgenic mice is associated with increased apoptosis.
Reduced insulin and islet content in RIP1-PolγExo(−) mice could result from reduced β-cell proliferation, increased β-cell death, or a combination of both processes. To investigate these possibilities, β-cell proliferation was determined by BrdU incorporation and apoptosis by the presence of cleaved or active caspase-3 on pancreatic sections prepared from 1-wk-old mice. Overall, there were no differences in BrdU incorporation when wild-type mice were compared with transgenic mice (Fig. 7, A and B); however, the incorporation of BrdU in β-cells was slightly higher in female mice than 1-wk-old male mice. In contrast, β-cell apoptosis is 7-fold higher in transgenic female and 11-fold higher in male transgenic mice compared with their wild-type littermate controls (Fig. 7, C and D). For these studies, β-cell apoptosis was quantified as the percentage of cleaved caspase-3-positive/insulin-positive cells. These results suggest that the glucose intolerance observed in the female mice and the glucose intolerance and diabetes development observed in male mice expressing a mutator PolγExo(−) under the control of RIP1 are associated with accelerated β-cell apoptosis.
Fig. 7.

Increased β-cell apoptosis in Tg RIP1-PolγExo(−) mice. Levels of β-cell proliferation and apoptosis were determined by immunostaining for insulin (green) and either 2′-deoxyuridine:5-fluoro-2′-deoxyuridine incorporation (BrdU; red; A) or cleaved caspase-3 (red; C) on sections prepared from wild-type and transgenic mice. B: quantification of BrdU-positive/insulin-positive cells showed no significant difference in β-cell proliferation in the Tg mice compared with WT (n = 3). D: quantification of cleaved caspase-3-positive/insulin-positive cells indicates that there is a 7- and 11-fold increase in the apoptosis of β-cells in 1-wk-old female and male Tg mice, respectively. Less than 0.1% of WT β-cells undergo apoptosis at this age (P < 0.05; n = 4–7).
DISCUSSION
Type 2 diabetes is an age- and obesity-related disease that follows years of insulin resistance leading to the eventual failure of β-cells. Prolonged exposures to elevated levels of glucose and lipid may contribute to diabetes development by the increased production of reactive oxygen species, leading to oxidative stress and damage to β-cells. Further, as we age, the insulin secretory function of β-cells is diminished (1). mtDNA is one potential molecular target that may participate in these age-related pathologies. Indeed, mtDNA mutations are known to increase with age (2), and mtDNA is sensitive to free radical-mediated damage largely because of its proximity to the primary source of free radical generation (26). To examine whether mtDNA mutations are a cause or consequence of age-related pathologies, mouse models have been generated using tissue-specific expression and gene knockin strategies of a proofreading-deficient mutant of mtDNA polymerase that permits the incorporation of random mutations during mtDNA replication (9, 24, 27). Tissue-specific expression of a proofreading-deficient mtDNA polymerase mutant under the control of the cardiac-specific α-myosin heavy chain promoter results in the rapid accumulation of mtDNA mutations at a frequency of 0.01% at 1 mo of age, and this mutational burden is associated with the development of four-chamber dilated cardiomyopathy (27). Expression of proofreading-deficient mtDNA polymerase in all tissues using a knockin strategy results in a phenotype of premature aging that is associated with hair loss, kyphosis, hearing loss, and reduced muscle mass (9, 24). These knockin mice also develop heart disease; however, it is less severe and develops at a much later age than transgenic mice with heart-specific expression.
Much like heart disease in mice with restricted expression of a proofreading-deficient mtDNA polymerase in cardiac tissues, transgenic expression in β-cells results in mtDNA mutations and diabetes development. Transgenic mice expressing a proofreading-deficient mutant of mtDNA polymerase under the control of the RIP1 accumulate random mtDNA mutations in islets to a level of 0.01% (∼1–2 mutations/mitochondrial genome). Male mice expressing mutator PolγExo(−) under the control of RIP1 are glucose intolerant and have a prevalence of overt diabetes of 52% by 6 wk of age. Female transgenic mice are glucose intolerant; however, their prevalence of overt diabetes is significantly lower (14%). Gender differences in the development of diabetes have been observed in rodent models. In many models, the development of type 2 diabetes is commonly observed in males, while the incidence of autoimmune diabetes is more prevalent in female mice such as the nonobese diabetic mouse. These differences may be due to increased insulin sensitivity in female mice or to the action of estrogen hormones (10). While the sex difference in disease incidence in our colony is of interest, ovariectomy or orchiectomy studies have yet to be performed on these mice.
Glucose-stimulated insulin secretion by isolated islets, in addition to demonstrating islet function, can also be used as an index for oxidative stress and/or impaired oxidative metabolism. Insulin secretion in response to glucose requires oxidation of glucose to CO2 to generate ATP to levels sufficient to close K+ channels (12), and this process is highly sensitive to oxidative stresses (18). Importantly, there is no impairment in the insulin secretory response of islets isolated from RIP1-PolγExo(−) male and female transgenic mice in response to glucose, direct depolarizing agents, or amino acids that are metabolized by the mitochondria. In fact, islets from female transgenic mice secrete more insulin in response to leucine than wild type. These findings suggest that diabetes development in RIP1-PolγExo(−) mice is not likely to be the result of oxidative stress or impaired oxidative metabolism by β-cells, as glucose-stimulated insulin secretion requires the oxidation of glucose to CO2 and insulin secretion is extremely sensitive to oxidative stress. Consistent with these conclusions is the absence of oxidative damage or impaired oxidative metabolism in the homozygous knockin mice and in the hearts of mice with cardiac-specific expression of PolγExo(−) (9, 16, 23).
Reduced β-cell mass due to enhanced levels of apoptosis appears to be the mechanism responsible for the development of diabetes in RIP1-PolγExo(−) mice. Apoptosis is increased 11-fold in male and 7-fold in female RIP1-PolγExo(−) transgenic mice at 1 wk of age compared with littermate controls. Importantly, β-cell proliferation is not modified in RIP1-PolγExo(−) mice, suggesting that reduced β-cell mass observed at later ages can be attributed to enhanced apoptosis in the absence of changes in proliferation. Enhanced apoptosis in the absence of oxidative damage has been observed in other mouse models of mtDNA mutation accumulation (9, 28, 29), suggesting that the disease pathology associated with the accumulation of low levels of somatic mtDNA mutations is associated with cell loss via apoptosis.
Although mice with β-cell-restricted expression of mutator PolγExo(−) develop diabetes, this phenotype was not identified in mice in which a similar mutant was expressed in all cells (9, 24). If, as observed for heart disease, a diabetes phenotype is mild, it is possible that this phenotype escaped detection in the mutator PolγExo(−) knockin mice. Also, any potential countervailing effect on other components of glucose regulation in these mice, such as insulin action or glucose uptake, is not known.
We have generated three additional lines of mice expressing a β-cell-specific myc-tagged mutator Polγ [RIP1-PolγExo(−)-myc; Supplemental Fig. 1], which are all glucose intolerant but do not develop overt diabetes. The differences in phenotype between the original line and the three new myc-tagged lines could be a consequence of mosaic transgene expression in islets (Supplemental Fig. S1D). Alternatively, the relative ratio of transgene to endogenous Polγ expression may play a role in the severity of the phenotype. Consistent with the latter interpretation, cardiac-specific expression of mutator PolγExo(−) results in a severe four-chamber cardiomyopathy, and these mice exclusively express the transgene. Diabetes observed in our original founder line was associated with the exclusive expression of the transgene in islets (Fig. 1C), where endogenous mtDNA Polγ was not expressed at levels sufficient for detection. The glucose intolerance phenotype observed in the newly generated myc-tagged lines (Supplemental Fig. 1C) is associated with lower levels of transgene expression compared with the original founder line and the expression of both endogenous and mutator PolγExo(−) in islets in the new lines (Supplemental Fig. 1B). Importantly, while the phenotype is mild in PolγExo(−)-myc expressing mice compared with the original founder, the impaired glucose tolerance observed in these new mouse lines provides evidence that diabetes associated with the original founder is not due to transgene insertion.
In this study, we show that expression of a proofreading-deficient mtDNA polymerase under the control of the RIP1 results in the accumulation of low levels (0.01%) of somatic mtDNA mutations in β-cells. Mutation accumulation correlates with impaired glucose tolerance and diabetes development that are the consequence of an approximately threefold reduction in β-cell mass (male mice). β-Cell loss is the result of enhanced apoptosis under conditions of normal β-cell proliferation early in islet development (1 wk of age). These findings suggest that diabetes development in these mice appears to be a consequence of enhanced β-cell apoptosis that occurs during islet remodeling and growth early in pancreas development (1–2 wk of age) leading to an inadequate β-cell mass.
The mechanisms by which low levels of mtDNA mutations induce apoptosis are unknown. Mott et al. (17) have proposed that mtDNA mutation accumulation occurs in cells that fail to initiate an anti-apoptotic response and that apoptosis may be mediated by a mitochondrial directed misfolded protein response. Someya et al. (21) have shown that knocking-in the mutator mtDNA polymerase leads to the enhanced expression of proapoptotic and stress response genes and reductions in the expression of genes associated with impairment of energy metabolism, suggesting that impaired energy metabolism induces apoptosis (21). In contrast, Dubec et al. (3) have observed that cardiac-specific expression of the mutator mtDNA polymerase leads to release of a peptide factor that correlates with the release of cytochrome c from mitochondria to direct apoptosis. While each of these proposed mechanisms has merit, it is clear that additional molecular-based studies are needed to determine the mechanisms responsible for the apoptosis of cells that have accumulated mtDNA mutations.
Taken together, these findings suggest that β-cells in these transgenic mice do not have the capacity to accelerate proliferation to levels sufficient to compensate for the apoptotic loss of β-cells. During the development of type 2 diabetes, β-cells compensate for increased insulin demand by enhanced proliferation and increased β-cell mass. Diabetes develops when β-cells fail to compensate for this increased demand for insulin. Could mtDNA mutation accumulation predispose β-cells to apoptosis when challenged to proliferate under conditions of enhanced insulin demand? Although it is not possible to directly address this question based on the data presented in this study, it is possible to correlate increased rates of β-cell apoptosis in mice expressing mutator PolγExo(−) under the control of RIP with active β-cell proliferation during development (1 wk of age). These findings suggest that mtDNA mutation accumulation may indeed predispose cells to apoptosis under conditions in which they are actively proliferating or forced to proliferate. Consistent with this interpretation is an approximately threefold increase in the rate of cardiac myocyte apoptosis during heart remodeling in mice expressing mutator Polγ under the control of the cardiac-specific promoter (29). Currently, we are examining whether PolγExo(−)-myc mice have the ability to compensate for a secondary stress such as obesity (Western high-fat diet), which normally stimulates β-cell hypertrophy and hyperplasia, in an effort to determine if mtDNA mutation accumulation contributes to β-cell loss and failure to compensate for insulin resistance.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-52194 (to J. A. Corbett), a Ruth L. Kirschstein National Research Service Award Predoctoral Fellowship through the National Institute on Aging (to K. G. Bensch), an American Diabetes Association Research (to J. A. Corbett) and Physician Scientist Awards (to W. de Graaf and J. L. Mott), and an American Heart Association predoctoral fellowship award (to K. T. Chambers). We also thank the Digestive Diseases Research and Development Center Molecular Pathology Core of the National Institutes of Health-funded Mucosal HIV and Immunobiology Center (DK-64400) for assistance with histology.
Supplementary Material
Acknowledgments
We thank Colleen Kelly Bratcher and Grace Denniger for expert technical assistance.
Present address of J. L. Mott: Miles and Shirley Fiterman Center for Digestive Diseases, Division of Gastroenterology and Hepatology, Mayo Clinic College of Medicine, Rochester, MN 55905.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Chang AM, Halter JB. Aging and insulin secretion. Am J Physiol Endocrinol Metab 284: E7–E12, 2003. [DOI] [PubMed] [Google Scholar]
- 2.Cortopassi GA, Wong A. Mitochondria in organismal aging and degeneration. Biochim Biophys Acta 1410: 183–193, 1999. [DOI] [PubMed] [Google Scholar]
- 3.Dubec SJ, Aurora R, Zassenhaus HP. Mitochondrial DNA mutations may contribute to aging via cell death caused by peptides that induce cytochrome c release. Rejuvenation Res 11: 611–619, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harman D Aging: a theory based on free radical and radiation chemistry. J Gerontol 11: 298–300, 1956. [DOI] [PubMed] [Google Scholar]
- 5.Harman D The biologic clock: the mitochondria? J Am Geriatr Soc 20: 145–147, 1972. [DOI] [PubMed] [Google Scholar]
- 6.Heitmeier MR, Scarim AL, Corbett JA. Interferon-gamma increases the sensitivity of islets of Langerhans for inducible nitric-oxide synthase expression induced by interleukin 1. J Biol Chem 272: 13697–13704, 1997. [DOI] [PubMed] [Google Scholar]
- 7.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444: 840–846, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Kelly C, Blair L, Corbett J, Scarim A. Isolation of islets of Langerhans from rodent pancreas. Methods Mol Med 83: 3–14, 2003. [DOI] [PubMed] [Google Scholar]
- 9.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309: 481–484, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Le May C, Chu K, Hu M, Ortega CS, Simpson ER, Korach KS, Tsai MJ, Mauvais-Jarvis F. Estrogens protect pancreatic beta-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc Natl Acad Sci USA 103: 9232–9237, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maassen JA, 't Hart LM, Janssen GMC, Reiling E, Romijn JA, Lemkes HH. Mitochondrial diabetes and its lessons for common type 2 diabetes. Biochem Soc Trans 34: 819–823, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic beta-cells. Nature 414: 807–812, 2001. [DOI] [PubMed] [Google Scholar]
- 13.Miquel J An update on the oxygen stress-mitochondrial mutation theory of aging: genetic and evolutionary implications. Exp Gerontol 33: 113–126, 1998. [DOI] [PubMed] [Google Scholar]
- 14.Mokdad AH, Serdula MK, Dietz WH, Bowman BA, Marks JS, Koplan JP. The continuing epidemic of obesity in the United States. JAMA 284: 1650–1651, 2000. [DOI] [PubMed] [Google Scholar]
- 15.Montana E, Bonner-Weir S, Weir GC. Beta-cell mass and growth after syngeneic islet cell transplantation in normal and streptozocin diabetic C57BL/6 mice. J Clin Invest 91: 780–787, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mott JL, Zhang D, Stevens M, Chang S, Denniger G, Zassenhaus HP. Oxidative stress is not an obligate mediator of disease provoked by mitochondrial DNA mutations. Mutat Res 474: 35–45, 2001. [DOI] [PubMed] [Google Scholar]
- 17.Mott JL, Zhang D, Zassenhaus HP. Mitochondrial DNA mutations, apoptosis, and the misfolded protein response. Rejuvenation Res 8: 216–226, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Poitout V, Robertson RP. Minireview: secondary beta-cell failure in type 2 diabetes–a convergence of glucotoxicity and lipotoxicity. Endocrinology 143: 339–342, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Prentki M, Nolan CJ. Islet beta-cell failure in type 2 diabetes. J Clin Invest 116: 1802–1812, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scaglia L, Cahill CJ, Finegood DT, Bonner-Weir S. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology 138: 1736–1741, 1997. [DOI] [PubMed] [Google Scholar]
- 21.Someya S, Yamasoba T, Kujoth GC, Pugh TD, Weindruch R, Tanokura M, Prolla TA. The role of mtDNA mutations in the pathogenesis of age-related hearing loss in mice carrying a mutator DNA polymerase gamma. Neurobiol Aging 29: 1080–1092, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spelbrink JN, Toivonen JM, Hakkaart GA, Kurkela JM, Cooper HM, Lehtinen SK, Lecrenier N, Back JW, Speijer D, Foury F, Jacobs HT. In vivo functional analysis of the human mitochondrial DNA polymerase POLG expressed in cultured human cells. J Biol Chem 275: 24818–24828, 2000. [DOI] [PubMed] [Google Scholar]
- 23.Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci USA 102: 17993–17998, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429: 417–423, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, Loeb LA. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet 40: 392–394, 2008. [DOI] [PubMed] [Google Scholar]
- 26.Wallace DC A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39: 359–407, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang D, Mott JL, Chang SW, Denniger G, Feng Z, Zassenhaus HP. Construction of transgenic mice with tissue-specific acceleration of mitochondrial DNA mutagenesis. Genomics 69: 151–161, 2000. [DOI] [PubMed] [Google Scholar]
- 28.Zhang D, Mott JL, Chang SW, Stevens M, Mikolajczak P, Zassenhaus HP. Mitochondrial DNA mutations activate programmed cell survival in the mouse heart. Am J Physiol Heart Circ Physiol 288: H2476–H2483, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Zhang D, Mott JL, Farrar P, Ryerse JS, Chang SW, Stevens M, Denniger G, Zassenhaus HP. Mitochondrial DNA mutations activate the mitochondrial apoptotic pathway and cause dilated cardiomyopathy. Cardiovasc Res 57: 147–157, 2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.