
Figure 1.
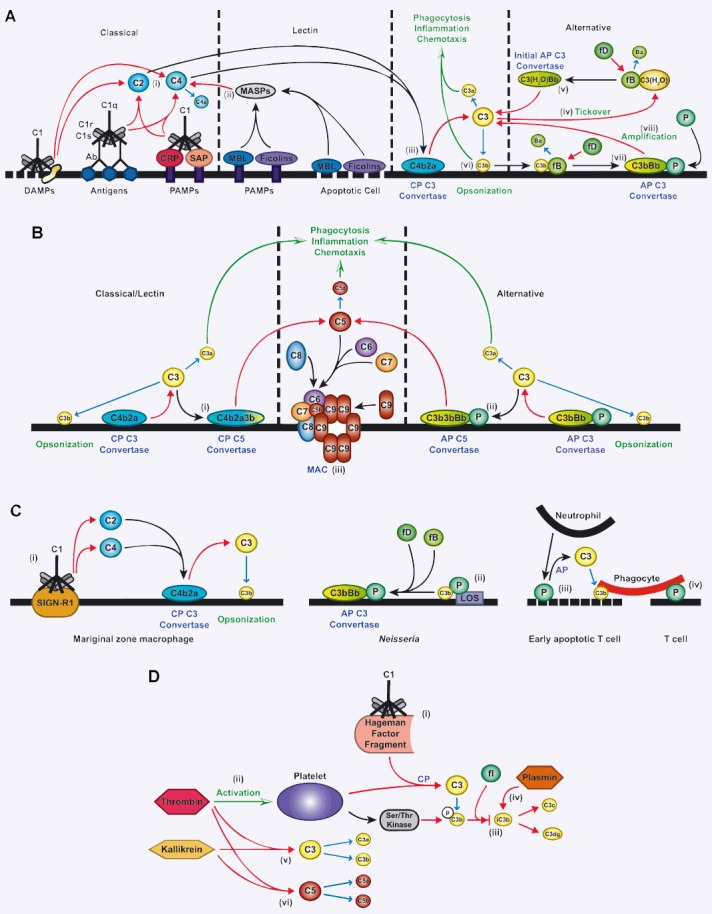
Activation of the complement system. (A) C1 can bind to various factors to initiate the classical pathway (CP). These include, most commonly antibody-antigen complexes, but also danger-associated molecular patterns (DAMPs) such as membrane fragments and proteins associated with tissue damage. C1 also binds to C-reactive protein (CRP) and serum amyloid protein (SAP), which recognize pathogen-associated molecular patterns (PAMPs) present on the surfaces of many pathogens. Binding by the C1 q subunit of C1 activates C1 r and C1 s, which results in the cleavage of C2 and C4 by C1s (i). Binding of mannose-binding lectin (MBL) or ficolins to PAMPs or apoptotic host cells activates MBL-associated serine proteases (MASPs), which cleave C4 (ii). The result of activation through either pathway is that C4a is released and C2a and C4b form the CP C3 convertase on the surface of the pathogen or apoptotic cell (iii), resulting in cleavage of C3 into C3a and C3b fragments. The alternative pathway (AP) can be initiated by spontaneous hydrolysis of C3 (‘tickover’) to form C3(H20) (iv). C3(H20) binds to factor B (fB), which is cleaved by factor D (fD) into Ba and Bb fragments, resulting in formation of the initial AP C3 convertase (v). Like the CP convertase, the AP C3 convertase cleaves C3 into C3a and C3b. The anaphylatoxin C3a induces chemotaxis and inflammation, while some C3b binds to the cell surface (opsonization) (vi), which promotes phagocytosis by CR3-bearing cells. Surface-bound C3b binds to factor B, and the resulting complex is cleaved by factor D to form the AP C3 convertase (vii). This convertase is stabilized by the binding of properdin (P). Through an amplification loop, the AP C3 convertase cleaves more C3 to augment complement activation induced by the classical or lectin pathways (viii). (B) After C3 is cleaved by the CP C3 convertase, C3b binds to the cell surface but also can bind to the C4b2a complex to form the CP C5 convertase (i). Similarly, C3b resulting from activity of the AP C3 convertase can bind to C3bBbP to form the AP C5 convertase (ii). These convertases cleave C5, leading to the generation of C5a, which acts similarly to C3a to promote inflammation and chemotaxis, and C5b. C5b is bound by C6 and C7, which can insert into the cell membrane and bind C8. One or multiple C9 molecules then bind, resulting in formation of the membrane attack complex (MAC) (iii). (C) C1 can bind to SIGN-R1 on marginal zone macrophages to result in formation of the CP C3 convertase, and subsequent C3 cleavage (i). P can bind directly to C3b through interaction with Neisseria lipo-oligosaccharide (LOS) and, in the presence of fB and fD, can form the AP C3 convertase (ii). P from neutrophils can bind to early apoptotic T cells to activate the AP and initiate C3b deposition, facilitating phagocytosis (iii). Phagocytosis can also be promoted through a direct interaction of P with phagocytes (without complement activation) when it binds to T cells (iv). (D) C1 can interact with the Hageman factor fragment, which results in complement activation through the CP (i). Thrombin induces C3 cleavage through the CP through the activation of platelets (ii). Activated platelets release a serine/threonine (ser/thr) kinase that can phosphorylate (p) C3b to block its cleavage into iC3b by Factor I (fl) (iii). Plasmin can directly cleave iC3b into C3c and C3dg (iv). Similarly, kallikrein and thrombin can directly cleave both C3 (v) and C5 (vi) to generate cleavage products.