

Abstract
Cerebral white matter injury during prenatal maternal infection characterized as periventricular leukomalacia (PVL) is the main substrate for cerebral palsy (CP) in premature infants. Previously, we reported that maternal LPS exposure causes oligodendrocyte (OL)-injury/hypomyelination in the developing brain which can be attenuated by an antioxidant agent, N-acetyl cysteine (NAC). Herein, we elucidated the role of peroxisomes in LPS-induced neuroinflammation and cerebral white matter injury. Peroxisomes are important for detoxification of reactive oxidative species (ROS) and metabolism of myelin-lipids in OLs. Maternal LPS exposure induced selective depletion of developing OLs in the fetal brain which was associated with ROS generation, glutathione depletion and peroxisomal dysfunction. Likewise, hypomyelination in the postnatal brain was associated with decrease in peroxisomes in OLs after maternal LPS exposure. Conversely, NAC abolished these LPS-induced effects in the developing brain. CP brains imitated these changes in peroxisomal/myelin proteins in the postnatal brain after maternal LPS exposure. In vitro studies revealed that pro-inflammatory cytokines cause OL-injury via peroxisomal dysfunction and ROS generation. NAC or WY14643 (peroxisome proliferators activated receptor (PPAR)-α agonist) reverses these effects of proinflammatory cytokines in the wild-type OLs, but not in PPAR-α (−/−) OLs. Similarly treated B12 oligodenroglial cells co-transfected with PPAR-α siRNAs/pTK-PPREx3-Luc, and LPS exposed PPAR-α(−/−) pregnant mice treated with NAC or WY14643 further suggested that PPAR-α activity mediates NAC-induced protective effects. Collectively, these data provide unprecedented evidence that LPS-induced peroxisomal dysfunction exacerbates cerebral white matter injury and NAC-induced protection is via a PPAR-α dependent mechanism expands therapeutic avenues for PVL and related demyelinating diseases.
Keywords: Lipopolysaccharide, periventricular leukomalacia, cerebral white matter injury, cerebral palsy, peroxisome proliferators-activated receptor-α, N-acetyl cysteine
INTRODUCTION
Periventricular leukomalacia (PVL) lesions result from white matter injury which can initiate cerebral palsy (CP) in premature infants (Bell and Hallenbeck, 2002). PVL consists of two main components i.e., focal and diffuse (Back and Rivkees, 2004). Hypoxia-ischemia is known to be one of the major causes of PVL, but maternal infection and inflammation are suggested as other important factors involved in the development of such lesions (Dammann and Leviton, 1997; Back and Rivkees, 2004). Systemic inflammation following maternal infection may have deleterious effects on the fetus. Excessive secretion of pro-inflammatory cytokines is known to be toxic to the developing fetal brain (Dammann and Leviton, 2000; Bell et al., 2004), which leads to astrogliosis affecting the maturation of myelin-forming oligodendrocytes (OLs) (Leviton and Gilles, 1996; Back et al., 2002) and contributes to neonatal brain injury and later developmental disability (Dammann and Leviton, 2000). Loss of developing oligodendrocytes (OLs) is the hallmark of the diffuse type of human PVL secondary to the production of reactive oxygen species (ROS) and nitrogen species, glutamate, cytokines, and adenosine in perinatal insults (Kinney and Back, 1998). The understanding of mechanisms involved in the increased vulnerability of developing OLs to various insults may be exploited for the development of strategies for neuroprotection against the effects of such insults. One of the popular therapeutic approaches for the inhibition of oxidant mediated injury is the use of glutathione-modulating agents such as N-acetyl cysteine (NAC). NAC is probably one of the most widely investigated agents that serves as a precursor of glutathione and has both antioxidant and anti-inflammatory properties. NAC has been shown to attenuate inflammation in various disease models such as ischemia-reperfusion injury in brain (Sekhon et al., 2003; Khan et al., 2004), lethal endotoxemia (Victor et al., 2003), animal model of multiple sclerosis (Lehmann et al., 1994; Stanislaus et al., 2005), and hypoxic-ischemic brain injury in neonatal brains (Jatana et al., 2006; Wang et al., 2007). In addition, we and others have reported the attenuation of brain white matter injury by NAC in the systemic maternal infection model of PVL (Cai et al., 2000; Paintlia et al., 2004).
Animal models are useful for understanding the mechanism of developing cerebral white matter injury underlying CP. Nevertheless, no model reliably replicates all aspect of the human disease. Some groups use the hypoxia/ischemia model (Back et al., 2002; McQuillen et al., 2003), whereas others use models of neuroinflammation (Cai et al., 2000; Bell and Hallenbeck, 2002; Paintlia et al., 2004), reflecting the two hypotheses regarding the etiology of PVL. In the last decade, several animal models have been developed which involve maternal intraperitoneal (ip) administration of lipopolysaccharide (LPS; the cell wall component of Gram-negative bacteria) to pregnant mothers (Wang et al., 2006). In adults, LPS exposure (ip) is known to induce cerebral effects (pyrogenic and inflammatory) via activation of liver kupfer cells and macrophages, including interaction of cytokines with vagus nerve (Perry et al., 2003). LPS administration (ip) at mid-gestation causes fetal death in a dose dependent manner via increase in COX-2 expression and production of prostanoids in the decidua (Silver et al., 1995). Also, maternal injection of LPS at embryonic gestation day 18 (E18) and E19 increases pro-inflammatory cytokine (TNF-α and IL-1β) expression in the maternal and fetal compartments including fetal brain (Cai et al., 2000; Paintlia et al., 2004; Wang et al., 2006). Secretion of proinflammatory cytokines in the amniotic fluid arising from intrauterine infection is shown to be associated with brain white matter injury (Yoon et al., 1997).
LPS induced effects are known to cause peroxisomal dysfunction (β-oxidation inhibition) in the liver when administered (ip) to adult animals (Khan et al., 2000). Peroxisomes are ubiquitous, metabolically active sub-cellular organelles responsible for metabolism of myelin lipids (i.e., plasmalogens, cholesterol and VLC-fatty acids) (Singh, 1997) and detoxification of ROS (Schrader and Fahimi, 2004). Interestingly, accumulation of very long chain fatty acids (VLC-fatty acids) due to impaired peroxisomal β-oxidation is reported to be associated with the induction of neuroinflammation/demyelination in human brain disease i.e., human X-adrenoleukodystrophy (X-ALD), hereditary peroxisomal disorder (Paintlia et al., 2003). Also, peroxisomal dysfunction has been linked with ROS generation in apoptosis (Baumgart et al., 2001), aging (Lavrovsky et al., 2000), and ischemia/reperfusion injury (Deplanque et al., 2003; Schrader and Fahimi, 2006) including pro-inflammatory disease processes (Poynter and Daynes, 1998). Peroxisomal dysfunctions manifested in inherited metabolic diseases due to one or more peroxisomal proteins deficiencies are thought to be responsible for ~ 18 unique fatal neurological disorders (Moser, 1996; Moser, 1999). Biochemical inhibition of VLC-fatty acid β-oxidation by thioridazine causing peroxisomal dysfunction has been shown to affect the rate of myelination in the developing brain (Van den Branden et al., 1989; Van den Branden et al., 1990). Moreover, neuroinflammation has been shown to cause peroxisomal dysfunction in the CNS of an animal model of multiple sclerosis (Singh et al., 2004). Recently we and others, using a systemic maternal infection PVL model, reported that LPS-induces inflammation and OL-injury in the developing rat brain which can be attenuated by NAC (Cai et al., 2000; Paintlia et al., 2004). The present study provides novel and unprecedented evidence that LPS-induced peroxisomal dysfunction depletes OLs and exacerbates cerebral white matter injury (hypomyelination) in premature infants and that this is related to prenatal maternal infections.
MATERIALS AND METHODS
Chemicals & reagents
LPS (Escherichia coli, serotype 055:B5), NAC, WY14643, bovine serum albumin (BSA) and other chemicals were purchased from Sigma-Aldrich. Rabbit anti-glial fibrillary acidic protein (GFAP) antibodies were purchased from Abcam (Cambridge, MA). Mouse and rabbit anti-NG2, -O4, -O1, -A2B5 (clone A2B5, 105), -integrin alphaM OX42 (microglia), -neuron specific enolase and –anti-oligodendrocyte (clone NS-1; RIP) antibodies were purchased from Chemicon International. Anti-myelin basic protein (MBP clone 1; 129–138) was purchased from Serotec. Anti-4-HNE antibodies were purchased from OxisResearch™. Anti-PMP70 and -anti-acyl CoA: dihydroxyacetonephosphate-acyltransferase (DHAP-AT) antibodies were developed as described earlier (Khan et al., 2005). Anti-peroxisome proliferators-proliferated receptor (PPAR)-α antibody was purchased from Santa Cruz. Goat anti-mouse or anti-rabbit IgG conjugated with FITC or Texas Red antibodies were purchased from Vector Lab.
Human brain tissues
Frozen cerebral brain tissue slices from six-human CP patients and age-matched controls were obtained from the institute of Brain and Tissue for Development Disorders at the University of Maryland (Baltimore, MD). Brain tissue slices were obtained from deceased CP patients (1–3 years-of-age) including age-matched controls, which were collected at 8–11 h postmortem (indicated in patient records). Cerebral brain slices were examined by a pathologist, and tissue was cut from the edge of cortical white matter lesion for purification of total RNA to determine myelin and peroxisomal protein transcripts.
Animals
All experiments were performed according to the NIH Guidelines for the Care and Use of Laboratory Animals (NIH publication #80-23) and were approved by the Medical University of South Carolina Animal Care and Use Committee. Animals were provided with food and water ad libitum. Timed-pregnant Sprague-Dawley (SD) rats at E16 used in this study was obtained from Harlan Laboratory (Harlan, IN). Animals were divided into three groups: 1) LPS group—received nonpyrogenic PBS (vehicle) injection, 2 h prior to systemic maternal injection of LPS (0.7 mg/kg, ip) at E18; 2) NAC + LPS group—received NAC (50 mg/kg in PBS, ip), 2 h prior to systemic maternal injection of LPS at E18; and 3) control group—received either PBS or NAC (50 mg/kg in PBS, ip) at E18 (Paintlia et al., 2004). Fetuses were removed at two time points (24 and 48 h) post-LPS administration to collect fetal brain tissues. Likewise, offspring from each group were sacrificed at specific postnatal days (PNDs) i.e., 9, 16, 23, and 30, to collect brain tissues. PPAR-α (−/−) mice (129S4/SvJAe-Pparatm1Gonz/j strain) with matching wild-type mice strain were purchased from Jackson Laboratory. PPAR-α (−/−) and wild-type mice were exposed to LPS (1 mg/kg) at E18 and pretreated with NAC (50 mg/kg) or PPAR-α agonist, WY14643 (1 mg/kg). Fetuses were removed at two time points (24 h and 48 h) following LPS administration. Tissues were either fixed in 10 % buffered neutral formalin immediately for immunohistochemistry or frozen in the liquid nitrogen and then stored at −70 °C until later use.
FACS analysis
Fetal brains were removed from 15 fetuses/group and hemispheres were separated, freed from meninges, and placed in 15 mL of ice-cold PBS containing 0.2% BSA. The minced tissue was treated with trypsin-EDTA at 37 °C for 30 min under agitation at 50 × g and passed through nylon mesh. Subsequently, cells were collected by centrifugation, washed twice in PBS, and loaded on Percoll gradient. After centrifugation at 20,000g for 20 min at 4°C, cells present below the myelin layer down to the RBC band were collected. Cells (1 × 106) were washed twice and suspended in PBS containing 3% BSA followed by incubation with 10 µg/ml non-immune mouse IgG for 15 min. After washing, cells were incubated with 2 µg/ml of anti-O4, -O1, -GFAP and -neuron specific enolase antibodies diluted 1:100 in PBS containing 3% BSA at 4 °C for 30 min. After washing, cells were incubated with FITC-conjugated anti-mouse IgM diluted at 1:200. Cells were washed before analysis and measured in a FL-1 channel (530 ± 15 nm band pass filter) on a FACS caliber flow cytometer (BD Biosciences) operating with Cell Quest™ software. Dead cells and debris were excluded from the analysis by gating live cells from size/structure density plots, with at least 10,000 events gated. For in vitro experiments, purified primary developing OLs were blocked in PBS containing 3% BSA followed by incubation with anti-MBP antibodies with respective secondary antibodies. Finally, cells were analyzed by FACS analysis as described above.
RNA purification and quantitative real-time-PCR analysis
Total RNA from brain tissues or cells was purified using TRIZOL reagent (Gibco BRL) as described earlier (Paintlia et al., 2003). Single-stranded cDNA was synthesized from total RNA by using the superscript pre-amplification system for first-strand cDNA synthesis and quantitative real-time-PCR was performed as described earlier (Paintlia et al., 2003). The primer sets (IDT, Coralville, IA, USA) used was as follows; catalase (CAT; Accession No, BC081853), forward: 5′-gagaggaaacgcctgtgtgag-3′, reverse: 5′-aagagcctggactcgggccc-3′; rat DHAP-AT (Accession No. AF218826), forward; 5′-catcatcctcacagacaaaggg-3′, reverse: 5′-cttcatgcaagaggcatttgga-3′; acyl CoA oxidase (AOX; Accession No. BC085743), forward: 5′-gaggtccatgaatcttaccaca-3′, reverse: 5′-gtgagtagaggaagaagttttctgtg-3′, rat and human PPAR-α (Accession Nos. NM_013196 and BC000052), forward; 5′-tcgggatgtcacacaatgcaatcc-3′, reverse: 5′-cgtgttcacaggtaaggatttctgcc-3′; rat and human MBP (Accession No. AF439750), forward: 5′-aatcggctcacaagggattcaagg-3′ and, reverse primer, 5′-gctgtctcttcctcccagcttaaa-3′; rat PMP-70 (Accession No. D90038), forward: 5’-acaccacgagtactacctgcacat-3’, reverse: 5’-ccgaactcaactgtctcttctgtg-3’; rat peroxisomal biogenesis factor 6 (Pex6; Accession No. NM_057125), forward: 5’-tgcagcctcacctttctcagtgta-3’. Reverse: 5’- cctggcaaacacttcccgaacatt-3’; Human PMP-70 (Accession No. BC068509), forward: 5’-ggttggcatcactctcttcactgt-3’, reverse: 5’-tcatagttgcctctgccatcca -3’; Human DHAP-AT (Accession No. AF218229), forward; 5′-tcactctcctcatgtgctcagctt-3′, reverse: 5′-atcctctttcctgaaccctggtgt -3′; human and rat GAPDH (Accession No. DQ403053), forward: 5′-cctacccccaatgtatccgttgtg-3′, reverse: 5′-ggaggaatgggagttgctgttgaa-3′; and 18S rRNA (for rat, human and mouse), forward: 5’-ccagagcgaaagcatttgccaaga-3’ and reverse: 5’-tcggcatcgtttatggtcggaact-3’. Thermal cycling conditions were as follows: activation of iTaq DNA polymerase (Bio-Rad) at 95 °C for 10 min, followed by 35 cycles of amplification at 95 °C for 30 s and 55–60 °C for 1 min. The detection of threshold was set above the mean baseline fluorescence determined by the first 20 cycles. Amplification reactions in which the fluorescence increased above the threshold were defined as positive. In addition, dissociation or temperature melting curve was run each time for confirmations of specific amplification. The quantity of target gene expression was normalized to the corresponding GAPDH mRNA quantity in respective test samples. Similar results were obtained when normalized with reference genes such as GAPDH or 18S rRNA.
Immunohistochemistry
Studies were performed using standard protocol as described previously (Paintlia et al., 2005). Briefly, tissue sections were incubated with anti-MBP, -DHAP-AT, -RIP, -GFAP, -PMP70 and -PPAR-α antibodies at 1:200 for overnight at 4°C for both single and double labeling. Then respective secondary IgG antibodies conjugated with FITC or Texas red were used at dilution 1: 200. Sections were also incubated with Texas red or FITC-conjugated IgG without primary antibody as a negative control. After thorough washings, slides were mounted and examined under immunofluorescence microscopy (Olympus BX-60) with an Olympus digital camera (Optronics, Goleta, CA) using a dual band-pass filter as described earlier (Paintlia et al., 2004). For 4-HNE immunostaining, sections were immunostained with anti-4-HNE antibodies (1:50 dilution) individually or double labeled with NG2 according to a standard protocol as described earlier (Paintlia et al., 2005). Immunofluorescence intensities were determined using Image Pro-Plus software as described earlier (Paintlia et al., 2005).
Assay for determination of peroxisomal enzyme activities
Enzymatic activities of long and VLC acyl-CoA synthetases were determined using [1-14C]-labeled fatty acids as substrate (C24:0, lignoceric acid or C16:0, palmitic acid (ARC Inc., St. Louis, MO); 150,000 dpm suspended in 0.25 mg of α–cyclodextrin/ assay). [1-14C]-labeled lignoceric acid was synthesized as described elsewhere (Sandhir et al., 1998). The reaction mixture was incubated 37 °C for 5 min and stopped with 1.25 ml of Dole’s solution (isopropanol: n-heptane: 1N sulfuric acid 40:10:1 v/v/v). After addition of 0.45 ml of water, the mixture was washed 3 times with 0.9 ml n-heptane and the radioactivity present in the aqueous solution was measure in a liquid scintillation counter (Beckman) (Lazo et al., 1991). The peroxisomal β-oxidation of fatty acids to acetate was determined using [1-14C]-labeled fatty acids as substrate (C24:0, lignoceric acid or C16:0, palmitic acid); 150,000 dpm suspended in 0.25 mg of α-cyclodextrin/assay) (Lazo et al., 1991). The reaction mixture was incubated at 37 °C for 1 h, and stopped with 0.625 ml of 1M KOH in methanol. The methanolic solution was then incubated at 60 °C for 1 h, neutralized with 0.125 ml of 6 N HCl and partitioned with 1.25 ml of chloroform. The amount of radioactivity in the aqueous phase represents the amount of [1-14C]-labeled fatty acid oxidized to acetate.
Immunoblot analysis
Tissues were homogenized in ice-cold lysis buffer containing protease inhibitors and protein was estimated using Bradford reagent (Bio-Rad, Hercules, CA) and separated by SDS-PAGE followed by immunoblotting as described earlier (Paintlia et al., 2005). Immunoreactivity was detected using the ECL detection method according to the manufacturer’s instructions (Amersham Biosciences) with subsequent exposure of the membrane to X-ray films.
Lipid peroxides Assay
Lipid peroxidation in the homogenate of fetal brains was determined by measuring malondialdehyde (MDA), as an indicator of lipid peroxidation, using Bioxytech ® MDA-586 kit (OxisResearch™, Portland, OR). Results were expressed as nano mole/mg of protein.
Determination of intracellular reduced GSH level
Reduced glutathione (GSH) levels were measured in fetal brain homogenates using a glutathione detection kit (Chemicon International) per product manual instructions. Data are presented as nano mole /mg of protein.
Generation of brain mixed cultures, generation of developing OLs and treatments
Brain mixed glial cultures were generated from PPAR-α (−/−) and wild-type mice (Jackson ‘s Lab) at PND 1–2 brains as described earlier for rats (Paintlia et al., 2005). For purification of OL progenitors, mixed glial cell cultures were shaken for 30 min at 200 × g at 37 °C to remove microglia, followed by further shaking for 8 h to collect OL progenitors. Then supernatants containing OL progenitors were centrifuged and plated in 100-mm plates after suspending them in fresh media. Cells were incubated for 30 min at 37 °C to remove remaining microglia and unattached OL progenitors were transferred to new plates. Cultures were examined by FACS using specific markers for different cell types. Ninety-seven percent of OL progenitors were A2B5+ containing approximately 3% of both OX42+ and GFAP+ cells. Developing OLs were generated from OL progenitors cultured in defined media (DMEM with 10 % FBS and supplemented with growth factors i.e., PDGF-α and FGF-2; 10 ng/ml each) for 96 h to enrich O4 population (confirmed by FACS analysis). Mixed glial cultures and developing OLs were treated in 100-mm plates at density 1 × 105 cells/ml.
Plasmids and small interfering RNA (siRNA) oligonucleotides and transfection
B12 oligodendroglial cells (kindly provided by Dr. D. Schubert from the Salk Institute, La Jolla, CA) were cultured in DMEM supplemented with 10 % FBS were cultured in 6-well plates at density of 1 × 105 cells/ml. The source of peroxisome proliferators-response element-containing reporter plasmids, pTK-PPREx3-Luc used in the study are the same as previously described (Paintlia et al., 2006). A pool of three siRNAs duplexes for PPAR-α i.e., nucleotide 940: ggaugucacacaaugcaau, nucleotide 1535: gaaguucaaugccuuagaa and nucleotide 2725: cagcuccuuugauaugaua, and respective scramble siRNA (sequences were not disclosed by the company) including transfection reagent and media, were purchased from Santa Cruz Biotechnology, Inc. The protocol for transient co-transfection used in these studies was described in the product manual.
VLC-fatty acid analysis
Fatty acid methyl ester (FAME) from astrocytes or primary OL progenitors was prepared and analyzed by gas chromatography as described earlier (Paintlia et al., 2005).
Measurement of ROS
ROS was determined using the membrane permeable dye 6-carboxy 2’,7’-dichlorodihydrofluorescein diacetate (DCFH2-DA) in serum-free medium as described earlier (Khan et al., 2005).
Luciferase reporter Assay
Luciferase reporter assay were performed with a luciferase assay reagent kit (Roche) as described earlier (Paintlia et al., 2006). The protein concentration was determined with the Bradford protein assay (Bio-Rad; Hurcules, CA) and used to normalize luciferase enzyme activities in samples.
Statistical analysis
Using the student’s unpaired t-test and ANOVA (Student-Newman-Keuls to compare all pairs of columns) p values were determined for the respective experiment from three identical experiments using GraphPad software (GraphPad Software Inc. San Diego, CA USA). The criterion for statistical significance was p<0.05.
RESULTS
Maternal LPS exposure causes targeted injury of developing OLs and leading to cerebral white matter injury (hypomyelination) in the postnatal brain
We have previously reported that a single systemic maternal LPS injection at E18 causes OLs injury and hypomyelination in the rat cerebral white matter (Paintlia et al., 2004). Next, to determine the vulnerability of OL lineages responsible for cerebral white matter injury, we first determined the fate of developing OLs in the fetal brain after maternal LPS exposure. LPS significantly reduced developing OLs (O4+ were reduced by ~ 60% and O1+ were reduced by ~ 80%) in the fetal brain after 48 h (E20) of LPS exposure compared to controls (Fig. 1 and Table 1). In contrast, NAC attenuated this LPS-induced depletion of developing OLs (Fig. 1 and Table 1). There was no observed loss of astrocytes or neurons in the fetal brain under those conditions (Table 1).
Figure 1. Maternal LPS administration at E18 causes the loss of developing OLs in the rat fetal brain which can be blocked by NAC.
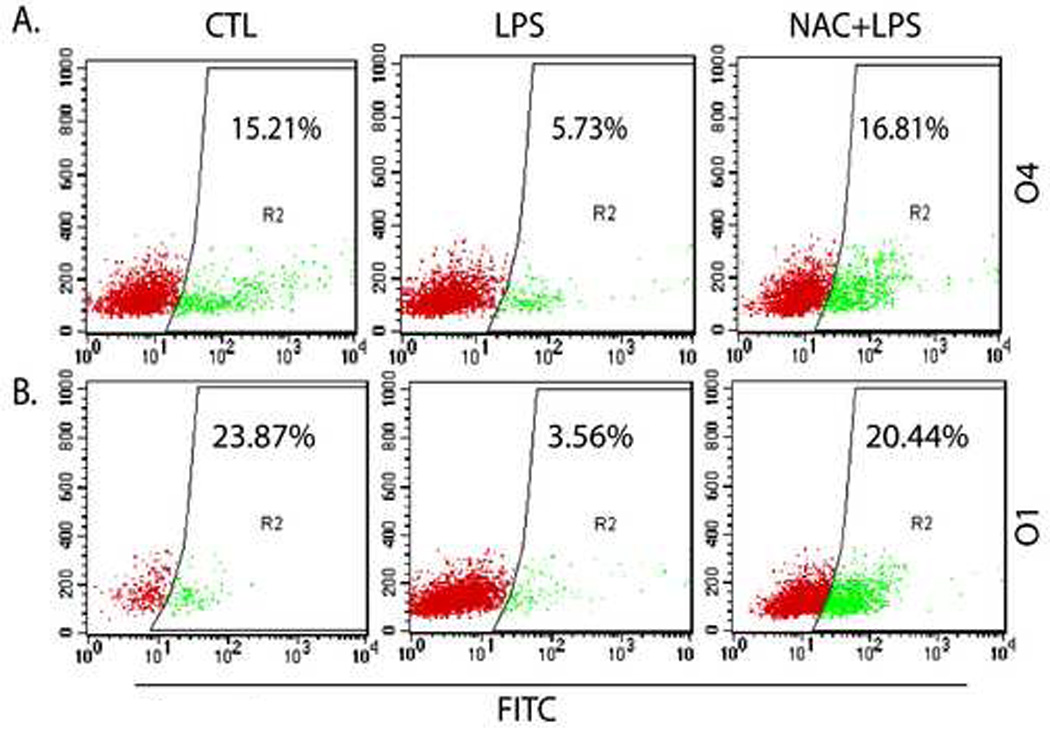
An injection of LPS (0.7 mg/kg; ip) was administered to SD rat female pregnant mothers on E18 in the LPS group, whereas NAC + LPS group animals were treated with NAC (50 mg/kg) prior to LPS administration. Animals were sacrificed at E20 (48 h post LPS-administration) to remove fetuses from each group including respective controls. Brains were pooled from similarly treated fetuses and processed for purification of brain glial cells as described under Experimental Procedures followed by FACS analysis. Representative scatter plots depict the percentage of O4+ (A) and O1+ (B) population of developing OLs in the fetal brains from each group. Scatter plots are the representative of three identical experiments run each time in duplicate.
Table 1.
Survival of Rat brain glial cells in the fetal brain at E20 after 48 h of LPS exposure to pregnant mothers at E18 and protection by NAC treatment, 2 h prior to LPS administration
| Groups | E20 | |||
|---|---|---|---|---|
| Cell Type | O4 | O1 | AS | N |
| CTL | 17.54 ± 0.02*** | 21.76 ± 0.02*** | 39.72 ± 0.23NS | 13.71 ± 0.12NS |
| LPS | 6.73 ± 0.01 | 4.06 ± 0.005 | 46.47 ± 0.456 | 12.57 ± 0.001 |
| NAC + LPS | 18.14 ± 0.015*** | 21.38 ± 0.01*** | 34.60 ± 0.265NS | 14.93 ± 0.23NS |
O4 and O1 (Developing OLs), AS; Astrocytes, N; Neuron Data are presented as Mean ± SD from 3 identical Experiments.
Statistical significance indicated as
p<0.001 and NS (non-significant) versus LPS
Next we observed myelination in the postnatal brain after maternal LPS exposure. As expected, MBP (principal myelin structural protein) transcripts were significantly decreased after maternal LPS exposure in the corpus callosum (Fig. 2A) and MBP protein was decreased (~10%) both in corpus-callosum and cingulum of the postnatal brain (Fig. 2B–D) at PNDs 23 and 30 compared to controls. Conversely, NAC attenuated this LPS-induced decrease in MBP in similar regions of postnatal brain (Fig. 2A–D). Myelination in the postnatal brain of NAC-treated controls was not changed (data not shown). Also, NAC enabled 100% fetal survival when administered simultaneously or 2 h prior to LPS administration. In therapeutic window evaluations of NAC therapy, fetal survival was reduced to 83.30%, 79.20% and 63.30%, when NAC was administered at 1 h, 4 h and 24 h, respectively, after maternal LPS administration. Thus, maternal LPS exposure at E18 significantly depleted developing OLs, thereby resulting in hypomyelination in the postnatal brain.
Figure 2. Systemic maternal injection of LPS at E18 causes hypomyelination in the postnatal rat brain which is attenuated by NAC.

Brains were collected from rat pups in the LPS, NAC + LPS, and control groups at two time points; PND 23 and 30. Brains were fixed for immunohistochemistry studies, or corpus callosum of the brain tissue was removed and pooled from similarly treated groups for real-time PCR analysis. Plot depicts the level of MBP transcripts in the corpus callosum of the brain (A). Plot depicts the immunofluorescence intensity (IF) presented as arbitrary units (A.U.) for MBP in the different regions of the brain at PND 23 (B). Representative sections show MBP immunostaining in the corpus callosum (C), cingulum (D) of the brain at PND 23: left panel; immunolabeled with the structural myelin protein MBP (red) and right panel; MBP positive OL with nuclei stained blue with Hoechst. Plot data are expressed as Mean ± SD from 3 independent experiments. Statistical significance is indicated as ***p<0.001 versus the LPS group. Bars ; 200 µM at original magnifications 400x.
Maternal LPS exposure at E18 induces oxidative-stress in the fetal brain
Next, to better understand the mechanism of OL depletion by maternal LPS exposure, we first measured ROS in the fetal brain which could be responsible for developing OL depletion. We measured oxidative stress markers (MDA, malondialdehyde and 4-HNE, 4-hydroxynonenal). As expected, MDA and 4-HNE were increased (Fig. 3A–C) in the fetal brain after maternal LPS exposure compared to controls. Furthermore, double-immunostaining revealed that OL-progenitors (NG2+) were 4-HNE+ in the fetal brain after maternal LPS exposure (Fig. 3D). In contrast, NG2+ cells were protected against LPS-induced ROS generation in the fetal brain by NAC (Fig. 3A–C). This increase in MDA or 4-HNE coincided with the depletion of reduced-GSH in the fetal brain after LPS exposure compared to controls at both E19 and E20 (Fig. 3E and F). NAC pretreatment replenished reduced-GSH and attenuated LPS-induced ROS generation in the fetal brain (Fig. 3E and F). These data suggest that exposure of pregnant mothers to LPS causes the depletion of reduced-GSH and induces ROS generation resulting in the targeted depletion of developing OLs in the fetal brain.
Figure 3. Systemic maternal injection of LPS at E18 induces oxidative stress and causes reduced-GSH depletion in the rat fetal brain which can be blocked by NAC pretreatment.

An injection of LPS (0.7 mg/kg; ip) was administered to SD rat female pregnant mothers on E18 in the LPS group, whereas NAC + LPS group animals were treated with NAC (50 mg/kg) prior to LPS administration. Animals were sacrificed at both E19 and E20 i.e., 24 h and 48 h LPS administration, respectively, to remove fetuses from each group including respective controls. Brains were pooled from similarly treated fetuses and homogenized for analysis. Plot depicts MDA (n mole/mg) of protein at both E19 and E20 in the each group (A). Plot depicts the immunofluorescence intensity (IF) presented as arbitrary units (A.U.) for 4-HNE in the fetal brain at E20 (B). Representative sections show 4-HNE immunostaining in the fetal brain at E20 (C). Representative section shows the double-immunostaining of OL progenoitors with anti-NG2 and 4-HNE antibodies in the fetal brain section from the LPS group (D). Level of the GSH (reduced) in the fetal brain at both E19 (E) and E20 (F) in each group. Plot data are expressed as Mean ± SD from 3 identical experiments. Statistical significance is indicated as **p<0.01 and ***p<0.001 versus LPS. Bars; 200 µM (C) 100 µM (D) at original magnifications 400x and 600x respectively.
Maternal LPS exposure suppresses peroxisomal proliferation/function in the fetal brain
Excessive generation of ROS is known to affect peroxisomal function in the cell (Schrader and Fahimi, 2006), so we investigated peroxisomal proliferation/function in the fetal brain after maternal LPS exposure. Surprisingly, peroxisomal proliferation was significantly reduced in the fetal brain 24 h (E19) and 48 h (E20) post-LPS administration compared to controls (Table 2) as indicated by decreases in transcripts for both peroxisomal enzymes (i.e., DHAP-AT, CAT and AOX), and proteins (i.e., PMP70 and Pex6). On the other hand, NAC pretreatment blocked this LPS-induced decrease in peroxisomal proliferation in the fetal brain (Table 2). Western blot also showed a corresponding decrease in CAT, PMP70, DHAP-AT, and thiolase proteins in the fetal brain 48 h (E20) after maternal LPS exposure compared to control, and these decreases were attenuated by NAC (Fig. 4A). Immunohistochemistry studies further revealed that maternal LPS exposure causes the targeted depletion of peroxisomes in OL-progenitors as indicated by a decrease in PMP70 puncta in NG2+ cells in the fetal brain (Fig. 4B). In contrast, no change in the PMP70 puncta was observed in neurons (neuron specific enolase+) and astrocytes (GFAP+) in the postnatal brain after maternal LPS exposure (Fig. 4C and D). In addition, GFAP immunostaining was significantly elevated (indicator of reactive astrogliosis) with a corresponding increase in PMP70 puncta in the postnatal brain after maternal LPS exposure compared with controls (Fig. 4D).
Table 2.
Level of peroxisomal proteins transcripts in the rat fetal brain at E19 and E20 after 24 h and 48 h of LPS administration at E18 or treatment with NAC, 2 h prior to LPS administration.
| 24 h (E19) | 48 h (E20) | |
|---|---|---|
|
Peroxisome Enzymes: | ||
| DHAP-AT | ||
| CTL | 34.4 ± 2.61*** | 46.4 ± 5.01*** |
| LPS | 17.21 ± 2.06 | 28.48 ± 0.19 |
| NAC+LPS | 25.4 ± 3.1** | 39.1 ± 3.16** |
| CAT | ||
| CTL | 17.2 ± 0.13*** | 23.8 ± 0.93*** |
| LPS | 7.41 ± 0.18 | 9.14 ± 0.39 |
| NAC+LPS | 11.6 ± 0.22** | 16.1 ± 1.6** |
| AOX | ||
| CTL | 11.4 ± 0.33*** | 13.3 ± 1.04*** |
| LPS | 5.58 ± 0.68 | 8.78 ± 0.1 |
| NAC+LPS | 9.64 ± 1.06** | 11.6 ± 0.9** |
|
Peroxisome Proteins: | ||
| PMP70 | ||
| CTL | 14.1 ± 0.4*** | 14.2 ± 0.36*** |
| LPS | 8.19 ± 0.67 | 9.84 ± 0.5 |
| NAC+LPS | 12.4 ± 1.76** | 13 ± 1.99** |
| Pex6 | ||
| CTL | 5.22 ± 0.4*** | 5.74 ± 0.13*** |
| LPS | 2.11 ± 0.08 | 2.9 ± 0.001 |
| NAC+LPS | 4.22 ± 0.78** | 4.89 ± 0.23** |
Data are presented as Mean ± SD from three identical experiments.
Statistical significance indicated as
* p<0.05,
p<0.01 and
p<0.001 versus LPS
Figure 4. Systemic maternal injection of LPS at E18 inhibits peroxisomal proliferation/function in the rat fetal brain which is blocked by NAC.

An injection of LPS (0.7 mg/kg; ip) was administered to SD female pregnant mothers on E18 in the LPS group, whereas NAC + LPS group animals were treated with NAC (50 mg/kg) prior to LPS administration. Animals were sacrificed at E19 and E20 i.e., 24 h and 48 h post LPS-administration, respectively, to remove fetuses from each group including respective controls. Brains were pooled from similarly treated fetuses and processed for Western blot or determination of β-oxidation. Representative immunoblot depicts levels of catalase, PMP70, DHAP-AT, and thiolase including reference protein β-actin in the fetal brain at E20 (A). Representative sections depict PMP70 punctate immunostaining in NG2+ cells in the fetal brain E20 (B) and neuron-specific enolase+ neurons (C) and GFAP+ astrocytes (D) in the rat postnatal day 16. Arrowhead depicts PMP70 puncta in double labeled cells. Plots depict β-oxidation activities for VLC-fatty acids (E) and palmitic acid (F) in the fetal brain at E19 and E20. Similarly, plots depict the activation of VLC-fatty acids (G) and palmitic acid (H) in the fetal brain homogenate at E19 and E20. Data in plots are expressed as Mean ± SD from 3 identical experiments. Statistical significance is indicated as *p<0.05, **p<0.01 and ***p<0.001 versus LPS. Bars; 100 µM at original magnifications 400x .
To investigate whether reduction of peroxisomal proliferation affects peroxisomal functions, we measured peroxisomal enzyme activities in the fetal brain. β-oxidation of lignoceric acid (24:0 fatty acids) and palmitic acid (16:0 fatty acids) including acyl-CoA synthetases (which activate VLC-fatty acids to their CoA-derived products which become substrates for β-oxidation) was measured (Fig. 4B–E). Data indicate that oxidation of both fatty acids was drastically inhibited in the fetal brain after maternal LPS exposure at 24 h (E19) and 48 h (E20) (Fig. 4E and F). β-oxidation was significantly decreased to 49.0% and 52.2% at E19 and 60.5% and 14% at E20 for lignoceric and palmitic acid, respectively. Correspondingly, enzymes that activate lignoceric and palmitic acids were also inhibited in the fetal brain after maternal LPS exposure at 24 and 48 h (Fig. 4G and H): 49.1% and 45.6% at E19 and 30.8% and 22% at E20 for lignoceric and palmitic acid, respectively. NAC pretreatment attenuated the LPS-induced decrease in both β-oxidation and synthetase activities at both time points in the fetal brains (Fig. 4E–H). No effect on peroxisomal function was observed in the fetal brain from the NAC-treated controls (data not shown). Together, these data suggest that maternal LPS exposure drastically affects peroxisomal proliferation/function in the developing brain which may contribute in the development of PVL.
Maternal LPS exposure causes reduction of peroxisomes in myelinating OLs thereby astrogliosis and hypomyelination in the postnatal brain
Peroxisomes are present in virtually all cell types and are essential for detoxification of ROS and metabolism of lipids i.e., plasmalogens, cholesterol and VLC-fatty acids (Lazarow, 1995; Schrader and Fahimi, 2004) and OLs are reported to have high number of peroxisomes (Kassmann et al., 2007), compared to other brain cells. So, we next measured peroxisomal and myelin proteins in the postnatal brains after maternal LPS exposure and compared these groups with NAC pretreatment groups and controls. Interestingly, DHAP-AT (an enzyme important for synthesis of plasmalogens) transcripts were significantly decreased in the corpus callosum of developing cerebral white matter at PND 9, 16, 23, and 30 after maternal LPS exposure compared to controls (Fig. 5A). Also, reduction of DHAP-AT transcripts after maternal LPS exposure was reversed by NAC (Fig. 5A). DHAP-AT protein was also reduced in the corpus callosum of postnatal brain after maternal LPS exposure both at PND 23 and 30 which was blocked by NAC (Fig. 5B). Moreover, a significant decrease in immunofluorescence for DHAP-AT punctuate (peroxisomal) and RIP (anti-RIP antibodies stain both early and mature OLs) staining was observed in the corpus callosum after maternal LPS exposure compared to controls (Fig. 5C and D). This LPS-induced decrease in immunofluorescence for DHAP-AT and RIP was attenuated by NAC (Fig. 5C and D). Interestingly, a decrease in number of DHAP-AT+/RIP+ OLs, as shown by overlaying of sections indicates that LPS-induced effects specifically reduce the number of myelinating OLs in the postnatal brain and this could be blocked by NAC (Fig. 5D). GFAP immunofluorescence as an indicator of reactive astrogliosis, was elevated in the corpus callosum of postnatal brain after maternal LPS exposure compared to controls (Fig. 5E and F). This LPS-induced increase in astrogliosis was attenuated by NAC in the postnatal brain (Fig. 5E and F).
Figure 5. Systemic maternal injection of LPS at E18 alters the expression of DHAP-AT and GFAP in the rat postnatal brain.

Brains were collected from pups at different time points (PND 9, 16, 23 and 30) from the LPS, NAC + LPS, and control groups. Brains were fixed for immunohistochemistry studies or corpus callosum was cut and pooled from similarly treated animals for real-time PCR or Western blot analyses. Plot depicts the level of DHAP-AT transcripts in the corpus callosum of the brain at different PNDs (A). Representative Immunoblot autoradiogram (insert) and plot depict the level of DHAP-AT protein presented as arbitrary units (A.U.) in the brain at both PND 23 and 30 (B). Plot depicts levels of both RIP and DHAP-AT immunofluorescence in the corpus callosum of the brain at PND 23 (C). Representative sections show DHAP-AT immunostaining in the corpus callosum of the brain at PND 23 (D). Plot depicts level of GFAP immunofluorescence (E) and representative sections (F) depict GFAP immunostaining in the corpus callosum of the brain at PND 23. Data in plot are expressed as Mean ± SD from 3 independent experiments (A, C and E), but data in plot B represent the average mean of densitometeric analysis of two identical experiments. Statistical significance is indicated as *p<0.05 and ***p<0.001 versus LPS. Bar; 200 µM at original magnification 400x. Immunofluorescence intensity (IF) presented as arbitrary units (A.U.).
Corresponding to these, PMP70 punctate (peroxisomal) staining reveled that peroxisomes were significantly reduced within both corpus callosum and cingulum of the postnatal brain after maternal LPS exposure compared with controls (Fig. 6A and B). This LPS-induced decrease in PMP70 puncta was abolished in similar regions of the postnatal brain with NAC pretreatment (Fig. 6A and B). Immunoblotting further showed that the levels of PMP70 were relatively reduced within the corpus callosum of postnatal brain after maternal LPS exposure and not in those pretreated with NAC at both PND 23 and 30 (Fig. 6C). Consistent with these findings, PMP70 transcripts were also significantly decreased in the corpus callosum of the postnatal brain at PND 9, 16, 23, and 30 after maternal LPS exposure compared with controls and that was reversed by NAC (Fig. 6D).
Figure 6. Systemic maternal injection of LPS at E18 alters the expression of PMP70, PPAR-α and MBP in the rat postnatal brain.

Brains were collected from pups at different time points PND 9, 16, 23 and 30) from the LPS, NAC + LPS, and control groups. Brains were fixed for immunohistochemistry studies or corpus callosum of the brain was cut and pooled from similarly treated groups for real-time PCR or Western blot analyses. Representative sections demonstrate PMP70 immunostaining in the different regions of the brain at 23 PNDs (A). Plot depicts the level of PMP70 in the different regions of the brain at PND 23 (B). Representative immunoblot autoradiogram shows the quantitative expression of PMP70 presented as arbitrary units (A.U.) with respect to β-actin in the corpus callosum of the brain at PND 23 and 30 (C). Plots depict the level of PMP70 (D) and PPAR-α (E) transcript in the corpus callosum of the brain. Plot depicts both MBP and PPAR-α protein in the corpus callosum of the brain at PND 23 (F). Representative sections demonstrate double-immunostaining for MBP/PPAR-α immunofluorescence intensity (IF) presented as arbitrary units (A.U.) in the corpus callosum of the brain at PND 23 (G). Plot data are expressed as Mean ± SD from 3 identical experiments. Statistical significance is indicated as ***p<0.001 versus LPS. Bars; 200 µM at original magnification 400x.
Similarly, PPAR-α transcripts, a transcription factor for expression of peroxisomal proteins, were significantly reduced at PND 9, 16, 23, and 30 in the corpus callosum of the postnatal brain after maternal LPS exposure compared to controls and this was attenuated by NAC (Fig. 6E). Interestingly, there was a corresponding decrease in both MBP and PPAR-α immunofluorescence within the corpus callosum of postnatal brain after maternal LPS exposure compared to controls, which was reversed by NAC (Fig. 6F and G). The overlaying of sections revealed that numbers of MBP+/PPAR-α+ OLs were reduced within the corpus callosum of postnatal brain after maternal LPS exposure, which was attenuated by NAC (Fig. 6G). No such effects were observed in similar regions of the postnatal brain in NAC-treated controls (data not shown). Taken together, these data revealed that maternal LPS exposure limits the number of myelin-forming OLs and thereby hypomyelination in the postnatal brain.
Human Cerebral Palsy (CP) brain has alteration like those observed in rat postnatal brain after maternal LPS exposure
Since, PVL lesions are believed to be initiators of CP (Bell and Hallenbeck, 2002); we investigated the significance of the observed decrease in numbers of OLs with reduction of peroxisomes in the postnatal brain after maternal LPS exposure with respect to human CP. Transcripts for both peroxisomal and myelin proteins were measured in the deep cerebral white matter lesion of CP brain and compared with normal cerebral white matter from age-matched controls. Interestingly, similar to the LPS-induced reduction of peroxisomes and OLs in the postnatal brain, transcripts for DHAP-AT, PMP70, and PPAR-α were significantly reduced in the CP brain compared to controls (Fig. 7A–C). Likewise, MBP transcripts were also significantly decreased in the CP brain compared to controls (Fig. 7D). Together, these results support our findings in animals and suggest that a decrease in the number of peroxisomes is associated with OL loss and establishes a possible link between peroxisomal dysfunction and PVL pathobiology.
Figure 7. Level of transcripts for peroxisomal/myelin proteins in the cerebral white matter region of human brain from CP patients and age-matched controls.

Frozen sections of the cerebral region of the brains from three human CP patients and three age-matched controls was obtained from the institute Brain and Tissue Bank for Development Disorders at the University of Maryland (Baltimore, MD) and included post-mortem details and patient history. Tissues were processed for real-time PCR analysis and transcripts for both peroxisomal and myelin proteins were analyzed. Plots depict transcript for DHAP-AT (A), PMP70 (B), PPAR-α (C), and MBP (D) in both human CP and age-matched normal brains. Data are expressed as Mean ± SD from 3 samples in each group. Statistical significance is indicated as **p<0.01 versus CP.
Pro-inflammatory cytokines cause peroxisomal dysfunction and ROS generation in the developing OLs
Because, maternal LPS exposure at E18 induces expression of pro-inflammatory cytokines i.e., IL-1β and TNF-α in the fetal brain (Cai et al., 2000; Paintlia et al., 2004), we next investigated the effect of these pro-inflammatory cytokines on peroxisomes and ROS generation in the developing OLs and astrocytes. A cytokine mixture (Cyt-Mix; TNF-α and IL-1β) significantly reduced peroxisomal β-oxidation activity (evidenced by increase in accumulation of VLC-fatty acids) in the developing OLs compared with controls (Fig. 8A). Conversely, NAC pretreatment reversed this Cyt-Mix-induced inhibition of peroxisomal activity in the developing OLs (Fig. 8A). Moreover, similar treatment of astrocytes did not significantly reduce β-oxidation activity in astrocytes (Fig. 8B). A Cyt-Mix treatment induced a significant increase in ROS generation in the developing OLs compared with controls (Fig. 8C). NAC pretreatment reversed this Cyt-Mix induced ROS generation in the developing OLs (Fig. 8C). Of note, similar treatment of astrocytes, however, significantly changed ROS generation compared with controls, but it was not as prominent as that was observed in the developing OLs (Fig. 8D). These data support our in vivo findings that LPS-induced inflammation in the fetal brain inhibits peroxisomal activities and oxidative-stress in the developing OLs.
Figure 8. Proinflammatory cytokines induce ROS generation and inhibit peroxisomal function in the developing OLs.

Mice developing OLs and astrocytes were treated separately with a cocktail of pro-inflammatory cytokines (Cyt-Mix: TNF-α and IL-1β; 10 ng/ml each) in the presence/absence of NAC (10 mM) for 48 h followed by determination of VLC-fatty acid accumulation. Plots depict VLC-fatty acids (C26:0/22:0 ratio) in the developing OLs (A) and astrocytes (B). Plots depict ROS generation in similarly treated developing OLs (C) and astrocytes (D). Plot data are presented as Mean ± SD of three identical experiments. Statistical significance is indicated as *p<0.05, **p<0.01, ***p<0.001 and NS (non-significant) versus Cyt-Mix.
Pro-inflammatory cytokines cause inactivation of PPAR-α in the developing OLs
Next we treated developing OLs generated from wild-type and PPAR-α (−/−) mice with Cyt-Mix in the presence/absence of NAC or WY14643 (PPAR-α agonist). FACS analysis revealed that pretreatment with NAC and WY14643 protects developing wild-type OLs against Cyt-Mix cytotoxicity as indicated by increased MBP+ OLs (Fig. 9A). Conversely, effects of NAC and WY14643 were diminished in similarly treated developing PPAR-α (−/−) OLs (Fig. 9A). Likewise, Cyt-Mix treated B12 oligodendroglial cells transiently co-transfected with pTK-PPREx3-Luc and scramble siRNAs showed a decrease in luciferase activity and that was blocked by both NAC and WY14643 pretreatment (Fig. 9B). This reversal of decreased luciferase activity by NAC or WY14643 did not occur in B12 cells co-transfected with pTK-PPREx3-Luc and PPAR-α siRNA (Fig. 9B). Together, these data suggest that LPS-induced inhibition of peroxisomal proliferation in the developing OLs is mediated via inhibition of PPAR-α activity and its attenuation by NAC exhibits its mechanism of action.
Figure 9. Pro-inflammatory cytokines inhibit peroxisomal function in the developing OLs.

Plot depicts percentage of MBP positive cells in NAC (10 mM) or WY14643 (WY; 50 µM) and Cyt-Mix treated developing OLs generated from WT and PPAR-α(−/−) mice 96 h post treatment analyzed by FACS analysis (A). Plot depicts relative luciferase activity in B12 oligodendroglial cells co-transfected with pTK-PPREx3-Luc and siRNAs for PPAR-α or scramble and treated with NAC (10 mM) or WY (50 µM) prior to incubation with Cyt-Mix as described under Experimental Procedures (B). An injection of LPS (1 mg/kg; ip) was administered to PPAR-α (−/−)/wild-type pregnant mice on E18 in the LPS group. NAC + LPS and WY14643 + LPS group animals were pretreated with NAC (50 mg/kg) and WY14643 (1 mg/kg) 24 h prior to LPS administration. Animals were sacrificed at E19 (24 h) E20 (48 h) following LPS-administration to remove fetuses. Plot depicts the survival rate of fetuses in each group from wild-type (WT) and PPAR-α (−/−) mice (C). Plot data are presented as Mean ± SD of three identical experiments (B and C) and average mean of two identical experiments (A). Statistical significance is indicated as **p<0.01 and NS (non-significant) versus LPS (C). ***p< 0.001 and NS (non-significant) versus Cyt-Mix (scramble siRNA), and * p< 0.05 versus Cyt-Mix and NS versus Cyt-Mix (PPAR-α siRNA) (B).
LPS-induced fetal morbidity was severe and not abolished by NAC or WY14643 in PPAR-α (−/−) pregnant mice
To further elucidate the role of LPS-induced peroxisomal dysfunction in cerebral white matter injury, we exposed PPAR-α (−/−) and wild-type pregnant mice to LPS at E18 and compared these with NAC and WY14643 (PPAR-α agonist) pretreatment. LPS exposure worsened fetal morbidity as shown by decrease in fetal survival rate in LPS exposed PPAR-α (−/−) versus wild-type mothers (Fig. 9C). NAC pretreatment, however, protected fetuses in wild-type pregnant mothers after maternal LPS exposure at both 24 h and 48 h, but not in PPAR-α (−/−) pregnant mothers (Fig. 9C). Furthermore, WY14643 mimicked an NAC-induced effect in LPS-exposed wild-type and PPAR-α (−/−) pregnant mothers (Fig. 9C). These data support our observations that the attenuation of LPS-induced fetal demise and developing OL depletion mediated white matter injury by NAC is via a PPAR-α dependent mechanism.
DISCUSSION
Maternal LPS exposure has been shown to stimulate the secretion of pro-inflammatory cytokines i.e., TNF-α, IL-1α and IL-6 in the maternal serum and amniotic fluid of pregnant mice (Fidel et al., 1994) mimicking maternal infection (Yoon et al., 1997). Results presented here describe the role of peroxisomes in prenatal maternal infection (LPS)-induced cerebral white matter injury. Our conclusions are based upon the following observations. First, maternal LPS exposure at E18 causes ROS generation (MDA and 4-HNE) in the fetal brain thereby causing targeted injury of developing OLs with relative sparing of other glial cells (Table 1 and Fig. 1A–B and Fig. 3A–F). Second, maternal LPS exposure inhibits peroxisomal proliferation/function in the fetal brain, especially in OL-progenitors (Table 2 and Fig. 4A–E). Third, maternal LPS exposure induced effects persist in the later life of offspring as exhibited by the decrease in peroxisomal (Fig. 5A–D and Fig. 6A–G) and myelin (Fig. 2A–D) proteins in the corpus-callosum and cingulum of the postnatal brain. These LPS-induced effects in the developing rat brain were abolished by NAC pretreatment. Like the postnatal brain, alterations in peroxisomal and myelin proteins were apparent in the cerebral white matter of human CP brain (Fig. 7A–D). Proinflammatory cytokines caused peroxisomal dysfunction and induced ROS generation in the developing OL which was attenuated by NAC (Fig. 8A–D). In addition, experiments with proinflammatory cytokine-exposed and NAC- or WY14643-treated developing PPAR-α (−/−)/wild-type OLs and transiently co-transfected B12 OLs (with anti-PPAR-α SiRNA and reporter plasmids) revealed that NAC maintains peroxisomal proliferation/function in the developing OLs via a PPAR-α dependent mechanism (Fig. 9A–B). These findings were further supported by LPS exposure and NAC or WY14643 treatment of pregnant PPAR-α (−/−) and wild-type pregnant mice (Fig. 9C). These findings describe biochemical effects of maternal LPS-induced peroxisomal dysfunction and exacerbation of cerebral white matter injury via targeted depletion of developing OLs. To our knowledge, this is the first report implicating peroxisomes in prenatal maternal infection (LPS)-induced cerebral white matter injury.
The pathogenesis of PVL appears to be multi-factorial. Hypoxia/ischemia and maternal-fetal infections are two important factors. Pro-inflammatory cytokines such as IL-1 and TNF-α and brain hypoxia/ischemia are shown to be responsible for the loss of immature OLs in PVL (Levison et al., 2001; Pang et al., 2005). The majority of PVL lesions occur between 23–32 weeks post conception (Back et al., 2001; Volpe, 2001; Back, 2006), a time when the predominant population of the OL lineage consists of late OL progenitors (O4+) in the human fetal brain (Back et al., 2001). Our results showed a significant decrease in O4+ (~ 60%) and O1+ (~ 80%) cells in the fetal brain at E20 after maternal LPS exposure, versus controls with a relative sparing of astrocytes and neurons (Fig. 1 and Table 1). In addition, OL progenitors were showed to have high oxidative-stress as showed by 4-HNE+/NG2+ cells in the fetal brain at E20 after LPS exposure (Fig. 3D). This suggests that LPS-induced secretion of factors by brain cells causes the targeted injury of developing OLs which is consistent with hypoxia/ischemia study models (Back et al., 2001; Levison et al., 2001). Consistent with our earlier study, which showed that an increase in PDGF-αR+/TUNEL+ OPs is corresponded with decreased O4+ and O1+ cells in the fetal brain at E20 after maternal LPS exposure (Paintlia et al., 2004). Concurrent with loss of OL progenitors, an observed 10% MBP immunostaining in the postnatal brain is probably attributed to the left over developing OLs that are not depleted by LPS maternal exposure (Fig. 2).
Several lines of evidence suggest that free radical injury to the developing OLs underlies (at least in part) the pathogenesis of PVL including hypomyelination seen in long-term survivors of preterm labor (Haynes et al., 2005). In the human CP brain, the presence of free radical injury is supported by the existence of both oxidative and nitrative stress markers of lipid peroxidation and nitrosylation/nitration, respectively (Haynes et al., 2003). LPS has been shown to cause the depletion of intracellular GSH and an increase in MDA, a byproduct of lipid peroxidation (Millan-Plano et al., 2003; Topal et al., 2004). This LPS-induced oxidative-stress and lipid peroxidation leads to the production of potentially toxic aldehydes such as 4-HNE in the cell (Siems et al., 1992). 4-HNE has been reported to cause cellular damage mainly by modification of intracellular proteins (Toyokuni et al., 1994; Uchida, 2003). In agreement with these, we observed an increase in level of MDA and 4-HNE with a corresponding decrease in reduced-GSH levels in the fetal brain after maternal LPS exposure (Fig. 3) suggesting that LPS-induced maternal pro-inflammatory cytokines or LPS itself activate microglia in the fetal brain (Haynes et al., 2005). The presence of microglia has been demonstrated in the rat brain starting from E17 or E18 (Dalmau et al., 1997). Activation of microglia in the fetal brain triggers events associated with reactive astrogliosis which depletes the number of myelin-forming OLs leading to hypomyelination, in the postnatal brain. In support to this, we observed co-localization of 4-HNE+/NG2+ in OL progenitors in the fetal brain at E20 after LPS exposure (Fig. 3C–D) which corresponds with the decrease in O4+ (late OL-progenitors) and O1+ (immature OLs) populations (Fig. 1 and Table 1) in the fetal brain. These findings suggest that LPS-induced activation of fetal brain glial cells results in ROS generation in OL-progenitors, leading to their targeted injury (Back et al., 2001; Levison et al., 2001).
Reactive astrogliosis is the hallmark of neuroinflammation induced by secretion of TNF-α and IL-1β as observed in the neonatal brain after a stab injury (Balasingam et al., 1994). Consistent with this report, we observed an increase in astrogliosis and hypomyelination in the corpus callosum at PND 23 following maternal LPS exposure (Fig. 2 and Fig. 5F). Supporting this result, clinical studies have also shown that PVL is closely associated with astrogliosis in the white matter lesions of infant brains (Deguchi et al., 1997), suggesting that astrogliosis is important in the cerebral white matter injury at the later age of offspring than previously thought: the previous hypothesis was that the vulnerability window of immature (O4+/O1−) OLs was between 23–32 weeks of gestation (Volpe, 2001; Back, 2006). Interestingly, astrogliosis in the cerebral white matter was accompanied by reduction of peroxisomes in OLs (DHAP-AT+/RIP+) in the corpus callosum of the postnatal brain (Fig. 5). DHAP-AT is an important enzyme required for synthesis of plasmalogens (Purdue et al., 1997), which are strong antioxidant phospholipids important for scavenging ROS and preventing cell membrane oxidation (Maeba and Ueta, 2003). Our study suggests that a decrease in DHAP-AT activity reduced plasmalogens, which likely increases the vulnerability of late OL-progenitors to oxidative-stress. Consequently, reduced DHAP-AT activity decreases the number of myelin-forming OLs resulting in cerebral white matter injury in premature infants. Of note, DHAP-AT deficiency has been reported to cause abnormal myelination, causing developmental delay (Sztriha et al., 1997) and growth failure in young children (Elias et al., 1998).
Interestingly, no significant loss of astrocytes and neurons was observed in the fetal brain after maternal LPS exposure (Table 1) suggesting that brain cells, but not OLs, have strong antioxidant systems. In line with these findings, in vitro studies have shown that TNF-α and IL-1β inhibit peroxisomal function and increase ROS generation in the developing OLs, while astrocytes showed modest change in peroxisomal function and ROS generation, which was attenuated by NAC (Fig. 8). Similarly, brain glial cells treated with antioxidants have less ROS generation and their cytotoxicity on OLs (Singh et al., 1998; Bahat-Stroomza et al., 2005). The observed LPS-induced ~ 60% reduction of O4+ cells in the fetal brain (Table 1) was due to inflammation-induced oxidative stress in OL-progenitors i.e., NG2+/4-HNE+ (Fig. 3D), which occur along with the depletion of MBP+ OLs in the postnatal brain at PND 23 and PND 30 (Fig. 2). At this point, it is still an open question whether peroxisomal defect occurs in the developing OLs and causes cell death or peroxisomal defect is the consequence of cell death. An indirect evidence of impaired β-oxidation and accumulation of VLC-fatty acids in brains of X-adrenoleukodystrophy and Krabbe’s patients suggested that peroxisomal dysfunctions are responsible for OL loss and demyelination (Singh, 1997; Paintlia et al., 2003; Khan et al., 2005). In addition, deficiency of peroxisomal enzyme DHAP-AT has been shown to cause abnormal myelination in youngest siblings (Sztriha et al., 1997). Recent study shows that impaired peroxisomal biogenesis in OLs is associated with axonal loss, demyelination and neuroinflammation in the adult brain suggesting that peroxisomes are vital in OL development (Kassmann et al., 2007). Future studies will be directed to distinguish between the cell death of mature OLs and OL-progenitors from peroxisomal dysfunction on cells that are not depleted by LPS exposure.
Peroxisomal dysfunction has been shown to contribute to the generation of ROS in the cell. Defects in peroxisomal functioning can exacerbate demyelination in the brain (Paintlia et al., 2003), resulting in neurological deficits in animals (Elias et al., 1998). Likewise, neuroinflammation has been shown to cause peroxisomal dysfunction and demyelination in acute experimental autoimmune encephalomyelitis model (Singh et al., 2004). Our findings suggest that LPS-induced oxidative-stress, in part, is contributed by peroxisomal dysfunction which increases the vulnerability of the developing OLs to targeted injury, perhaps due to their high lipid environment and a decrease in synthesis of plasmalogens. In addition, oxidative stress-induced decrease in GSH-peroxidase and an increase in iron release has been shown to increase the vulnerability of OLs (Hemdan and Almazan, 2007). LPS-induced oxidative-stress increases the activation of microglia and astrocytes, albeit peroxisomal functioning remains normal, likely due to the availability of strong endogenous antioxidant-systems in these cells (Min et al., 2006). Notably, we observed that LPS exposure profoundly affects both peroxisomal and mitochondrial β-oxidation in the fetal brain as shown by down-regulation of VLC acyl-CoA ligase (peroxisomes) and long-chain acyl-CoA ligase present both in peroxisomes and mitochondria, especially at E20. This is suggesting that maternal LPS exposure affects more cellular processes than just peroxisomes possibly due to the global effects on cellular functions as a result of LPS-induced neuroinflammation and ROS generation leading to mitochondrial dysfunction (Goossens et al., 1995) and apoptotic death (Fiers et al., 1995) in the fetal brain.
The major function of peroxisomes in the myelin-forming OLs is the metabolism of ROS and myelin lipids (Sztriha et al., 1997; Paintlia et al., 2003). Proliferation of peroxisomes is regulated by PPAR-α in combination with the retinoic acid receptor by acting on PPREs present in the promoter region of most of the genes of peroxisomal proteins (Qi et al., 2000). Increases in ROS generation and NF-κB activation have been shown to inhibit the expression of both AOX and PPAR-α thus peroxisomal proliferation in skeletal muscle cells which contributes to cardiac hypertrophy (Cabrero et al., 2002; Cabrero et al., 2003). In line with these findings, WY14643 has been shown to protect hippocampal neurons against β-amyloid peptide induced degenerative changes in Alzheimer disease via peroxisomal proliferation (Santos et al., 2005). Mechanistically, NAC replenishes the level of reduced-GSH and thereby provides protection by quenching ROS in the fetal brain after maternal LPS exposure. In addition, anti-inflammatory effects of NAC are attributed to the suppression of pro-inflammatory cytokine expression/release (Tsuji et al., 1999), adhesion molecule expression and activation of NF-κB in endothelial cells (Rahman et al., 1998), and activation of glial cells (Moynagh et al., 1994). The underlying detailed mechanism of NAC-induced restoration of peroxisomal proliferation/function in the developing OLs via a PPAR-α dependent mechanism is currently under investigation in our laboratory.
In summary, these studies reveal that prenatal maternal infection (LPS) induced inflammation mediated inhibition of peroxisomal dysfunction contributes significantly to OL injury and thereby exacerbates cerebral white matter injury (hypomyelination) in later life of the offspring. On the other hand, abnormal peroxisomal biogenesis in OLs has been shown to cause axonal loss, reactive astrogliosis, inflammation and demyelination in the adult brain (Kassmann et al., 2007). More detailed investigations are warranted to distinguish between OL injury due to peroxisomal dysfunction and impaired peroxisomal biogenesis, but both studies signify the role of peroxisomes in OL development. NAC pretreatment attenuates this LPS-induced cerebral white matter injury by replenishment of reduced-GSH, ROS quenching and maintenance of peroxisomal proliferation/function via a PPAR-α dependent mechanism. Together, these data show that LPS-induced peroxisomal dysfunction exacerbates cerebral white matter injury in the developing brain and suggests new therapeutic interventions to prevent the debilitating effects of prenatal maternal infections.
ACKNOWLEDGEMENT
We thank all members of our laboratory for their valuable comments and help during the course of this study. We thank especially Dr. Jennifer G. Schnellmann for critical reading of this manuscript and Ms. Joyce Brian and Ms. Carrie Barnes for their technical assistance. This study was supported in part by grants from the National Institutes of Health: NS-22576, NS-34741, NS-37766, NS-40810 and, C06 RR018823 and C06 RR015455 from the Extramural Research Facilities Program of the National Center fro Research Resources.
Abbreviations
- PVL
periventricular leukomalacia
- CP
cerebral palsy
- PPAR-α
peroxisome proliferators activated receptor-α
- OL
oligodendrocyte
- ROS
reactive oxygen species
- NAC
N-acetyl cysteine
- CAT
catalase
- AOX
acyl-CoA oxidase
- DHAP-AT
dihydroxyacetonephosphate acyltransferase
- PMP70
peroxisomal membrane protein 70
- PND
postnatal day
- Cyt-Mix
cytokine mixture and VLC-fatty acids, very long chain fatty acids.
Contributor Information
Manjeet K. Paintlia, Department of Pediatrics, Medical University of South Carolina.
Ajaib S. Paintlia, Department of Pediatrics, Medical University of South Carolina.
Miguel Contreras, Department of Pediatrics, Medical University of South Carolina
Inderjit Singh, Department of Pediatrics, Medical University of South Carolina
Avtar K. Singh, Department of Pathology and Laboratory Medicine, Ralph H. Johnson VA Medical Center, Charleston, South Carolina, USA..
REFERENCES
- Back SA. Perinatal white matter injury: the changing spectrum of pathology and emerging insights into pathogenetic mechanisms. Ment Retard Dev Disabil Res Rev. 2006;12:129–140. doi: 10.1002/mrdd.20107. [DOI] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci. 2001;21:1302–1312. doi: 10.1523/JNEUROSCI.21-04-01302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Rivkees SA. Emerging concepts in periventricular white matter injury. Semin Perinatol. 2004;28:405–414. doi: 10.1053/j.semperi.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Bahat-Stroomza M, Gilgun-Sherki Y, Offen D, Panet H, Saada A, Krool-Galron N, Barzilai A, Atlas D, Melamed E. A novel thiol antioxidant that crosses the blood brain barrier protects dopaminergic neurons in experimental models of Parkinson's disease. Eur J Neurosci. 2005;21:637–646. doi: 10.1111/j.1460-9568.2005.03889.x. [DOI] [PubMed] [Google Scholar]
- Balasingam V, Tejada-Berges T, Wright E, Bouckova R, Yong VW. Reactive astrogliosis in the neonatal mouse brain and its modulation by cytokines. J Neurosci. 1994;14:846–856. doi: 10.1523/JNEUROSCI.14-02-00846.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart E, Vanhorebeek I, Grabenbauer M, Borgers M, Declercq PE, Fahimi HD, Baes M. Mitochondrial alterations caused by defective peroxisomal biogenesis in a mouse model for Zellweger syndrome (PEX5 knockout mouse) Am J Pathol. 2001;159:1477–1494. doi: 10.1016/S0002-9440(10)62534-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell MJ, Hallenbeck JM. Effects of intrauterine inflammation on developing rat brain. J Neurosci Res. 2002;70:570–579. doi: 10.1002/jnr.10423. [DOI] [PubMed] [Google Scholar]
- Bell MJ, Hallenbeck JM, Gallo V. Determining the fetal inflammatory response in an experimental model of intrauterine inflammation in rats. Pediatr Res. 2004;56:541–546. doi: 10.1203/01.PDR.0000139407.89883.6B. [DOI] [PubMed] [Google Scholar]
- Cabrero A, Alegret M, Sanchez RM, Adzet T, Laguna JC, Carrera MV. Increased reactive oxygen species production down-regulates peroxisome proliferator-activated alpha pathway in C2C12 skeletal muscle cells. J Biol Chem. 2002;277:10100–10107. doi: 10.1074/jbc.M110321200. [DOI] [PubMed] [Google Scholar]
- Cabrero A, Merlos M, Laguna JC, Carrera MV. Down-regulation of acyl-CoA oxidase gene expression and increased NF-kappaB activity in etomoxir-induced cardiac hypertrophy. J Lipid Res. 2003;44:388–398. doi: 10.1194/jlr.M200294-JLR200. [DOI] [PubMed] [Google Scholar]
- Cai Z, Pan ZL, Pang Y, Evans OB, Rhodes PG. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr Res. 2000;47:64–72. doi: 10.1203/00006450-200001000-00013. [DOI] [PubMed] [Google Scholar]
- Dalmau I, Finsen B, Tonder N, Zimmer J, Gonzalez B, Castellano B. Development of microglia in the prenatal rat hippocampus. J Comp Neurol. 1997;377:70–84. [PubMed] [Google Scholar]
- Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res. 1997;42:1–8. doi: 10.1203/00006450-199707000-00001. [DOI] [PubMed] [Google Scholar]
- Dammann O, Leviton A. Role of the fetus in perinatal infection and neonatal brain damage. Curr Opin Pediatr. 2000;12:99–104. doi: 10.1097/00008480-200004000-00002. [DOI] [PubMed] [Google Scholar]
- Deguchi K, Oguchi K, Takashima S. Characteristic neuropathology of leukomalacia in extremely low birth weight infants. Pediatr Neurol. 1997;16:296–300. doi: 10.1016/s0887-8994(97)00041-6. [DOI] [PubMed] [Google Scholar]
- Deplanque D, Gele P, Petrault O, Six I, Furman C, Bouly M, Nion S, Dupuis B, Leys D, Fruchart JC, Cecchelli R, Staels B, Duriez P, Bordet R. Peroxisome proliferators-activated receptor-alpha activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. J Neurosci. 2003;23:6264–6271. doi: 10.1523/JNEUROSCI.23-15-06264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias ER, Mobassaleh M, Hajra AK, Moser AB. Developmental delay and growth failure caused by a peroxisomal disorder, dihydroxyacetonephosphate acyltransferase (DHAP-AT) deficiency. Am J Med Genet. 1998;80:223–226. [PubMed] [Google Scholar]
- Fidel PL, Jr, Romero R, Wolf N, Cutright J, Ramirez M, Araneda H, Cotton DB. Systemic and local cytokine profiles in endotoxin-induced preterm parturition in mice. Am J Obstet Gynecol. 1994;170:1467–1475. doi: 10.1016/s0002-9378(94)70180-6. [DOI] [PubMed] [Google Scholar]
- Fiers W, Beyaert R, Boone E, Cornelis S, Declercq W, Decoster E, Denecker G, Depuydt B, De Valck D, De Wilde G, Goossens V, Grooten J, Haegeman G, Heyninck K, Penning L, Plaisance S, Vancompernolle K, Van Criekinge W, Vandenabeele P, Vanden Berghe W, Van de Craen M, Vandevoorde V, Vercammen D. TNF-induced intracellular signaling leading to gene induction or to cytotoxicity by necrosis or by apoptosis. J Inflamm. 1995;47:67–75. [PubMed] [Google Scholar]
- Goossens V, Grooten J, De Vos K, Fiers W. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc Natl Acad Sci U S A. 1995;92:8115–8119. doi: 10.1073/pnas.92.18.8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes RL, Baud O, Li J, Kinney HC, Volpe JJ, Folkerth DR. Oxidative and nitrative injury in periventricular leukomalacia: a review. Brain Pathol. 2005;15:225–233. doi: 10.1111/j.1750-3639.2005.tb00525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda LI, Rosenberg PA, Volpe JJ, Kinney HC. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol. 2003;62:441–450. doi: 10.1093/jnen/62.5.441. [DOI] [PubMed] [Google Scholar]
- Hemdan S, Almazan G. Deficient peroxide detoxification underlies the susceptibility of oligodendrocyte progenitors to dopamine toxicity. Neuropharmacology. 2007;52:1385–1395. doi: 10.1016/j.neuropharm.2007.01.019. [DOI] [PubMed] [Google Scholar]
- Jatana M, Singh I, Singh AK, Jenkins D. Combination of systemic hypothermia and N-acetylcysteine attenuates hypoxic-ischemic brain injury in neonatal rats. Pediatr Res. 2006;59:684–689. doi: 10.1203/01.pdr.0000215045.91122.44. [DOI] [PubMed] [Google Scholar]
- Kassmann CM, Lappe-Siefke C, Baes M, Brugger B, Mildner A, Werner HB, Natt O, Michaelis T, Prinz M, Frahm J, Nave KA. Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat Genet. 2007;39:969–976. doi: 10.1038/ng2070. [DOI] [PubMed] [Google Scholar]
- Khan M, Contreras M, Singh I. Endotoxin-induced alterations of lipid and fatty acid compositions in rat liver peroxisomes. J Endotoxin Res. 2000;6:41–50. doi: 10.1177/09680519000060010601. [DOI] [PubMed] [Google Scholar]
- Khan M, Haq E, Giri S, Singh I, Singh AK. Peroxisomal participation in psychosine-mediated toxicity: implications for Krabbe's disease. J Neurosci Res. 2005;80:845–854. doi: 10.1002/jnr.20529. [DOI] [PubMed] [Google Scholar]
- Khan M, Sekhon B, Jatana M, Giri S, Gilg AG, Sekhon C, Singh I, Singh AK. Administration of N-acetylcysteine after focal cerebral ischemia protects brain and reduces inflammation in a rat model of experimental stroke. J Neurosci Res. 2004;76:519–527. doi: 10.1002/jnr.20087. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Back SA. Human oligodendroglial development: relationship to periventricular leukomalacia. Semin Pediatr Neurol. 1998;5:180–189. doi: 10.1016/s1071-9091(98)80033-8. [DOI] [PubMed] [Google Scholar]
- Lavrovsky Y, Chatterjee B, Clark RA, Roy AK. Role of redox-regulated transcription factors in inflammation, aging and age-related diseases. Exp Gerontol. 2000;35:521–532. doi: 10.1016/s0531-5565(00)00118-2. [DOI] [PubMed] [Google Scholar]
- Lazarow PB. Peroxisome structure, function, and biogenesis--human patients and yeast mutants show strikingly similar defects in peroxisome biogenesis. J Neuropathol Exp Neurol. 1995;54:720–725. doi: 10.1097/00005072-199509000-00015. [DOI] [PubMed] [Google Scholar]
- Lazo O, Singh AK, Singh I. Postnatal development and isolation of peroxisomes from brain. J Neurochem. 1991;56:1343–1353. doi: 10.1111/j.1471-4159.1991.tb11431.x. [DOI] [PubMed] [Google Scholar]
- Lehmann D, Karussis D, Misrachi-Koll R, Shezen E, Ovadia H, Abramsky O. Oral administration of the oxidant-scavenger N-acetyl-L-cysteine inhibits acute experimental autoimmune encephalomyelitis. J Neuroimmunol. 1994;50:35–42. doi: 10.1016/0165-5728(94)90212-7. [DOI] [PubMed] [Google Scholar]
- Levison SW, Rothstein RP, Romanko MJ, Snyder MJ, Meyers RL, Vannucci SJ. Hypoxia/ischemia depletes the rat perinatal subventricular zone of oligodendrocyte progenitors and neural stem cells. Dev Neurosci. 2001;23:234–247. doi: 10.1159/000046149. [DOI] [PubMed] [Google Scholar]
- Leviton A, Gilles F. Ventriculomegaly, delayed myelination, white matter hypoplasia, and "periventricular" leukomalacia: how are they related? Pediatr Neurol. 1996;15:127–136. doi: 10.1016/0887-8994(96)00157-9. [DOI] [PubMed] [Google Scholar]
- Maeba R, Ueta N. Ethanolamine plasmalogens prevent the oxidation of cholesterol by reducing the oxidizability of cholesterol in phospholipid bilayers. J Lipid Res. 2003;44:164–171. doi: 10.1194/jlr.m200340-jlr200. [DOI] [PubMed] [Google Scholar]
- McQuillen PS, Sheldon RA, Shatz CJ, Ferriero DM. Selective vulnerability of subplate neurons after early neonatal hypoxia-ischemia. J Neurosci. 2003;23:3308–3315. doi: 10.1523/JNEUROSCI.23-08-03308.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan-Plano S, Garcia JJ, Martinez-Ballarin E, Reiter RJ, Ortega-Gutierrez S, Lazaro RM, Escanero JF. Melatonin and pinoline prevent aluminium-induced lipid peroxidation in rat synaptosomes. J Trace Elem Med Biol. 2003;17:39–44. doi: 10.1016/S0946-672X(03)80044-5. [DOI] [PubMed] [Google Scholar]
- Min KJ, Yang MS, Kim SU, Jou I, Joe EH. Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J Neurosci. 2006;26:1880–1887. doi: 10.1523/JNEUROSCI.3696-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser HW. Peroxisomal disorders. Semin Pediatr Neurol. 1996;3:298–304. doi: 10.1016/s1071-9091(96)80033-7. [DOI] [PubMed] [Google Scholar]
- Moser HW. Peroxisomal disorders: classification and overview of biochemical abnormalities. Rev Neurol. 1999;28 Suppl 1:S45–S48. [PubMed] [Google Scholar]
- Moynagh PN, Williams DC, O'Neill LA. Activation of NF-kappa B and induction of vascular cell adhesion molecule-1 and intracellular adhesion molecule-1 expression in human glial cells by IL-1. Modulation by antioxidants. J Immunol. 1994;153:2681–2690. [PubMed] [Google Scholar]
- Paintlia AS, Gilg AG, Khan M, Singh AK, Barbosa E, Singh I. Correlation of very long chain fatty acid accumulation and inflammatory disease progression in childhood X-ALD: implications for potential therapies. Neurobiol Dis. 2003;14:425–439. doi: 10.1016/j.nbd.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Khan M, Vollmer T, Singh AK, Singh I. HMG-CoA reductase inhibitor augments survival and differentiation of oligodendrocyte progenitors in animal model of multiple sclerosis. Faseb J. 2005;19:1407–1421. doi: 10.1096/fj.05-3861com. [DOI] [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Singh AK, Stanislaus R, Gilg AG, Barbosa E, Singh I. Regulation of gene expression associated with acute experimental autoimmune encephalomyelitis by Lovastatin. J Neurosci Res. 2004;77:63–81. doi: 10.1002/jnr.20130. [DOI] [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Singh I, Singh AK. IL-4-induced peroxisome proliferators-activated receptor gamma activation inhibits NF-kappaB trans activation in central nervous system (CNS) glial cells and protects oligodendrocyte progenitors under neuroinflammatory disease conditions: implication for CNS-demyelinating diseases. J Immunol. 2006;176:4385–4398. doi: 10.4049/jimmunol.176.7.4385. [DOI] [PubMed] [Google Scholar]
- Paintlia MK, Paintlia AS, Barbosa E, Singh I, Singh AK. N-acetylcysteine prevents endotoxin-induced degeneration of oligodendrocyte progenitors and hypomyelination in developing rat brain. J Neurosci Res. 2004;78:347–361. doi: 10.1002/jnr.20261. [DOI] [PubMed] [Google Scholar]
- Pang Y, Cai Z, Rhodes PG. Effect of tumor necrosis factor-alpha on developing optic nerve oligodendrocytes in culture. J Neurosci Res. 2005;80:226–234. doi: 10.1002/jnr.20450. [DOI] [PubMed] [Google Scholar]
- Perry VH, Newman TA, Cunningham C. The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci. 2003;4:103–112. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- Poynter ME, Daynes RA. Peroxisome proliferator-activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production in aging. J Biol Chem. 1998;273:32833–32841. doi: 10.1074/jbc.273.49.32833. [DOI] [PubMed] [Google Scholar]
- Purdue PE, Zhang JW, Skoneczny M, Lazarow PB. Rhizomelic chondrodysplasia punctata is caused by deficiency of human PEX7, a homologue of the yeast PTS2 receptor. Nat Genet. 1997;15:381–384. doi: 10.1038/ng0497-381. [DOI] [PubMed] [Google Scholar]
- Qi C, Zhu Y, Reddy JK. Cell Biochem Biophys. Vol. 32. Spring: 2000. Peroxisome proliferator-activated receptors, coactivators, and downstream targets; pp. 187–204. [DOI] [PubMed] [Google Scholar]
- Rahman A, Kefer J, Bando M, Niles WD, Malik AB. E-selectin expression in human endothelial cells by TNF-alpha-induced oxidant generation and NF-kappaB activation. Am J Physiol. 1998;275:L533–L544. doi: 10.1152/ajplung.1998.275.3.L533. [DOI] [PubMed] [Google Scholar]
- Sandhir R, Khan M, Chahal A, Singh I. Localization of nervonic acid beta-oxidation in human and rodent peroxisomes: impaired oxidation in Zellweger syndrome and X-linked adrenoleukodystrophy. J Lipid Res. 1998;39:2161–2171. [PubMed] [Google Scholar]
- Santos MJ, Quintanilla RA, Toro A, Grandy R, Dinamarca MC, Godoy JA, Inestrosa NC. Peroxisomal proliferation protects from beta-amyloid neurodegeneration. J Biol Chem. 2005;280:41057–41068. doi: 10.1074/jbc.M505160200. [DOI] [PubMed] [Google Scholar]
- Schrader M, Fahimi HD. Mammalian peroxisomes and reactive oxygen species. Histochem Cell Biol. 2004;122:383–393. doi: 10.1007/s00418-004-0673-1. [DOI] [PubMed] [Google Scholar]
- Schrader M, Fahimi HD. Peroxisomes and oxidative stress. Biochim Biophys Acta. 2006;1763:1755–1766. doi: 10.1016/j.bbamcr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Sekhon B, Sekhon C, Khan M, Patel SJ, Singh I, Singh AK. N-Acetyl cysteine protects against injury in a rat model of focal cerebral ischemia. Brain Res. 2003;971:1–8. doi: 10.1016/s0006-8993(03)02244-3. [DOI] [PubMed] [Google Scholar]
- Siems WG, Grune T, Beierl B, Zollner H, Esterbauer H. The metabolism of 4-hydroxynonenal, a lipid peroxidation product, is dependent on tumor age in Ehrlich mouse ascites cells. Exs. 1992;62:124–135. doi: 10.1007/978-3-0348-7460-1_13. [DOI] [PubMed] [Google Scholar]
- Silver RM, Edwin SS, Trautman MS, Simmons DL, Branch DW, Dudley DJ, Mitchell MD. Bacterial lipopolysaccharide-mediated fetal death. Production of a newly recognized form of inducible cyclooxygenase (COX-2) in murine decidua in response to lipopolysaccharide. J Clin Invest. 1995;95:725–731. doi: 10.1172/JCI117719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh I. Biochemistry of peroxisomes in health and disease. Mol Cell Biochem. 1997;167:1–29. doi: 10.1023/a:1006883229684. [DOI] [PubMed] [Google Scholar]
- Singh I, Pahan K, Khan M, Singh AK. Cytokine-mediated induction of ceramide production is redox-sensitive. Implications to proinflammatory cytokine-mediated apoptosis in demyelinating diseases. J Biol Chem. 1998;273:20354–20362. doi: 10.1074/jbc.273.32.20354. [DOI] [PubMed] [Google Scholar]
- Singh I, Paintlia AS, Khan M, Stanislaus R, Paintlia MK, Haq E, Singh AK, Contreras MA. Impaired peroxisomal function in the central nervous system with inflammatory disease of experimental autoimmune encephalomyelitis animals and protection by lovastatin treatment. Brain Res. 2004;1022:1–11. doi: 10.1016/j.brainres.2004.06.059. [DOI] [PubMed] [Google Scholar]
- Stanislaus R, Gilg AG, Singh AK, Singh I. N-acetyl-L-cysteine ameliorates the inflammatory disease process in experimental autoimmune encephalomyelitis in Lewis rats. J Autoimmune Dis. 2005;2:4. doi: 10.1186/1740-2557-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztriha LS, Nork MP, Abdulrazzaq YM, al-Gazali LI, Bakalinova DB. Abnormal myelination in peroxisomal isolated dihydroxyacetonephosphate acyltransferase deficiency. Pediatr Neurol. 1997;16:232–236. doi: 10.1016/s0887-8994(97)00014-3. [DOI] [PubMed] [Google Scholar]
- Topal T, Oter S, Korkmaz A, Sadir S, Metinyurt G, Korkmazhan ET, Serdar MA, Bilgic H, Reiter RJ. Exogenously administered and endogenously produced melatonin reduce hyperbaric oxygen-induced oxidative stress in rat lung. Life Sci. 2004;75:461–467. doi: 10.1016/j.lfs.2004.01.014. [DOI] [PubMed] [Google Scholar]
- Toyokuni S, Uchida K, Okamoto K, Hattori-Nakakuki Y, Hiai H, Stadtman ER. Formation of 4-hydroxy-2-nonenal-modified proteins in the renal proximal tubules of rats treated with a renal carcinogen, ferric nitrilotriacetate. Proc Natl Acad Sci U S A. 1994;91:2616–2620. doi: 10.1073/pnas.91.7.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji F, Miyake Y, Aono H, Kawashima Y, Mita S. Effects of bucillamine and N-acetyl-L-cysteine on cytokine production and collagen-induced arthritis (CIA) Clin Exp Immunol. 1999;115:26–31. doi: 10.1046/j.1365-2249.1999.00749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- Van den Branden C, Dacremont G, Hooghe R, Roels F. Inhibition of peroxisomal beta-oxidation and brain development in rats. Glia. 1989;2:260–265. doi: 10.1002/glia.440020407. [DOI] [PubMed] [Google Scholar]
- Van den Branden C, Leeman J, Dacremont G, Collumbien R, Roels F. Experimental inhibition of peroxisomal beta-oxidation in rats: influence on brain myelination. Glia. 1990;3:458–463. doi: 10.1002/glia.440030604. [DOI] [PubMed] [Google Scholar]
- Victor VM, Rocha M, De la Fuente M. N-acetylcysteine protects mice from lethal endotoxemia by regulating the redox state of immune cells. Free Radic Res. 2003;37:919–929. doi: 10.1080/1071576031000148727. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Perinatal brain injury: from pathogenesis to neuroprotection. Ment Retard Dev Disabil Res Rev. 2001;7:56–64. doi: 10.1002/1098-2779(200102)7:1<56::AID-MRDD1008>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Wang X, Rousset CI, Hagberg H, Mallard C. Lipopolysaccharide-induced inflammation and perinatal brain injury. Semin Fetal Neonatal Med. 2006;11:343–353. doi: 10.1016/j.siny.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Wang X, Svedin P, Nie C, Lapatto R, Zhu C, Gustavsson M, Sandberg M, Karlsson JO, Romero R, Hagberg H, Mallard C. N-acetylcysteine reduces lipopolysaccharide-sensitized hypoxic-ischemic brain injury. Ann Neurol. 2007;61:263–271. doi: 10.1002/ana.21066. [DOI] [PubMed] [Google Scholar]
- Yoon BH, Jun JK, Romero R, Park KH, Gomez R, Choi JH, Kim IO. Amniotic fluid inflammatory cytokines (interleukin-6, interleukin-1beta, and tumor necrosis facto-ralpha), neonatal brain white matter lesions, and cerebral palsy. Am J Obstet Gynecol. 1997;177:19–26. doi: 10.1016/s0002-9378(97)70432-0. [DOI] [PubMed] [Google Scholar]