

Abstract
Tenascin-C (TN-C) is a key component of extracellular matrix (ECM) and its expression process is poorly understood during rheumatic heart valvular disease (RHVD). In this study, we found that interferon (IFN)-γ, tumour necrosis factor (TNF)-α and TN-C concentrations in patients with RHVD were significantly higher than in normal controls. More IFN-γ receptors and TNF receptors were found being expressed on rheumatic aortic valves interstitial cells than on non-rheumatic ones and their expression was patients’ sera dependent. Antibodies neutralizing IFN-γ or TNF-α could attenuate patients’ sera-induced TN-C transcription by isolated rheumatic aortic valves interstitial cells. By application with different protein kinase inhibitors, we found that combined with cyclic strain, TNF-α and IFN-γ induced TN-C transcription through the RhoA/ROCK signalling pathway. At the same time, p38 mitogen-activated protein kinase was involved in TNF-α and IFN-γ induced TN-C transcription. TNF-α also increased TN-C mRNA level by additional PKC and ERK 1/2 activation. Our finding revealed a new insight into ECM remodelling during RHVD pathogenesis and new mechanisms involved in the clinical anti-IFN-γ and anti-TNF-α therapy.
Keywords: cyclic strain, IFN-γ, RHVD, TN-C, TNF-α
Introduction
Tenascin-C (TN-C), a hexameric extracellular matrix (ECM) glycoprotein, is a major component of ECM. TN-C was reported to be transiently present during embryo maturation, ECM remodelling, inflammation and neoplasias. TN-C was involved in cell differentiation, proliferation and migration [1–4]. TN-C was among few genes that were induced upon dynamic mechanical stress [5]. Regulation of TN-C expression induced by mechanical signals depended on integrin engagement with ECM and subsequent activation of the small GTPase RhoA and thereby induced Rho-dependent kinase (ROCK)-dependent actin assembly [6]. TN-C was identified to be induced by transforming growth factor-β, tumour necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and platelet-derived growth factor within various cell types [7–10]. Expression of TN-C in heart valves differed under physiological and pathological conditions. In normal valves, TN-C was mainly distributed on the basement membrane beneath the endothelial cells, whereas no such expression but rather immunoreactivity was found in the deeper layers of the valves in stenotic valves [11]. TN-C combined with matrix metalloproteinase-2, thymosin β-4 and osteopontin was associated with heart valve calcification [12–14].
Group A streptococcal infection was taken as the pathogenesis of rheumatic heart disease and rheumatic valvulitis because of the shared antigen epitopes between group A streptococcal M peptides and valve components [15]. Th1-like proinflammatory cytokines interferon (IFN)-γ and TNF-α were predominant in RHD other than IL-4 [16].
Until now, no data were revealed about the relationship between rheumatic heart valvular disease (RHVD) pathogenesis and TN-C expression and whether other RHD-related factors were involved in TN-C production besides the local valves strain. In this study, we found that RHD-specific Th1-like proinflammatory cytokines and mechanical stress had synergistic effects on TN-C production by aortic valves interstitial fibroblast isolated from patients with RHVD.
Materials and methods
Patients and normal subjects
The sera were collected from 80 patients (24 female and 56 male; median age, 42.7years; range, 26–58 years) with RHVD, 50 patients (28 female and 22 male; median age, 40.6 years; range, 20–53 years) with congenital valvular malformation and 30 healthy volunteers (20 female and 10 male; median age, 41.3 years; range, 28–55 years), who served as controls, and kept at −70°C for storage. Human aortic valve specimens were obtained from those undergoing valve exchange operations and with RHVD (13 subjects) and congenital valvular malformation (eight subjects), or from normal autopsy (three subjects). All patients gave informed consent according to institutional guidelines.
Immunohistochemistry
Formalin-fixed and paraffin-embedded normal, malformed or rheumatic valves were obtained from the tissue bank of the Department of Pathology at the Xijing Hospital. All tissues were sampled from surgical specimens within 2 h of resection. Three-micrometre thick sections were mounted on poly-L-lysine-coated slides, followed by deparaffinization in xylene and dehydration in graded alcohol. The endogenous peroxidase activity was blocked by an incubation with 3 µl/ml H2O2 in methanol for 30 min. Tissue sections were subjected to antigen retrieval by boiling in 0·01 mol/l sodium-citrate (pH 6) for 10 min in a microwave oven. After blocking with 15 µg/ml normal goat serum for 1 h, immunohistochemistry assay was carried out with an Elivision plus staining kit (Maixub-bio, Fuzhou, China) according to the manufacturer's instructions. In brief, tissue sections were stained with polyclonal antibodies against TN-C (Santa Cruz Biotechnology, Santa Cruz, CA, USA) with a concentration of 50 µg/ml for 1 h at room temperature and washed three times, after which an enhancer was added for 30 min, followed by three washes before HRP-conjugated goat-anti-rabbit IgG were added and washed another three times. Finally, diaminobenzidine was added for colouring.
Mechanical ventricular assist device and equi-biaxial cyclic strain production
The mechanical ventricular assist device was designed and manufactured by us and our cooperators from Xi'an JiaoTong University. It was composed of nine main parts (Fig. 1b) and used as a cyclic strain producer in our system. The corresponding cyclic strain accessory device with special culture dishes was an imitation of Chiquet's [10]. The difference in our system was the appliance of a clinical machine that mimicked the physiological cyclic strain of valve fibroblast. Similarly, a sheet of silicone membrane (0·05 U, gloss/gloss; Specialty Manufacturing Inc.) was sandwiched between the scaffold and the base plate of the culture dish to form the bottom surface of the wells (Fig. 1c). The isolated valve fibroblasts were seeded on the collagen-coated silicone membrane. Linear strains over the x- and y-axis were measured the same.
Fig. 1.
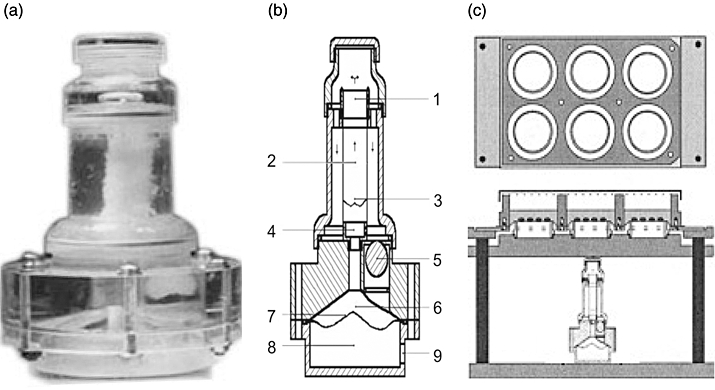
Mechanical ventricular assist device (VAD) and multiwelled equi-biaxial strain device. (a) Appearance of mechanical right ventricular assist device (RVAD). (b) Structure details of RVAD. Arrows indicate flow direction. (1) Upper adaptor. (2) Pulmonary artery frame with valves. (3) Valves. (4) Lower adaptor. (5) One-way valve. (6) Mimic right ventricle. (7) Diaphragm. (8) Air dynamic chamber. (9) Motivity entrance. (c) Multiwelled device to generate equi-biaxial strain on a fibronectin-coated silicone membrane onto which cells have been plated. The membrane is fitted between the frame and base plate of the six-well dish, forming the bottom of the wells (top view, upper of figure). The dish with cultured cells is mounted on the upper platform of the device (side view, bottom of figure). Frequency and amplitude of cyclic strain can be adjusted.
Coating of silicone membrane with human collagen I
Silicone membrane was coated with human collagen I as described [10]. Briefly, sterile human collagen I (frozen in 10 mM acetic acid, Becton Dickinson) was neutralized and diluted to final 30 µg/ml with ice-cold PBS. A drop of 100 µl human collagen I was carefully placed onto the silicone membrane in the centre of each well and covered with a plastic bottle cap (20 mm diameter) at room temperature for at least 1 h to allow collagen fibril assembly. Collagen coated wells were dried for 30 min under a UV lamp (254 nm, 20 W, 15-cm distance) to achieve mild protein cross-linking.
Isolation and culture of human valve interstitial cells
The endothelial cell layers on the dissected valves were removed and the remaining interstitial layers were minced and digested with 0·2% collagenase I (Sigma) in M199 medium (Invitrogen) containing 0·1% BSA at 37°C for 2 h. Cells were then seeded and maintained on culture dishes with M199 medium containing 10% valves corresponding to patients’ sera, supplemented with penicillin and streptomycin (Invitrogen). Cells used in the experiments were between passages 3 and 10. Cells were harvested by trypsinization and plated either (for cytokines co-culture) into normal six-well plastic dishes (Nunc; 6 × 105 cells per well in 4-ml DMEM with indicated sera), or (for cyclic strain assays) onto the silicone membrane of the specially designed six-well dishes. After 4 h of seeding, the medium in both normal and silicone dishes was replaced by 4-ml DMEM supplemented with 10% corresponding patients’ serum or 3% FCS. Cultures were left in the CO2-incubator.
Cytokines and cyclic strain assay
Cytokines (all purchased from Sigma) were used on the subjects’ corresponding sera concentration. The inhibitor (all purchased from Calbiochem) concentrations were chosen from the literature [17,18] or from the manufacturer's data sheets. The final standard concentrations of MEK-1 inhibitor PD 98059 (blocking ERK 1/2, PD), p38 MAPK inhibitor SB 203580 (SB), PKC inhibitor GF109203X (GFX) and ROCK-I/II inhibitor Y 27632 (Y) were all 10 µM.
Cultures in normal six-well dishes were washed twice with DMEM/0·3% FCS before cytokines assays. Cells were then pretreated with 2 ml of control or inhibitor containing medium for 60 min. Then, 2 ml of media without or with cytokines were added to each well. After being cultured in the CO2-incubator for the designed time course, cells were lysed and total RNA was extracted. The mAb (clone 3A8) neutralizing IFN-γ and the mAb (clone 1G7) neutralizing TNF-α were gifted by professor LiHua Chen from the Fourth Military Medical University.
Cultures in the special six-well dishes with silicone membrane were subjected to cyclic strain (10%, 0·3 Hz) in the CO2-incubator for the indicated time course. Similarly, cells were treated with protein kinase inhibitors as described above. Cells from strained and resting cultures were lysed and total RNA was isolated.
RNA isolation and single-step quantitative real-time RT-PCR
Cells were lysed with TRIzol (Invitrogen) and collected with the Qiagen RNeasy mini kit according to the manufacturer's instructions. All RNA samples isolated were treated with DNase (Qiagen). We used the SYBR® Green PCR Master Mix and RT-PCR kit in one-step RT-PCR according to the manufacturer's instructions. The sequence of primers was: β-actin sense, 5′-AGAAGGATTCCTATGTGGGCG-3′, antisense 5′-CATGTCGTCCCAGTTGGTGAC-3′; TN-C forward primer, 5′-CAGCTCCACACTCCAGGTAC-3′, antisense 5′-CTTTCGCTGG-GCTCTGAAGG-3′ The sizes of PCR products for β-actin and TN-C were 101 and 448 bp respectively. The amplification conditions were 30 min at 48°C, 10 min at 95°C, 40 cycles of denaturation at 95°C for 15 s, and annealing/extension at 60°C for 1 min. For relative quantification, we calculated the n-fold differential expression by the ΔCt method (Ct denoting the threshold cycle of PCR amplification at which product is first detected by fluorescence), which compares the amount of target gene amplification, normalized to the β-actin endogenous reference. The correct sizes of the amplified products were checked by electrophoresis on 1·5% agarose gels, and visualized by ethidium bromide staining.
Flow cytometry
Cells were incubated with FITC-conjugated anti-IFN-γ receptor 1 (IFN-γR1, clone 92101, R&D systems), FITC-conjugated anti-TNF receptor I (TNF RI, clone 16803, R&D systems) mAbs or their corresponding control mAbs with the same isotypes (R&D systems) at 4°C for 30 min respectively. After washing twice, cells were fixed and then analysed.
Sandwich ELISA
Levels of IFN-γ, TNF-α and TN-C were assessed using the corresponding ELIASA kits (IFN-γ and TNF-α ELISA kits from R&D systems, TN-C Large variants ELISA kit form IBL) according to the manufacturer's instructions. The final results were read at a 450 nm wavelength.
Immunoprecipitation and Western blot analysis
The collected sera were supplemented with protease inhibitor cocktail (Sigma) and incubated overnight at 4°C with 50 µl of protein A-Sepharose together with 2 µg of rabbit anti-TN-C IgG (H-300, Santa Cruz) per 200 µg total protein. Each mixture was then centrifuged at 10 000 g for 15 min, washed at least three times with PBS containing protease inhibitors, resuspended and boiled directly in SDS sample buffer. After electrophoresis and membrane transferring, TN-C protein was detected by incubation with 1:200 anti-TN-C mAb (6A194, Santa Cruz) followed by incubation with 1:5000 goat anti-mouse IgG-HRP. TN-C expression was detected by enhanced chemiluminescence according to the manufacturer's protocol (Amersham).
Statistics
All assays were performed in triplicate. Data are expressed as mean ± s.e.m. Statistical comparisons were performed by one-way analysis of variance followed by the Student-Newman-Keuls test. A P-value less than 0·05 was considered statistically significant.
Results
IFN-γ, TNF-α and TN-C concentrations in subjects
Concentrations of IFN-γ, TNF-α and TN-C in patients with RHVD were 24.6 ± 8.3 pg/ml, 19.3 ± 5.2 pg/ml and 93.4 ± 32.6 ng/ml respectively, which was significantly higher than those in congenital valvular malformation (7.6 ± 3.7 pg/ml, 3.8± 1.7 pg/ml and 29.6 ± 7.3 ng/ml respectively) and normal controls (6.9 ± 3.8 pg/ml, 3.7 ± 1.8 pg/ml and 32.6 ± 7.2 ng/ml respectively) (P < 0.01). No significant difference was found between those in sera of congenital valvular malformation and normal controls (P > 0.05) (Table 1).
Table 1.
IFN-γ, TNF-α and TN-C concentrations in subjects.
| Molecules | Normal controls | CVM patients | RHVD patients |
|---|---|---|---|
| IFN-γ (pg/ml) | 6·9 ± 3·8 | 7·6 ± 3·7* | 24·6 ± 8·3** |
| TNF-α (pg/ml) | 3·7 ± 1·8 | 3·8 ± 1·7* | 19·3 ± 5·2** |
| TN-C (ng/ml) | 32·6 ± 7·2 | 29·6 ± 7·3* | 93·4 ± 32·6** |
P > 0.05 versus the normal controls;
P < 0.01 versus the normal controls. CVM, congenital valvular malformation; RHVD, rheumatic heart valvular disease.
The TN-C in sera and their expression patterns in normal, malformed and rheumatic valves
In sera from the normal controls and patients with the malformed valves, the visible TN-C bands were almost the same and about 210-KDa (Fig. 2, lane 1 and lane 2), which matched the smallest TN-C variants (S). In contrast, TN-C distribution in RHVD patients (Fig. 2, lane 3) was among the largest variants (L, about 350-KDa) and smallest. TN-C abundance was much higher than that in normal and malformed valves groups and the most TN-C was larger variants forms.
Fig. 2.
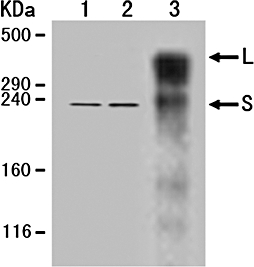
TN-C distribution in sera from the normal subjects (lane 1), patients with the malformed valves (lane 2) and with RHVD (lane 3) with method of immunoprecipitation and Western blot analysis. L, largest variants; S, smallest variants.
The morphology and TN-C expression patterns of the normal (Fig. 3A and B), the malformed (Fig. 3C and D) and rheumatic (Fig. 3E and F) valves were found different in immunohistochemistry results. The endothelial cells of the normal and malformed valves were unilaminar and the whole valve structure was tighter. The endothelial cells layer and whole bodies of rheumatic valves became thickened and loose. TN-C was mainly distributed on the basement membrane beneath the endothelial cells on the normal and malformed valves and throughout interstitial tissues and richer on the basement membrane on the rheumatic valves.
Fig. 3.
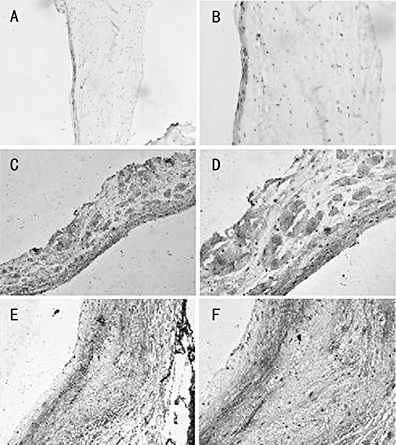
TN-C expression patterns in the normal, malformed and rheumatic valves. The normal (Fig. 2A and B), malformed (Fig. 2C and D) and rheumatic (Fig. 2E and F) valves were stained with polyclonal anti-TN-C antibodies as described in Materials and Methods. Magnification of 2A, 2C and 2E is 100-fold and that of 2B, 2D and 2F is 200-fold.
The IFN-γ receptor and TNF receptor expression pattern and effects of antibodies neutralizing IFN-γ or TNF-α on TN-C transcription
By flow cytometry analysis, we found IFN-γ R1 and TNF RI slightly expressed on aortic valve interstitial cells isolated from patients with congenital valvular malformation (Fig. 4a and b) and expressed with higher density on those from patients with RHVD (Fig. 4c and d). If the rheumatic valve interstitial cells were starved by 3% FCS instead of 10% corresponding patients’ sera, IFN-γ R1 and TNF RI expression was down-regulated 36 h later (Fig. 4e and f).
Fig. 4.
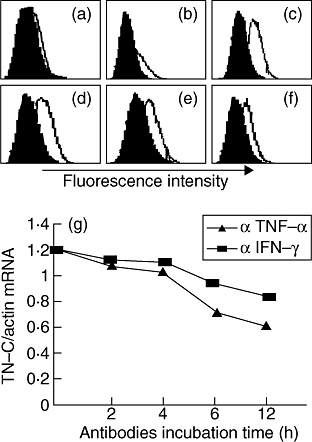
IFN-γ receptor 1 and TNF receptor I expression pattern and effects of antibodies neutralizing IFN-γ or TNF-α on TN-C transcription. (a) IFN-γ R1 on malformed valve interstitial cells (M). (b) TNF RI on M. (c) IFN-γ R1 on rheumatic valve interstitial cells (R). (d) TNF RI on R. (e) IFN-γ R1 on R after starvation. (f) TNF RI on R after starvation. (g) Antibodies neutralizing IFN-γ or TNF-α blocked TN-C transcription. Antibody against IFN-γ or TNF-α were added and incubated for the indicated time course according to antibody: targeting molecule mole ratio equal to 2:1 respectively. TN-C mRNA level was tested by quantitative real-time RT-PCR.
In the normal six-well culture system, the isolated rheumatic aortic valves interstitial cells were cultured in corresponding sera and TN-C transcription was analysed by quantitative RT-PCR. After antibodies neutralizing IFN-γ or TNF-α were added, TN-C transcription was attenuated. The obvious down-regulation started from approximate 6 h after antibodies were added. Figure 4g was representative of the assays. The decrease extent differed between antibodies neutralizing IFN-γ and TNF-α. The neutralization targeting TNF-α decreased TN-C transcription greater than targeting IFN-γ.
The TN-C transcription induced by cyclic stress and synergistic effects of mechanical strain and cytokines
The TN-C mRNA constitutively existed within isolated interstitial cells and the cyclic stress of 10% and 0·3 Hz increased its abundance from the second to the third hour. Although the constitutive TN-C mRNA was different between the tested groups, their increases were both found after cyclic strain stimulation. (Fig. 5a). The kinase inhibitor assays showed that ROCK rather than ERK 1/2, p38 MAPK or PKC pathways was involved in strain-induced TN-C transcription after strain for 6 h (Fig. 5b). When cyclic stress and cytokines were combined together targeting rheumatic valve interstitial cells for 6 h, synergistic effects were found on TN-C mRNA production (Fig. 5c).
Fig. 5.
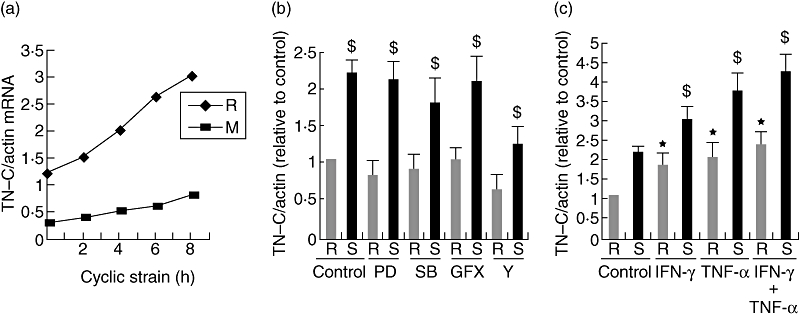
Cyclic strain increased TN-C mRNA level through the RhoA/ROCK pathway and its synergistic effects with cytokines. (a) TN-C mRNA was induced by cyclic strain within isolated malformed (M) or rheumatic (R) valve interstitial cells for the indicated time course. TN-C mRNA level was tested by quantitative real-time RT-PCR. (b) Induction of tenascin-C mRNA by cyclic strain was attenuated by ROCK antagonist. After inhibitors were added for 60 min, cells were stimulated by cyclic strain (10%, 0·3 Hz) (S) or left at rest (R) for 6 h. Data represent the average of five independent experiments; error bars indicate standard errors of the mean. $(P < 0·01), significant difference to the resting control (R, no inhibitor); *(P < 0·01), significant difference to the other four strained groups. (c) Cyclic strain and cytokines had synergistic effects on TN-C transcription. $(P < 0·01), significant difference to the stimulated control (S, no cytokines); *(P < 0·01), significant difference to resting control (R, no cytokines).
The TN-C transcription induced by IFN-γ
No obvious increased TN-C mRNA level was observed after IFN-γ stimulation within aortic valve interstitial cells isolated from patients with congenital valvular malformation even after 12 h. We found TN-C mRNA was increased after 4–6 h (varying among individuals) in the rheumatic valve interstitial cells after IFN-γ addition (Fig. 6a). The kinase inhibitor assays showed that both ROCK and p38 MAPK pathways other than ERK 1/2 and PKC were involved in IFN-γ-inducing TN-C transcription after stimulation (Fig. 6b).
Fig. 6.
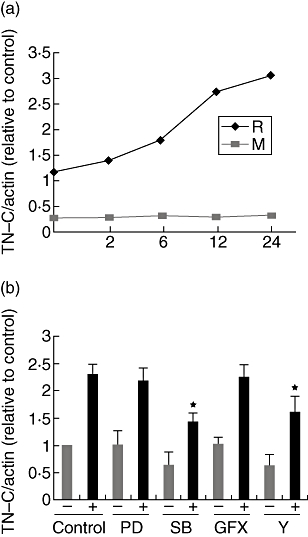
RhoA/ROCK and p38 MAPK pathways were involved in IFN-γ-inducing TN-C transcription. (a) TN-C mRNA was induced by IFN-γ within isolated rheumatic (R) other than malformed (M) valve interstitial cells. The Figure is representative of the assays. (b) Induction of tenascin-C mRNA by IFN-γ was attenuated by ROCK and p38 MAPK antagonist. After inhibitors were added for 60 min, cells were stimulated by IFN-γ for 12 h (+), the others left untreated (−). Data represent the average of five independent experiments; error bars indicate standard errors of the mean. *(P < 0·01), significant difference to the other three IFN-γ-treated groups.
The TN-C transcription induced by TNF-α
Similarly, no obvious increased TN-C mRNA level was observed after TNF-α stimulation within aortic valve interstitial cells isolated from patients with congenital valvular malformation. We found TN-C mRNA was increased after 3–5 h (varying among individuals) in the rheumatic valve interstitial cells after TNF-α addition (Fig. 7a). The kinase inhibitor assays showed that all the tested signal pathways including ROCK, p38 MAPK, ERK 1/2 and PKC were involved in TNF-α-inducing TN-C transcription after stimulation (Fig. 7b).
Fig. 7.
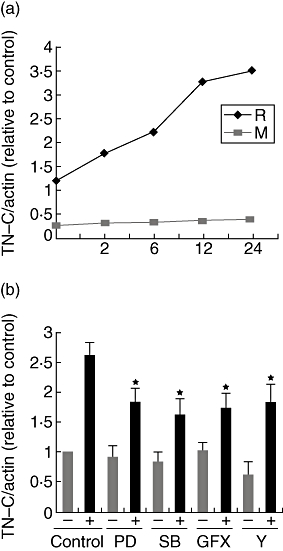
RhoA/ROCK, p38 MAPK, ERK 1/2 and PKC pathways were involved in TNF-α-inducing TN-C transcription. (a) TN-C mRNA was induced by TNF-α within isolated rheumatic (R) other than malformed (M) valve interstitial cells. The figure is representative of the assays. (b) Induction of tenascin-C mRNA by TNF-α was attenuated by ROCK, p38 MAPK, ERK 1/2 and PKC antagonist. After inhibitors were added for 60 min, cells were stimulated by TNF-α for 12 h (+), the others left untreated (−). Data represent the average of five independent experiments; error bars indicate standard errors of the mean. *(P < 0·01), significant difference to the TNF-α-treated control group.
Discussion
The ECM was composed of various proteins and collagens and its components were altered by stimuli such as mechanical strain, proinflammatory factors and cellular communication, etc. TN-C was supposed to be a profibrotic molecule, also involved in cell-ECM interaction and valve calcification. It was a unique six-armed multidomain macromolecule with a molecular mass of 210–400 kDa. Each arm consisted of 14 + 1/2 EGF-like domains, 8–15 FNIII domains, depending on alternative RNA splicing, and a single fibrinogen-like domain. The universal FNIII domains (FNIII repeats 1–5 and FNIII repeats 6–8) were present in all TN-C variants. The largest TN-C variant also contained nine alternatively spliced domains (FNIII repeats A1–D), which were missing in the shortest splice variant. The large variants, containing the alternatively spliced FNIII domains in various combinations, were predominantly expressed in pathological tissues, which undergo remodelling, as with regeneration, inflammation and tumorigenesis. TN-C tested in our assay belonged to its large variant and TN-C was also found in other rheumatic diseases and taken as a biomarker of disease progression [19]. In our assay, we found TN-C large variants were elevated in sera of patients with RHVD, which proved it's also a biomarker of RHVD. Rheumatic heart diseases were Th1-dominant, and here we found undoubtedly, Th1-like cytokines were abundant in patients with RHVD compared with those with congenital valvular malformation or normal controls.
In China, cadaver valves were hard to obtain because of Chinese traditional culture and incomplete organ donation laws. Therefore, in this assay, the selected non-rheumatic valves were from young patients diagnosed as having a bicuspid aortic valve. Their valves had relatively normal morphology and no obvious calcification. In the IHC results, we could see that the TN-C distribution pattern on our selected valves was similar to the previous report [11] on normal valves. TN-C was mainly on the basement membrane beneath the endothelial cells.
Our results revealed new evidence for the different aetiology of aortic stenosis under rheumatic non-rheumatic conditions. IFN-γ interacted with dimerized IFN-γ receptor 1 (CDw119) and TNF-α interacted with the trimerized TNF receptor I, and then the downstream signal pathways were activated. IFN-γR1 and TNF-RI were just slightly expressed on the malformation valve cells. However, co-culture with IFN-γ or TNF-α induced no TN-C transcription, which may be due to incomplete or ineffective cytokine receptor sensor systems. Interestingly, the expression of IFN-γR1 and TNF-RI on rheumatic valve cells was patients’ sera dependent and even if the sera were replaced by normal AB serum, a similar decrease was found (data not shown). It may be because the receptors expression needed persistent stimulation by certain molecules in patients’ sera with RHVD.
A previous study proved that fibroblasts sensed strain by cytoskeleton through integrins within cell-matrix adhesions, which then triggered MAPK and NK-κB activation [20].Therefore, collagen I was used as ECM on silicone membrane. In our study, it was found, although constitutive TN-C mRNA levels in both the rheumatic and malformation groups were up-regulated after cyclic strain no matter what the cell origin, but the induction extent and tendency was distinct. Cells from rheumatic valves were easily activated and we presumed this was due to the RHVD pathogenesis. Strain force led to the stiffening of the actin cytoskeleton mediated by RhoA and ROCK. As with effects on other fibroblasts, the mechanical strain induced TN-C through the RhoA/ROCK signalling pathway and could be attenuated by the chemical inhibitor Y 27632. Also, we found the mechanical strain could not active ERK 1/2, p38 MAPK or PKC pathways.
Several reports revealed that TNF-α stimulated TN-C expression in other cell types through several pathways, including: (i) NF-κB signalling with RelA activation, (ii) p38 MAPK signaling, and (iii) via the transcription factor Ets-1 [21–23]. In our assay, after inhibition of ROCK, PKC, ERK 1/2 and p38 MAPK, we found IFN-γ induced TN-C transcription through RhoA/ROCK and p38 MAPK pathways. TNF-α acted through all four pathways of ROCK, PKC, ERK 1/2 and p38 MAPK. Our finding is shown in Fig. 8.
Fig. 8.
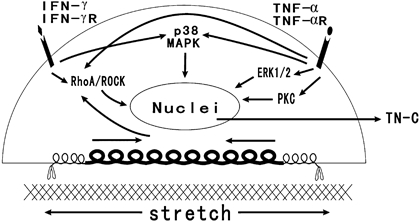
Signalling pathways and synergistic effects induced by cyclic strain, IFN-γ and TNF-α on tenascin-C production by rheumatic aortic valve interstitial cells.
Likewise, we inferred that TNF-α induced RhoA/ROCK activation and then the changes in cytoskeleton molecules such as F-actin. A similar phenomenon was found in the endothelial cell model [24]. The PKC family of proteins was comprised of at least 11 isozymes with diverse functions that were involved in important cellular processes [25]. It has been found that IL-13 could induce TN-C production through the PKC signalling pathway [26] and thrombin stimulated TN-C expression via opposing PKC-ε signalling mechanisms in systemic sclerosis and normal lung fibroblasts [27]. β3 integrin induced TN-C gene expression in vascular smooth muscle cell through the ERK1/2 pathway, which activated a 122-base pair TN-C promoter element [28]. Correspondingly, blocking of IFN-γ or TNF-α attenuated TN-C mRNA levels. The decease extent of blocking TNF-α was stronger than that of blocking IFN-γ. Concerning the downstream signal pathways, TNF-α blockade interfered with more down-stream molecules. In the clinic, anti-IFN-γ and anti-TNF-α therapy was undertaken [29,30] and our results provide new evidence for the potential mechanism of these effective targeting therapies.
In conclusion, within valve interstitial cells, TN-C was constitutively expressed, but at a low level, and also induced by cyclic strain under normal and malformed conditions. During RHVD, IFN-γ, TNF-α and cyclic strain had synergistic effects on increasing TN-C mRNA and protein level through the common and distinct signal pathways.
Acknowledgments
This work was supported by the Natural Science Foundation of ShaanXi Province (No. 2005c222).
References
- 1.Jones PL, Jones FS. Tenascin-C in development and disease: gene regulation and cell function. Matrix Biol. 2000;19:581–96. doi: 10.1016/s0945-053x(00)00106-2. [DOI] [PubMed] [Google Scholar]
- 2.Schellings MW, Pinto YM, Heymans S. Matricellular proteins in the heart: possible role during stress and remodeling. Cardiovasc Res. 2004;64:24–31. doi: 10.1016/j.cardiores.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Hasegawa M, Nakoshi Y, Muraki M, et al. Expression of large tenascin-C splice variants in synovial fluid of patients with rheumatoid arthritis. J Orthop Res. 2007;25:563–8. doi: 10.1002/jor.20366. [DOI] [PubMed] [Google Scholar]
- 4.Franz M, Hansen T, Borsi L, et al. A quantitative co-localization analysis of large unspliced tenascin-C(L) and laminin-5/gamma2-chain in basement membranes of oral squamous cell carcinoma by confocal laser scanning microscopy. J Oral Pathol Med. 2007;36:6–11. doi: 10.1111/j.1600-0714.2006.00492.x. [DOI] [PubMed] [Google Scholar]
- 5.Chapados R, Abe K, Ihida-Stansbury K, et al. ROCK controls matrix synthesis in vascular smooth muscle cells: coupling vasoconstriction to vascular remodeling. Circ Res. 2006;99:837–44. doi: 10.1161/01.RES.0000246172.77441.f1. [DOI] [PubMed] [Google Scholar]
- 6.Chiquet M, Tunç-Civelek V, Sarasa-Renedo A. Gene regulation by mechanotransduction in fibroblasts. Appl Physiol Nutr Metab. 2007;32:967–73. doi: 10.1139/H07-053. [DOI] [PubMed] [Google Scholar]
- 7.Liu Z, Lu X, Wang H, Gao Q, Cui Y. The up-regulated expression of tenascin C in human nasal polyp tissues is related to eosinophil-derived transforming growth factor beta1. Am J Rhinol. 2006;20:629–33. doi: 10.2500/ajr.2006.20.2918. [DOI] [PubMed] [Google Scholar]
- 8.Nakoshi Y, Hasegawa M, Sudo A, Yoshida T, Uchida A. Regulation of tenascin-C expression by tumor necrosis factor-alpha in cultured human osteoarthritis chondrocytes. J Rheumatol. 2008;35:147–52. [PubMed] [Google Scholar]
- 9.Chevillard G, Derjuga A, Devost D, Zingg HH, Blank V. Identification of interleukin-1beta regulated genes in uterine smooth muscle cells. Reproduction. 2007;134:811–22. doi: 10.1530/REP-07-0289. [DOI] [PubMed] [Google Scholar]
- 10.Chiquet M, Sarasa-Renedo A, Tunç-Civelek V. Induction of tenascin-C by cyclic tensile strain versus growth factors: distinct contributions by Rho/ROCK and MAPK signaling pathways. Biochim Biophys Acta. 2004;1693:193–204. doi: 10.1016/j.bbamcr.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 11.Satta J, Melkko J, Pöllänen R, et al. Progression of human aortic valve stenosis is associated with tenascin-C expression. J Am Coll Cardiol. 2002;39:96–101. doi: 10.1016/s0735-1097(01)01705-3. [DOI] [PubMed] [Google Scholar]
- 12.Jian B, Jones PL, Li Q, Mohler ER, III, Schoen FJ, Levy RJ. Matrix metalloproteinase-2 is associated with tenascin-C in calcific aortic stenosis. Am J Pathol. 2001;159:321–27. doi: 10.1016/S0002-9440(10)61698-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li QY, Jones PL, Lafferty RP, Safer D, Levy RJ. Thymosin beta4 regulation, expression and function in aortic valve interstitial cells. J Heart Valve Dis. 2002;11:726–35. [PubMed] [Google Scholar]
- 14.Ghazvini-Boroujerdi M, Clark J, Narula N, et al. Transcription factor Egr-1 in calcific aortic valve disease. J Heart Valve Dis. 2004;13:894–903. [PubMed] [Google Scholar]
- 15.Guilherme L, Ramasawmy R, Kalil J. Rheumatic fever and rheumatic heart disease: genetics and pathogenesis. Scand J Immunol. 2007;66:199–207. doi: 10.1111/j.1365-3083.2007.01974.x. [DOI] [PubMed] [Google Scholar]
- 16.Guilherme L, Cury P, Demarchi LM, et al. Rheumatic heart disease: proinflammatory cytokines play a role in the progression and maintenance of valvular lesions. Am J Pathol. 2004;165:1583–91. doi: 10.1016/S0002-9440(10)63415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee CK, Lee HM, Kim HJ, et al. Syk contributes to PDGF-BB-mediated migration of rat aortic smooth muscle cells via MAPK pathways. Cardiovasc Res. 2007;74:159–68. doi: 10.1016/j.cardiores.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 18.Durlu-Kandilci NT, Brading AF. Involvement of Rho kinase and protein kinase C in carbachol-induced calcium sensitization in beta-escin skinned rat and guinea-pig bladders. Br J Pharmacol. 2006;148:376–84. doi: 10.1038/sj.bjp.0706723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasegawa M, Nakoshi Y, Muraki M, et al. Expression of large tenascin-C splice variants in synovial fluid of patients with rheumatoid arthritis. J Orthop Res. 2007;25:563–8. doi: 10.1002/jor.20366. [DOI] [PubMed] [Google Scholar]
- 20.Sun HW, Li CJ, Chen HQ, et al. Involvement of integrins, MAPK, and NF-kappaB in regulation of the shear stress-induced MMP-9 expression in endothelial cells. Biochem Biophys Res Commun. 2007;353:152–8. doi: 10.1016/j.bbrc.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 21.Nakoshi Y, Hasegawa M, Sudo A, Yoshida T, Uchida A. Regulation of tenascin-C expression by tumor necrosis factor-alpha in cultured human osteoarthritis chondrocytes. J Rheumatol. 2008;35:147–52. [PubMed] [Google Scholar]
- 22.Scherberich A, Tucker RP, Degen M, Brown-Luedi M, Andres AC, Chiquet-Ehrismann R. Tenascin-W is found in malignant mammary tumors, promotes alpha8 integrin-dependent motility and requires p38MAPK activity for BMP-2 and TNF-alpha induced expression in vitro. Oncogene. 2005;24:1525–32. doi: 10.1038/sj.onc.1208342. [DOI] [PubMed] [Google Scholar]
- 23.Nakamura Y, Esnault S, Maeda T, Kelly EA, Malter JS, Jarjour NN. Ets-1 regulates TNF-alpha-induced matrix metalloproteinase-9 and tenascin expression in primary bronchial fibroblasts. J Immunol. 2004;172:1945–52. doi: 10.4049/jimmunol.172.3.1945. [DOI] [PubMed] [Google Scholar]
- 24.McKenzie JA, Ridley AJ. Roles of Rho/ROCK and MLCK in TNF-alpha-induced changes in endothelial morphology and permeability. J Cell Physiol. 2007;213:221–8. doi: 10.1002/jcp.21114. [DOI] [PubMed] [Google Scholar]
- 25.Lee MR, Duan W, Tan SL. Protein kinase C isozymes as potential therapeutic targets in immune disorders. Expert Opin Ther Targets. 2008;12:535–52. doi: 10.1517/14728222.12.5.535. [DOI] [PubMed] [Google Scholar]
- 26.Jinnin M, Ihn H, Asano Y, Yamane K, Trojanowska M, Tamaki K. Upregulation of tenascin-C expression by IL-13 in human dermal fibroblasts via the phosphoinositide 3-kinase/Akt and the protein kinase C signaling pathways. J Invest Dermatol. 2006;126:551–60. doi: 10.1038/sj.jid.5700090. [DOI] [PubMed] [Google Scholar]
- 27.Tourkina E, Hoffman S, Fenton JW, II, Lipsitz S, Silver RM, Ludwicka-Bradley A. Depletion of protein kinase Cepsilon in normal and scleroderma lung fibroblasts has opposite effects on tenascin expression. Arthritis Rheum. 2001;44:1370–81. doi: 10.1002/1529-0131(200106)44:6<1370::AID-ART230>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 28.Jones PL, Jones FS, Zhou B, Rabinovitch M. Induction of vascular smooth muscle cell tenascin-C gene expression by denatured type I collagen is dependent upon a beta3 integrin-mediated mitogen-activated protein kinase pathway and a 122-base pair promoter element. J Cell Sci. 1999;112:435–45. doi: 10.1242/jcs.112.4.435. [DOI] [PubMed] [Google Scholar]
- 29.Ackermann C, Kavanaugh A. Tumor necrosis factor as a therapeutic target of rheumatologic disease. Expert Opin Ther Targets. 2007;11:1369–84. doi: 10.1517/14728222.11.11.1369. [DOI] [PubMed] [Google Scholar]
- 30.Sigidin YA, Loukina GV, Skurkovich B, Skurkovich S. Randomized, double-blind trial of anti-interferon-gamma antibodies in rheumatoid arthritis. Scand J Rheumatol. 2001;30:203–7. doi: 10.1080/030097401316909530. [DOI] [PubMed] [Google Scholar]