

Abstract
Reduction of carbon dioxide by a diiron(I) complex gives μ-carbonato-κ3
O:O′,O′′-bis{[2,2,6,6-tetramethyl-3,5-bis(2,4,6-triisopropylphenyl)heptane-2,5-diiminate(1−)-κ2
N,N′]iron(II)} toluene disolvate, [Fe2(C41H65N)2(CO3)]·2C7H8, a diiron(II) species with a bridging carbonate ligand. The asymmetric unit contains one diiron complex and two cocrystallized toluene solvent molecules that are distributed over three sites, one with atoms in general positions and two in crystallographic 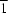 sites. Both FeII atoms are η2-coordinated to diketiminate ligands, but η1- and η2-coordinated to the bridging carbonate ligand. Thus, one FeII center is three-coordinate and the other is four-coordinate. The bridging carbonate ligand is nearly perpendicular to the iron–diketiminate plane of the four-coordinate FeII center and parallel to the plane of the three-coordinate FeII center.
sites. Both FeII atoms are η2-coordinated to diketiminate ligands, but η1- and η2-coordinated to the bridging carbonate ligand. Thus, one FeII center is three-coordinate and the other is four-coordinate. The bridging carbonate ligand is nearly perpendicular to the iron–diketiminate plane of the four-coordinate FeII center and parallel to the plane of the three-coordinate FeII center.
Comment
The reduction of CO2 is of great interest in the scientific community because of its relevance to biological (Ragsdale & Kumar, 1996 ▶) and proposed manmade (Lewis & Nocera, 2006 ▶) energy conversions. CO2 is also a promising one-carbon chemical feedstock (Aresta & Dibenedetto, 2007 ▶). Recently, the Peters group reported an interesting reductive cleavage of CO2 to bridging oxide (O2−) and carbonyl (CO) groups in a diiron complex (Lu et al., 2007 ▶). Our group reported a related reaction, where eight equivalents of CO2 react with five equivalents of the diiron(I) complex [L t-BuFe]2(μ-N2) to give two equivalents of an iron(I) dicarbonyl complex and four equivalents of an unusual diiron(II) complex, (II), with a bridging carbonate ligand (Sadique et al., 2008 ▶). In that work, L t-Bu represents the anionic bidentate β-diketiminate ligand 2,2,6,6-tetramethyl-3,5-bis(2,4,6-triisopropylphenylimido)-4-heptyl. We report here the structure of a very similar carbonate complex that is derived from CO2 using an identical method, but uses a differently substituted β-diketiminate ligand.
The work described here uses L
t-Bu′, a supporting β-diketiminate ligand containing an aryl substituent with isopropyl groups in the 2, 4 and 6 positions. This ligand, shown in Fig. 1 ▶, has been described previously, including its synthesis (Sadique et al., 2007 ▶). Here, we used this ligand in place of L
t-Bu, the 2,6-diisopropylphenyl β-diketiminate ligand from the previous CO2 study (Sadique et al., 2008 ▶). The diiron(I) dinitrogen precursor was synthesized in close analogy with the synthesis of its L
t-Bu analog, and it reacted rapidly with gaseous CO2. The title carbonate product, (I), crystallized as orange blocks. The 1H NMR spectrum of (I) is very similar to that of (II), except for the presence of additional peaks due to the 4-isopropyl groups.
Figure 1.

The synthesis and connectivity of (I) (R = iPr) and (II) (R = H). Thus, compound (II) differs from (I) only in the lack of isopropyl groups in the para positions of the aryl rings in the β-diketiminate ligands.
A displacement ellipsoid plot of the structure of (I) is shown in Fig. 2 ▶ and the core of the molecule is shown in Fig. 3 ▶. The β-diketiminate–Fe1 unit, which is η2-coordinated to the carbonate bridge, is essentially perpendicular to the carbonate plane and to the other β-diketiminate ligand, with interplanar angles of 85.49 (5) and 89.74 (4)°, respectively. It is interesting to compare the structures of the new compound (I) with that of (II) reported earlier (Sadique et al., 2008 ▶). The triclinic unit cell and intensity statistics of (I) indicate the space group P
with Z = 2. Therefore, there is only one molecule per asymmetric unit, in contrast with (II), which has two crystallographically independent molecules with different carbonate binding modes. In (II), one molecule has η2 binding of carbonate to each Fe atom, while the other molecule has η2 binding to one Fe atom and η1 binding to the other. In (I), on the other hand, only the latter bridging mode is seen. This is not surprising, because the larger β-diketiminate ligand in (I) would be expected to prevent the more sterically crowded η2:η2 bridging.
Figure 2.

The structure of (I), showing the atom-labelling scheme. Displacement ellipsoids are drawn at the 50% probability level and H atoms have been omitted for clarity. Only one orientation of each of the disordered components is shown.
Figure 3.

The core of (I), showing the perpendicular arrangement of the β-diketiminate rings.
Despite the increased steric hindrance in (I), the Fe2—O3 distance of 1.8563 (11) Å is shorter than the analogous distance of 1.881 (1) Å in the η2:η1 form of (II). This correlates with a significantly more obtuse angle at O3 of 138.24 (11)° in (I) versus 128.64 (11)° in (II), suggesting that (I) has more π-bonding contribution in the Fe2—O3 bond. Other bond lengths and angles are similar between (I) and (II).
Experimental
All manipulations were carried out using standard Schlenk or high-vacuum techniques to prevent reaction with oxygen or moisture. Solvents were dried by passing them through activated alumina, or by vacuum distillation from sodium benzophenone ketyl. Carbon dioxide was freed from oxygen by three cycles of degassing at 77 K, and dried over activated molecular sieves. The precursor L t-Bu′FeNNFeL t-Bu′ was synthesized from L t-Bu′FeCl by addition of KC8 under an N2 atmosphere, using the same procedure as that reported for analogs with other β-diketiminate ligands (Smith et al., 2006 ▶). For the preparation of (I), L t-Bu′FeNNFeL t-Bu′ (12 mg, 9.8 µmol) was dissolved in benzene-d 6 (0.5 ml), and the tube was cooled in liquid nitrogen and the headspace evacuated. Carbon dioxide (7.75 ml, 65 mbar) was condensed into the tube and the tube warmed to room temperature. The 1H NMR spectrum showed the formation of (I), with singlets (relative integrations in parentheses) at δ 51 (1H), 37 (18H), 24 (2H), 9 (4H), −11 (12H), −54 (12H) and −58 (4H). The solution was transferred into a glove-box, dried under vacuum, redissolved in pentane and cooled to 233 K to give orange crystals of (I).
Crystal data
[Fe2(C41H65N)2(CO3)]·2C7H8
M r = 1527.88
Triclinic,
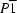
a = 14.7703 (7) Å
b = 15.5535 (8) Å
c = 21.6991 (11) Å
α = 77.533 (3)°
β = 79.815 (3)°
γ = 70.941 (3)°
V = 4569.9 (4) Å3
Z = 2
Mo Kα radiation
μ = 0.37 mm−1
T = 100 K
0.27 × 0.25 × 0.16 mm
Data collection
Bruker SMART APEXII Platform CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 2008a ▶) T min = 0.660, T max = 0.746
71339 measured reflections
26363 independent reflections
19303 reflections with I > 2σ(I)
R int = 0.034
Refinement
R[F 2 > 2σ(F 2)] = 0.042
wR(F 2) = 0.105
S = 1.02
26363 reflections
1199 parameters
275 restraints
H-atom parameters constrained
Δρmax = 0.42 e Å−3
Δρmin = −0.36 e Å−3
H atoms were positioned geometrically, with C—H = 0.95–1.00 Å, and refined with relative isotropic displacement parameters of U iso(H) = 1.2U eq(C) for methine, methylene and aromatic H atoms, and 1.5U eq(C) for methyl H atoms. There are three disordered moieties in the diiron molecule, two isopropyl groups and one tert-butyl group. The isopropyl group containing atoms C103, C113 and C123 was modeled over three positions, and the site occupancies refined to 0.511 (4), 0.275 (4) and 0.215 (4), while their sum was constrained to unity. A two-position disorder model proved insufficient due to the presence of a third site in the difference Fourier map, which manifested itself by residual peaks in an isotropic refinement or by elongated displacement ellipsoids in an anisotropic refinement of the same. All three positions of the group were restrained to have similar bond lengths and angles, and the anisotropic displacement parameters of the positionally similar atoms were constrained to be equal. The disorder of the second isopropyl group (atoms C134, C145, and C155) was modeled over two positions with site occupancies of 0.542 (7) and 0.458 (7). The bond lengths and angles were restrained to be similar and the anisotropic displacement parameters were constrained to be equal. The disorder of the tert-butyl group (atoms C14, C64, C74 and C84) was modeled over two positions and refined to site occupancies of 0.845 (2) and 0.155 (2). Despite the small percentage of the minor component of the disorder, its neglect left three residual peaks of electron density of 1.0–1.2 e Å−3 in positions geometrically appropriate for another orientation of the group. The bond lengths and angles of the two groups were restrained to be similar, and the anisotropic displacement parameters of related atoms were constrained to be equal.
There are three independent cocrystallized toluene solvent molecule sites, all of which required disorder modeling. The first (C17–C77) was modeled over two general positions with site occupancies of 0.608 (5) and 0.392 (5). Both orientations were restrained to have the same bond lengths and angles, and the minor component of the disorder was restrained to be flat. Additionally, the anistropic displacement parameters between one pair of promixal atoms (C37 and C77′) were restrained to be similar. The second toluene molecule (C18–C78) was modeled over a crystallographic inversion center with component occupancies of 0.50 each by symmetry. The molecule was restrained to be flat and the anisotropic displacement parameters of positionally close symmetry-equivalent atoms (C18 and C58, and C38 and C78) were constrained to be equal. The disorder model for the third toluene molecule (C19–C79) was over a crystallographic inversion center (0.50 each by symmetry) and additionally over two general positions, giving unique atom occupancies of 0.313 (8) and 0.187 (8). The latter model was required to account for two positions of electron density on each side of the inversion center. The two unique molecular positions were restrained to be flat, and the bond lengths and angles of both were restrained to be similar. A variety of restraints and constraints were also imposed on the anisotropic displacement parameters, due to the proximity of atomic positions between atoms of the general position disorder and those at symmetry-related positions.
Data collection: APEX2 (Bruker, 2004 ▶); cell refinement: SAINT (Bruker, 2008 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008b ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008b ▶); molecular graphics: SHELXTL (Sheldrick, 2008b ▶); software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S0108270109011275/mx3010sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S0108270109011275/mx3010Isup2.hkl
Table 1. Selected geometric parameters (Å, °).
| Fe1—N11 | 1.9754 (11) |
| Fe1—N21 | 1.9763 (11) |
| Fe1—O2 | 2.0293 (10) |
| Fe1—O1 | 2.0692 (10) |
| Fe2—O3 | 1.8563 (11) |
| Fe2—N14 | 1.9427 (11) |
| Fe2—N24 | 1.9816 (11) |
| C1—O1 | 1.2715 (17) |
| C1—O2 | 1.2745 (17) |
| C1—O3 | 1.2897 (17) |
| N11—Fe1—N21 | 97.44 (4) |
| N11—Fe1—O2 | 126.06 (5) |
| N21—Fe1—O2 | 118.41 (5) |
| N11—Fe1—O1 | 125.21 (4) |
| N21—Fe1—O1 | 125.81 (4) |
| O2—Fe1—O1 | 64.41 (4) |
| O3—Fe2—N14 | 152.19 (5) |
| O3—Fe2—N24 | 111.46 (5) |
| N14—Fe2—N24 | 96.34 (5) |
| O1—C1—O2 | 118.19 (12) |
| O1—C1—O3 | 122.12 (13) |
| O2—C1—O3 | 119.68 (13) |
| C1—O1—Fe1 | 87.85 (8) |
| C1—O2—Fe1 | 89.54 (8) |
| C1—O3—Fe2 | 138.24 (11) |
| C21—N11—Fe1 | 123.30 (9) |
| C12—N11—Fe1 | 108.05 (8) |
| C41—N21—Fe1 | 123.05 (9) |
| C13—N21—Fe1 | 110.10 (8) |
| C24—N14—Fe2 | 124.38 (9) |
| C15—N14—Fe2 | 109.48 (8) |
| C44—N24—Fe2 | 124.31 (9) |
| C16—N24—Fe2 | 106.86 (8) |
Acknowledgments
This work was supported by the National Institutes of Health (grant No. GM65313).
Footnotes
Supplementary data for this paper are available from the IUCr electronic archives (Reference: MX3010). Services for accessing these data are described at the back of the journal.
References
- Aresta, M. & Dibenedetto, A. (2007). Dalton Trans. pp. 2975–2992. [DOI] [PubMed]
- Bruker (2004). APEX2 Version 1.0-22. Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2008). SAINT Version 7.60A. Bruker AXS Inc., Madison, Wisconsin, USA.
- Lewis, N. S. & Nocera, D. G. (2006). Proc. Natl Acad. Sci. USA, 103, 15729–15735. [DOI] [PMC free article] [PubMed]
- Lu, C. C., Saouma, C. T., Day, M. W. & Peters, J. C. (2007). J. Am. Chem. Soc.129, 4–5. [DOI] [PMC free article] [PubMed]
- Ragsdale, S. W. & Kumar, M. (1996). Chem. Rev.96, 2515–2539. [DOI] [PubMed]
- Sadique, A. R., Brennessel, W. W. & Holland, P. L. (2008). Inorg. Chem.27, 784–786. [DOI] [PMC free article] [PubMed]
- Sadique, A. R., Gregory, E. A., Brennessel, W. W. & Holland, P. L. (2007). J. Am. Chem. Soc.129, 8112–8121. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008a). SADABS Version 2008/1. University of Göttingen, Germany.
- Sheldrick, G. M. (2008b). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Smith, J. M., Sadique, A. R., Cundari, T. R., Rodgers, K. R., Lukat-Rodgers, G., Lachicotte, R. J., Flaschenriem, C. J., Vela, J. & Holland, P. L. (2006). J. Am. Chem. Soc.128, 756–769. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S0108270109011275/mx3010sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S0108270109011275/mx3010Isup2.hkl
Crystal structure: contains datablocks I, global. DOI: 10.1107/S0108270109011275/mx3010sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S0108270109011275/mx3010Isup2.hkl