

Abstract
Objective:
The identification of susceptibility alleles to risk of Alzheimer disease (AD) is a major public health priority. Using apolipoprotein E genotype (APOE), we examined whether neuropathologic intermediate phenotypes, the pathology underlying clinical AD that presumably lies intermediate in the causal chain, would increase power for genetic associations.
Methods:
More than 700 older persons underwent annual evaluation and organ donation as part of the Religious Orders Study or Rush Memory and Aging Project. A total of 536 autopsied persons with clinical AD or without dementia with APOE genotyping and a quantitative measure of AD pathology were analyzed. Regression analyses were used to examine the relation of APOE to clinical AD, to the level of cognitive function proximate to death, and to measures of AD neuropathology.
Results:
APOE ɛ4 was associated with increased odds of clinical AD (p = 3 × 10−7), and its association with level of cognition was stronger (p = 8 × 10−12). However, the use of quantitative measures of AD pathology markedly enhanced the association (p = 9 × 10−24). The APOE ɛ2 was not associated with either AD (p = 0.69) or level of cognition (p = 0.82). However, its association with AD pathology (p = 1 × 10−5) was sufficiently strong that it would have warranted follow-up if discovered in a genome-wide association study. Power calculations demonstrate that a sample size of 625 subjects with our measure of AD pathology would be required to meet genome-wide significance of p = 5 × 10−8 for ɛ2.
Conclusion:
Discovery efforts for susceptibility loci for Alzheimer disease could benefit from the use of neuropathologic intermediate phenotypes as a complement to other approaches.
GLOSSARY
- AD
= Alzheimer disease;
- CERAD
= Consortium to Establish a Registry for Alzheimer's Disease;
- CI
= confidence interval;
- MCI
= mild cognitive impairment;
- NIA
= National Institute on Aging;
- OR
= odds ratio.
Genetic factors play an important role in the development of Alzheimer disease (AD).1 While variants in four genes are accepted as causing or increasing risk of AD, they only explain a small proportion of disease occurrence, suggesting that other genetic variants remain to be identified. However, identifying additional genetic variants has proven difficult. One reason relates to phenotypic heterogeneity. While the pathology of AD is often expressed as a dementia syndrome, AD pathology is common in persons without dementia.2 Further, cerebrovascular disease and Lewy bodies also impair cognition and contribute to the AD dementia phenotype.3 We believe that genetic analysis of neuropathologic phenotypes lying intermediate in the causal pathway linking allele status to clinical disease will enhance associations for susceptibility alleles. An overall conceptual model illustrating this is outlined in the figure.

Figure Putative pathways linking genetic and nongenetic risk factors to Alzheimer disease (AD) and other pathology, neurodegeneration, cognitive decline, and clinically diagnosed AD
Note that two or more of these pathways typically operate simultaneously.
We used clinical and postmortem data from the Religious Orders Study and Rush Memory and Aging Project to examine the relation of APOE to clinically diagnosed AD, to the level of cognitive function proximate to death, and to dichotomous and quantitative measures of AD pathology. We focused our assessment on the APOE locus because it is a well-validated susceptibility locus for sporadic, late-onset AD and because the alleles associated with disease risk (ɛ2 and ɛ4) are relatively common in human populations.4,5 We found much more robust associations and larger effect sizes between APOE and level of cognition than for clinically diagnosed disease and far more robust associations with the quantitative postmortem measures of AD pathology than for the clinical outcomes.
METHODS
Study participants.
Clinical and postmortem data came from participants in the Religious Orders Study and Rush Memory and Aging Project.6 In both studies, participants without known dementia at baseline agreed to annual clinical evaluation and brain donation at the time of death. Written informed consent and an Anatomic Gift Act were signed after the procedures were fully explained. Both studies were approved by the Institutional Review Board of Rush University Medical Center. Since 1993, more than 2,300 persons agreed to participate. The overall follow-up rate exceeds 90% of survivors and the autopsy rate is 90%. Clinical and postmortem evaluation procedures allowed for data to be pooled for analyses. More than 700 autopsies have been performed and the neuropathologic evaluation completed on the first 578. We excluded 15 persons with a diagnosis of dementia due to a clinical condition other than AD, 17 persons with APOE ɛ2/4, and 10 with missing genotype. This left 536 persons for analyses, of whom 317 (59.1%) were without dementia, including 137 with mild cognitive impairment (MCI), and 219 (40.9%) with probable or possible AD.
Clinical evaluation.
The clinical diagnoses of AD followed National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association criteria,7 as described.8 At the time of death, clinical data were reviewed by a neurologist without access to postmortem data and a summary diagnostic opinion rendered regarding the most likely clinical diagnosis at the time of death. Level of cognition was based on cognitive testing performed proximate to death. The studies have 19 cognitive performance tests in common. Mini-Mental State Examination9 was used to describe the cohort and one test was used for diagnostic classification purposes only. The remaining 17 tests have been previously described (table 1).6 Tests were converted to z scores, using the mean and SD from the baseline evaluation of all participants, and averaged to yield summary measures of global cognition and five cognitive domains: episodic memory, semantic memory, working memory, perceptual speed, and visuospatial ability. Summary measures minimize floor and ceiling effects and other sources of random variability.
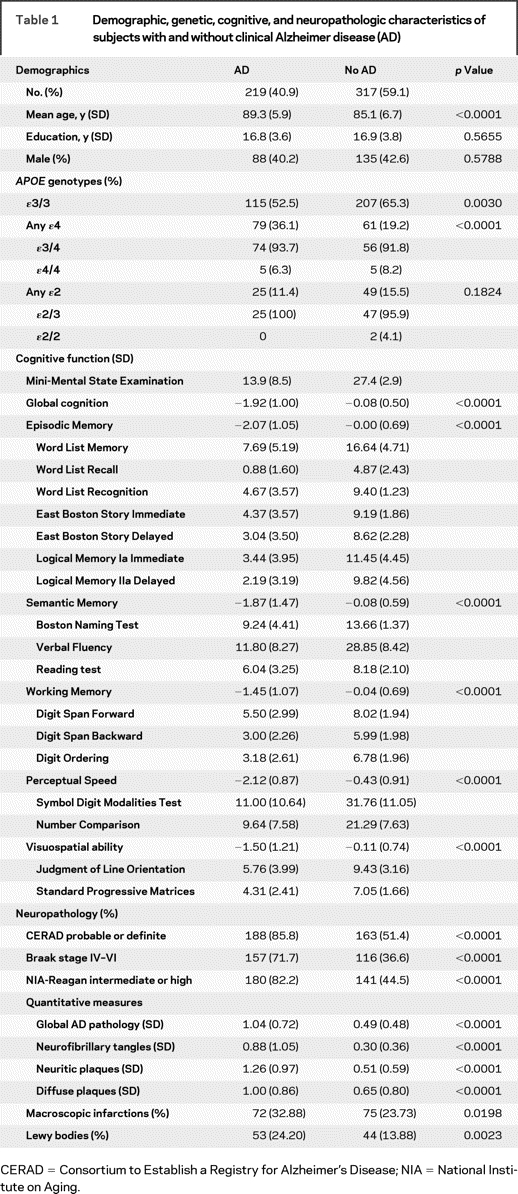
Table 1 Demographic, genetic, cognitive, and neuropathologic characteristics of subjects with and without clinical Alzheimer disease (AD)
Postmortem examination.
Bielschowsky silver stain was used to visualize neuritic plaques, diffuse plaques, and neurofibrillary tangles in tissue sections from the midfrontal, middle temporal, inferior parietal, and entorhinal (proper) cortices, and the hippocampal CA1 sector. Neuropathologic diagnoses of AD were made without access to clinical data. We classified persons as having pathologic AD three ways: the presence of probable or highly probable AD by Consortium to Establish a Registry for Alzheimer's Disease (CERAD) based on semiquantitative estimates of highest neuritic plaque density,10 Braak stage IV–VI based on the distribution and severity of neurofibrillary tangle pathology,11 and intermediate or high likelihood of AD by National Institute on Aging (NIA)-Reagan criteria based on CERAD estimates and Braak staging,12 as described.6 The quantitative composite AD pathology score was based on counts of neuritic plaques, diffuse plaques, and neurofibrillary tangles, as described.13,14 Because the means, standard deviations, and ranges of the data varied widely, we converted the raw counts to a standard distribution by dividing each person's count by the SD for that particular count and formed a summary measure by averaging the scaled scores. Because the data were skewed, square root of the scaled score was used in analyses. Separate summary measures of neurofibrillary tangles and neuritic and diffuse plaques were made. Macroscopic cerebral infarctions and Lewy bodies were determined as described.6
Apolipoprotein E genotyping.
APOE genotyping was performed by Agencourt Bioscience Corporation (Beverly, MA) utilizing high throughput sequencing of codon 112 (position 3937) and codon 158 (position 4075) of exon 4 of the APOE gene on chromosome 19.15
Statistical analysis.
χ2, t, and Wilcoxon tests were used to compare demographics, genotypes, and clinical and neuropathologic variables between those with and without AD. Logistic regression was used to examine the odds of dichotomous outcomes as a function of APOE, and linear regression was used to examine continuous measures as a function of APOE. All analyses were adjusted for age, sex, and education; APOE ɛ3/ɛ3 was the reference group. Analyses were carried out using SAS/STAT software V9 (SAS Institute Inc, Cary, NC). Model assumptions were evaluated analytically and graphically. p Values less than 0.0001 were determined via cumulative distribution functions (PROBCHI and PROBNORM) in SAS DATA steps. Power calculations were performed using the Genetic Power Calculator.16
RESULTS
Table 1 shows the demographics, APOE genotypes, and clinical and neuropathologic indices of the subjects considered in our analyses. As expected, those with clinical AD were older, were more likely to have an ɛ4 allele, scored lower on all cognitive tests, and had more AD and other neuropathologies.
Relation of APOE to clinical AD.
We first examined the expected relation of APOE alleles to the clinical diagnosis of AD proximate to death. The presence of the ɛ4 allele was associated with more than a threefold increase in the odds of clinical AD (odds ratio [OR] = 3.17, 95% confidence interval [CI] = 2.03–4.93, p = 3 × 10−7). By contrast, the ɛ2 allele was associated with only a slight reduction in the odds of clinical AD (OR = 0.89, 95% CI = 0.51–1.6, p = 0.692).
Relation of APOE to level of cognition.
We next examined the relation of APOE to the level of cognition proximate to death (table 2). Note that the p value for ɛ4 in the model with global cognition (p = 8 × 10−12) is clearly below the generally accepted level for genome-wide significance of p < 5 × 10−8.17 The ɛ2 allele remained nonsignificant. Overall, the core model which includes age, sex, and education explained about 11% of the variance of cognition, and APOE allele status explained nearly 9% additional variance. To ensure that the results were not due to those with ɛ4/4, we conducted separate analyses comparing those with ɛ3/4 to those with ɛ3/3 (there were too few cases of ɛ4/4 for meaningful analyses). The results were comparable to the results of the analyses considering any subject with an ɛ4 allele. We next examined the relation of allele status to level of function in five cognitive domains. The association of the ɛ4 allele was most robust for episodic memory (p = 5 × 10−14), where it explained more than 9% of the variance. The association with semantic memory also achieved genome-wide significance whereas the associations with working memory and perceptual speed nearly reached this level of significance. The least robust association in our sample was visuospatial ability, for which ɛ4 explained 2% of the variance, but demonstrated substantial evidence of association (p = 0.0009). By contrast, the ɛ2 allele was not associated with any cognitive outcome.
Table 2 Relation of APOE to level of global cognition and function in five different cognitive systems

Relation of APOE to neuropathologic AD phenotypes.
We next examined the relation of APOE to measures of AD neuropathology, which we hypothesize would be more robustly associated as they are more proximate to genetic variation in the causal chain to clinical AD. We first used three dichotomous measures of the neuropathologic diagnosis of AD that are available in many datasets (table 3). In each case, the effect of APOE ɛ4 exceeded the threshold for genome-wide significance and was more robust than with level of cognition. Further, the ɛ2 allele was marginally significant with the strongest association with CERAD (p = 0.0054).
Table 3 Relation of APOE to dichotomous indices of the neuropathologic diagnosis of AD by CERAD, Braak stage, and NIA-Reagan, and quantitative measures of global AD pathology and separate measures of neurofibrillary tangles and neuritic and diffuse plaques
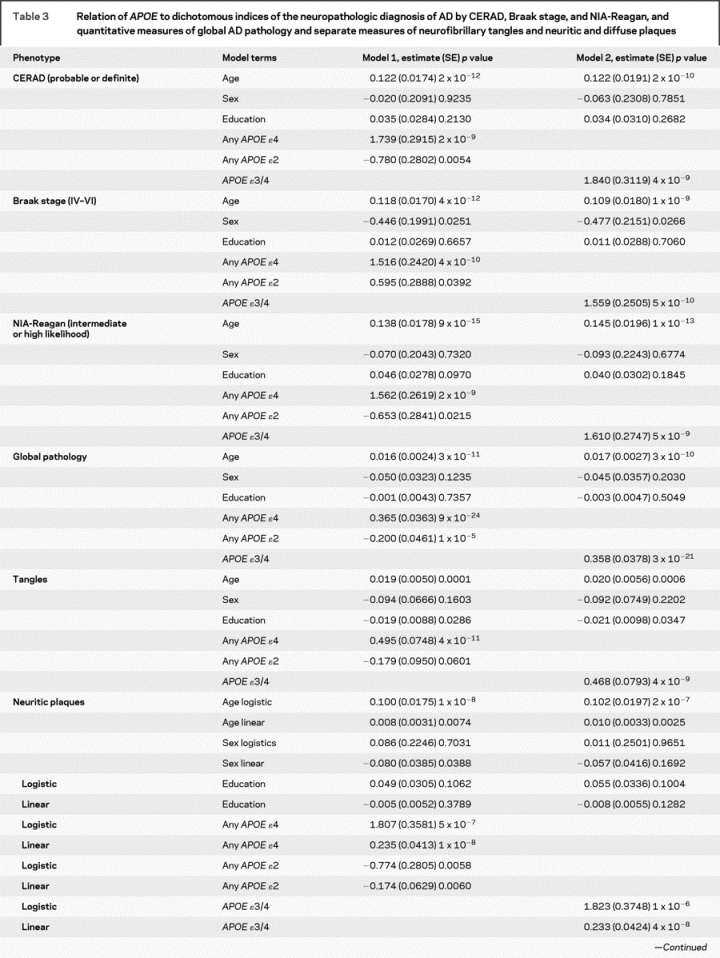
Table 3 Continued
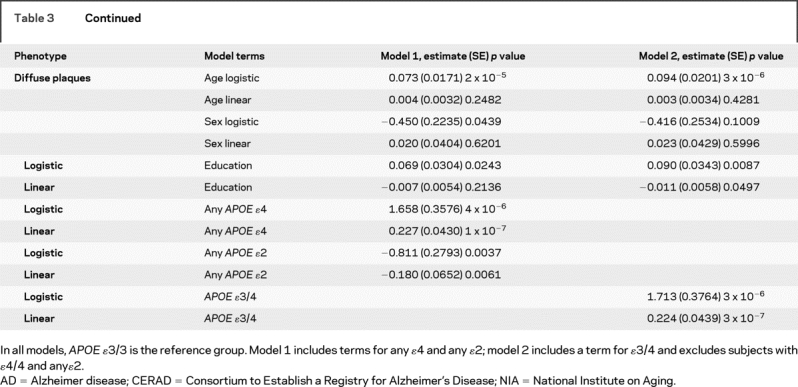
Next, we examined the relation of APOE to a continuous quantitative measure of AD pathology. Again, the effect of APOE ɛ4 far exceeded the threshold for genome-wide significance, and the ɛ2 allele also achieved substantial evidence of association (p = 1 × 10−5) and would warrant further evaluation in a genome-wide study which is often set at 5 × 10−8 < p < 10−3. Overall, the core model which included age, sex, and education explained around 5% of the variance of the global measures of AD pathology whereas allele status explained more than 20% of the variance. Separate analyses revealed that the associations were not due to those subjects with an ɛ4/4 genotype.
Additional analyses were conducted with the counts of three neuropathologic indices that comprise the global measure of AD pathology. The ɛ4 allele was strongly associated with neurofibrillary tangles but ɛ2 was not significant. Because the number of persons without plaques precluded the use of linear regression in the entire sample, analyses of plaque counts proceeded in two stages. First, we used logistic regression to examine the relation of allele status to the presence of plaques. This was followed by linear regression to examine the relation of allele status to the number of plaques among those with non-zero plaque counts. While it is difficult to compare these models directly with the neurofibrillary tangle model, the associations were more robust than the associations with tangles.
Relation of APOE to intermediate phenotypes in persons without and with clinical AD.
Because some genetic factors may influence endophenotypes in the disease population only whereas others may be related to endophenotypes among those with and without disease, we repeated the models separately among the 317 persons without clinical AD and the 219 persons with AD (table 4). Among persons without AD, the ɛ4 allele was marginally related to global cognition but the ɛ2 allele was not. By contrast, the association of the ɛ4 allele with global AD pathology in these subjects greatly exceeded the threshold for genome-wide significance and the ɛ2 allele was also significant. Overall, among persons without AD, allele status explained around 17% of the variance of AD pathology. The results of analyses with a term for ɛ3/4 were also highly significant (data not shown).
Table 4 Relation of APOE to global cognition and global AD pathology in persons without and with clinical AD
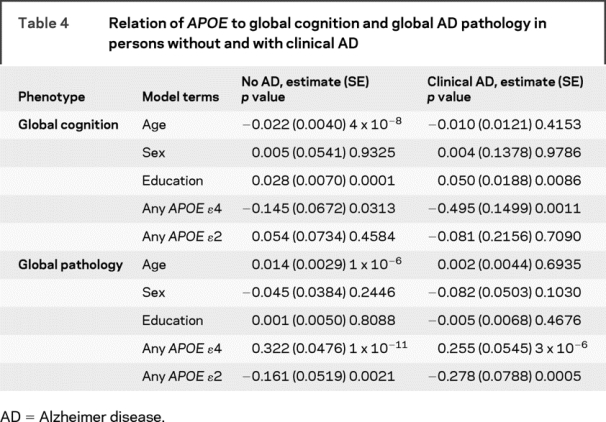
Among those with clinical AD, the ɛ4 allele was also related to global cognition whereas ɛ2 was not. By contrast, the association of the ɛ4 allele with global AD pathology approached the threshold for genome-wide significance, and the ɛ2 allele was also highly significant. Overall, among persons with clinical AD, allele status explained 18% of the variance of AD pathology. Results of analyses with ɛ3/4 were again highly significant (data not shown).
Power calculation for APOE ɛ2.
The results of the ɛ2 allele analyses are interesting since the effect size is similar to those described for loci influencing other complex disease traits (e.g., Type II diabetes). Therefore, we conducted a power calculation to determine the sample size needed to attain significance for a genome-wide study of the ɛ2 allele with the global measure of AD pathology as the study outcome. Assuming an allele frequency of 0.07 that explained 7% of the variance and a model without dominance, only 625 subjects would be required to achieve p = 5 × 10−8 with 90% power.
DISCUSSION
The application of genome-wide association methods to the analysis of human disease has met with recent success. Studies are also being performed for AD, but results have been variable.18–21 In view of the ongoing investment in large genetics consortia by the NIH, empirical data on the use of quantitative intermediate phenotypes for gene discovery, as a complement to other strategies, seems warranted.18,22
Our results demonstrate that phenotypes intermediate in the causal pathway linking APOE to clinical AD provide considerably more power to detect genetic variants compared with the more distal clinical diagnosis. With the current sample size, genome-wide significance was not achieved with clinical AD as the outcome, however, the ɛ4 allele achieved genome-wide significance with level of cognition and was far more robust with quantitative measures of AD pathology. While the ɛ2 allele was not associated with clinical AD or level of cognition with the present sample size, it was strongly associated with AD pathology and power calculations suggest that a sample size of only 625 would be needed to reach genome-wide significance with a quantitative measure of AD pathology as the study outcome. Finally, the associations with ɛ4 were not due solely to those subjects with the ɛ4/4 genotype, were present in persons with and without clinical AD, and were evident across multiple domains of cognition and different measures of AD pathology.
We suspect that our findings result in part from phenotypic heterogeneity. Our conceptual model (figure) is supported by several findings. First, all known susceptibility loci for AD (APP, PSEN1, PSEN2, and APOE) alter the metabolism of the amyloid-β peptide and result in the accumulation of neuritic plaques and neurofibrillary tangles.1 Second, while persons meeting rigorous clinical criteria for AD nearly always meet pathologic criteria for the disease, many persons without dementia, especially those with MCI, have significant AD neuropathology.2,3 In prior work in these two cohorts, most persons with MCI and a third of those without cognitive impairment met neuropathologic criteria for AD.6,23 Third, AD pathology appears to mediate the association of APOE with cognition and clinical disease.13 The model also illustrates additional pathways leading to clinical AD such as other genes associated with AD pathology, and genetic variants for clinical AD associated with cerebral infarctions, Lewy bodies, other neuropathologies, or ones that directly lead to neurodegenerative changes. For example, APOE has been related to measures of cerebrovascular disease and to neural repair mechanisms.24–26 Ultimately, it is important to understand all of these pathways as the prevention of the clinical dementia syndrome is of prime interest. Further, although presented as independent pathways, two or more operate simultaneously.3,27
Prior studies of genetic variants have employed AD neuropathology as the phenotype. For example, among persons with AD, some studies find an association between the presence of one or more ɛ4 alleles and neuritic plaques whereas other studies only find the association among persons with two copies of this allele.24,28 Less data are available from persons without dementia where the presence of the ɛ4 allele has been associated with amyloid deposition.29 AD pathology has also been examined in relation to other polymorphisms. CYP46 showed associations with amyloid and tau among those with and without AD in some studies, but not in a separate study of persons with AD.30,31 Interestingly, in one study, IDE polymorphisms were associated with measures of AD pathology but not cognitive status among persons with AD.32 Thus, finding an association with neuropathology does not guarantee that one has identified a risk factor for clinical disease and separate validation efforts for the clinical phenotype are warranted. Nonetheless, an allele associated with AD-related neuropathology would have a high likelihood of association with clinical AD, and the analyses presented here suggest that we should extend current studies that have used clinical and neuropathologic data to assign case status in whole genome studies to explore neuropathology itself as the phenotype of interest.19
There are other intermediate phenotypes that could be targeted by this strategy for gene discovery. Indeed, biomarkers of disease including structural and functional imaging, and CSF studies, have been examined in relation to susceptibility alleles.33–35 The best studied AD intermediate phenotype is rate of cognitive decline. We and others have reported strong associations between APOE and cognitive decline.36–38 Some of these studies, restricted to persons without dementia, have found associations between APOE and cognitive decline, and others have found associations with cognitive decline when there was insufficient power to discover association to incident AD. Nonetheless, statistical concerns remain regarding the ability of genome scans to find associations with change in quantitative traits.39
Clinical AD, level of cognition, and neuropathologic AD are all closely related variables. Thus, analyses including persons with and without AD may capitalize on these associations and appear to represent factors related to disease risk that may not be causal. While these analyses cannot prove causality, a number of findings raise the possibility that APOE influences both the accumulation of AD pathology and risk of clinical AD. First, APOE is a well-established risk factor for incident AD in a number of populations separate from the cohorts used in these analyses.1,4 Second, analyses were conducted separately among those with and without clinical AD and the ɛ4 allele was related to both level of cognition and measures of neuropathology in both groups. Third, we previously demonstrated that measures of AD pathology mediated the association of allele status to level of cognition and clinical AD.13 By contrast, the actual measures of AD pathology used in these analyses may not directly be in the causal chain. For example, molecularly specific antibodies might provide additional information regarding biologic pathways linking risk alleles to clinical disease. Some preclinical data suggest that the known genetic variants associated with clinical AD work through soluble oligomers of amyloid.40 Thus, the neuropathologic indices used in this study may be biologic markers of the true causal indices, and even more power might be obtained in analyses of other proteins that are more causally related.
There are a number of strengths to this study. Subjects in both cohorts had high rates of follow-up and high autopsy rates, both of which reduce bias. The volunteer nature of both cohorts probably served to enrich the sample with APOE as the presence of AD in a close family member often motivated participation. Finally, structured clinical and pathologic procedures and quantitative measures of AD pathology with excellent metric properties for these types of analyses were used in analyses.
AUTHOR CONTRIBUTIONS
Statistical analyses were performed by P.L.D. and S.E.L.
ACKNOWLEDGMENT
The authors thank the participants in the Religious Orders Study and the Memory and Aging Project and the staff of the Rush AD Center.
Address correspondence and reprint requests to Dr. David A. Bennett, Rush Alzheimer's Disease Center, 600 S. Paulina, Suite 1028, Chicago, IL 60062 dbennett@rush.edu
Supported by NIH grants P30AG10161, R01AG15819, R01AG17917, and K08NS46341.
Disclosure: The authors report no disclosures.
Received October 9, 2008. Accepted in final form February 2, 2009.
REFERENCES
- 1.Ertekin-Taner N. Genetics of Alzheimer's disease: a centennial review. Neurol Clin 2007;25:611–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petersen RC, Parisi JE, Dickson DW, et al. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol 2006;63:665–672. [DOI] [PubMed] [Google Scholar]
- 3.Sonnen JA, Larson EB, Crane PK, et al. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol 2007;62:406–413. [DOI] [PubMed] [Google Scholar]
- 4.Evans DA, Beckett LA, Field TS, et al. Apolipoprotein E epsilon4 and incidence of Alzheimer disease in a community population of older persons. JAMA 1997;277:822–824. [PubMed] [Google Scholar]
- 5.Daw EW, Payami H, Nemens EJ, et al. The number of trait loci in late-onset Alzheimer disease. Am J Hum Genet 2000;66:196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based clinical-pathologic studies. Neurology 2006;66:1837–1844. [DOI] [PubMed] [Google Scholar]
- 7.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan E. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 8.Bennett DA, Schneider JA, Aggarwal NT, et al. Decision rules guiding the clinical diagnosis of Alzheimer's disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study. Neuroepidemiology 2006;27:169–176. [DOI] [PubMed] [Google Scholar]
- 9.Folstein MF, Folstein SE, McHugh PR. ‘Mini-Mental State’: a practical method for grading the mental state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 10.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD): II: Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486. [DOI] [PubMed] [Google Scholar]
- 11.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 12.Consensus recommendations for the postmortem diagnosis of Alzheimer's disease: The National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging 1997;18(suppl 4):S1–S2. [PubMed] [Google Scholar]
- 13.Bennett DA, Wilson RS, Schneider JA, et al. Apolipoprotein E4 allele, Alzheimer's disease pathology, and the clinical expression of Alzheimer's disease. Neurology 2003;60:246–252. [DOI] [PubMed] [Google Scholar]
- 14.Bennett DA, Schneider JA, Arnold SE, Tang Y, Wilson RS. The effect of social networks on the relation between Alzheimer's disease pathology and level of cognitive function in old people: a longitudinal cohort study. Lancet Neurol 2006;5:406–412. [DOI] [PubMed] [Google Scholar]
- 15.Buchman AS, Boyle PA, Wilson RS, Beck TL, Kelly JF, Bennett DA. Apolipoprotein E ɛ4 allele is associated with more rapid motor decline in older persons. Alzheimer Dis Assoc Disord 2009;23:63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 2003;19:149–150. [DOI] [PubMed] [Google Scholar]
- 17.Pe'er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol 2008;32:381–5. [DOI] [PubMed] [Google Scholar]
- 18.Lee JH, Cheng R, Graff-Radford N, Foroud T, Mayeux R. National Institute on Aging Late-Onset Alzheimer's Disease Family Study Group. Analyses of the National Institute on Aging Late-Onset Alzheimer's Disease Family Study: implication of additional loci. Arch Neurol 2008;65:1518–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reiman EM, Webster JA, Myers AJ, et al. GAB2 alleles modify Alzheimer's risk in APOE epsilon4 carriers. Neuron 2007;54:713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer's disease. J Clin Psychiatry 2007;68:613–618. [DOI] [PubMed] [Google Scholar]
- 21.Grupe A, Abraham R, Li Y, et al. Evidence for novel susceptibility genes for late onset Alzheimer's disease from a genome-wide association study of putative functional variants. Hum Mol Genet 2007;16:865–873. [DOI] [PubMed] [Google Scholar]
- 22.Blacker D, Albert MS, Bassett SS, Go RC, Harrell LE, Folstein MF. Reliability and validity of NINCDS-ADRDA criteria for Alzheimer's disease: The National Institute of Mental Health Genetics Initiative. Arch Neurol 1994;51:1198–1204. [DOI] [PubMed] [Google Scholar]
- 23.Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS. Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology 2005;64:834–841. [DOI] [PubMed] [Google Scholar]
- 24.Tiraboschi P, Hansen LA, Masliah E, Alford M, Thal LJ, Corey-Bloom J. Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology 2004;62:1977–1983. [DOI] [PubMed] [Google Scholar]
- 25.Schneider JA, Bienias JL, Wilson RS, Berry-Kravis E, Evans DA, Bennett DA. Relation of the apolipoprotein E ɛ4 allele to cerebral infarction in older persons. Stroke 2005;36:954–959. [DOI] [PubMed] [Google Scholar]
- 26.Ji Y, Gong Y, Gan W, Beach T, Holtzman DM, Wisniewski T. Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer's disease patients. Neuroscience 2003;122:305–315. [DOI] [PubMed] [Google Scholar]
- 27.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community dwelling older persons. Neurology 2007;69:2197–2204. [DOI] [PubMed] [Google Scholar]
- 28.Yip AG, McKee AC, Green RC, et al. APOE, vascular pathology, and the AD brain. Neurology 2005;65:259–265. [DOI] [PubMed] [Google Scholar]
- 29.Polvikoski T, Sulkava R, Haltia M, et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med 1995;333:1242–1247. [DOI] [PubMed] [Google Scholar]
- 30.Papassotiropoulos A, Streffer JR, Tsolaki M, et al. Increased brain beta-amyloid load, phosphorylated tau, and risk of Alzheimer disease associated with an intronic CYP46 polymorphism. Arch Neurol 2003;60:29–35. [DOI] [PubMed] [Google Scholar]
- 31.Ingelsson M, Jesneck J, Irizarry MC, Hyman BT, Rebeck GW. Lack of association of the cholesterol 24-hydroxylase (CYP46) intron 2 polymorphism with Alzheimer's disease. Neurosci Lett 2004;367:228–231. [DOI] [PubMed] [Google Scholar]
- 32.Blomqvist ME, Chalmers K, Andreasen N, et al. Sequence variants of IDE are associated with the extent of beta-amyloid deposition in the Alzheimer's disease brain. Neurobiol Aging 2005;26:795–802. [DOI] [PubMed] [Google Scholar]
- 33.Fleisher A, Grundman M, Jack CR Jr, et al. Sex, apolipoprotein E epsilon 4 status, and hippocampal volume in mild cognitive impairment. Arch Neurol 2005;62:953–957. [DOI] [PubMed] [Google Scholar]
- 34.Reiman EM, Chen K, Alexander GE, et al. Correlations between apolipoprotein E epsilon4 gene dose and brain-imaging measurements of regional hypometabolism. Proc Natl Acad Sci USA 2005;102:8299–8302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peskind ER, Li G, Shofer J, et al. Age and apolipoprotein Eɛ4 allele effects on cerebrospinal fluid beta-amyloid 42 in adults with normal cognition. Arch Neurol 2006;63:936–939. [DOI] [PubMed] [Google Scholar]
- 36.Wilson RS, Bienias JL, Berry-Kravis E, Evans DA, Bennett DA. The apolipoprotein E epsilon 2 allele and decline in episodic memory. J Neurol Neurosurg Psychiatry 2002;73:672–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McQueen MB, Bertram L, Lange C, et al. Exploring candidate gene associations with neuropsychological performance. Am J Med Genet B Neuropsychiatr Genet 2007;144B:987–991. [DOI] [PubMed] [Google Scholar]
- 38.Caselli RJ, Reiman EM, Locke DE, et al. Cognitive domain decline in healthy apolipoprotein E epsilon4 homozygotes before the diagnosis of mild cognitive impairment. Arch Neurol 2007;64:1306–1311. [DOI] [PubMed] [Google Scholar]
- 39.Gauderman WJ, Macgregor S, Briollais L, et al. Longitudinal data analysis in pedigree studies. Genet Epidemiol 2003;25 suppl 1:S18–S28. [DOI] [PubMed] [Google Scholar]
- 40.Lesné S, Koh MT, Kotilinek L, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006;440:352–357. [DOI] [PubMed] [Google Scholar]