

Abstract
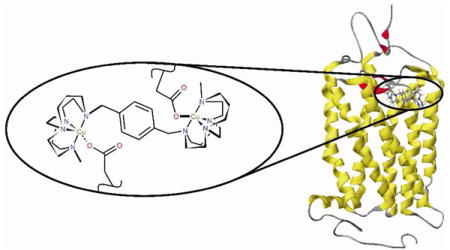
A new copper(II) containing bis-macrocyclic CXCR4 chemokine receptor antagonist is shown to have improved binding properties to the receptor protein in comparison to the drug AMD3100 (Plerixafor, Mozobil™). The interaction of the metallodrug has been optimized by using ultra rigid chelator units that offer an equatorial site for coordination to the amino acid side chains of the protein. Binding competition assays with anti-CXCR4 antibodies show that the new compound stays bound longer and it has improved anti-HIV potency in vitro (EC50 = 4.3 nM). X-ray structural studies using acetate as a model for carboxylate amino acid side chains indicate the nature of the coordination interaction.
The selectivity and binding affinity to the biological target are traditionally considered in selecting a potential drug candidate. In the case of G-protein coupled receptors (GPCRs) the drug residence time is also thought to be a key factor.1 The metal ions in metallodrugs can interact with donor atoms on a biological target via the formation of coordinate bonds rather than a combination of weaker intermolecular forces such as H-bonding, and there is an opportunity to tune this coordination chemistry by appropriate pairing of the metal ion and chelator, thus increasing the residence time. The chelator should form a high stability complex to retain the metal ion in vivo and exchangeable ligands must be present to allow coordination of amino acid side chains. The chemokine receptor CXCR4 is the GPCR target in this work. It has a surface that is rich in aspartate and glutamate residues which bind strongly to transition metals.
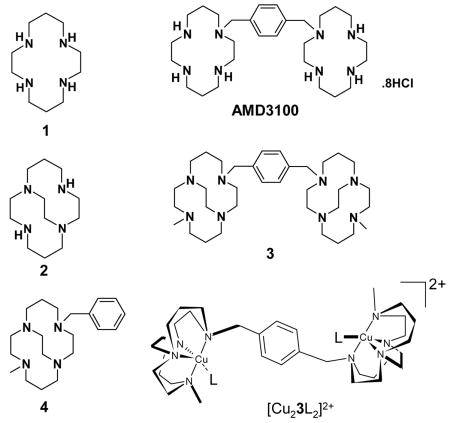
The drug candidates we have investigated are bis-aza-macrocyclic compounds. The CXCR4 antagonist AMD3100 is the best known drug in this class (also known as Mozobil™ or Plerixafor).2 New drug applications have been submitted in both the US and Europe (17 June 2008) for its use as a stem cell mobilizing agent to allow harvesting and transplantation of hematopoietic stem cells in patients with lymphoma and multiple myeloma3 and it has previously been investigated as an anti-HIV cell entry inhibitor.4 The AMD3100 binding site on CXCR4 has been well characterized5 and its metal complexes studied; making it an excellent starting point for development of a novel optimized metallodrug.6,7
We have previously investigated the effect of configurational fixing of metallodrugs to provide only one chelator ‘shape’ for binding.8 The aim of this work is to apply the combined effects of optimized coordinate bond formation and structural rigidity to improve drug potency by increasing the time the compound stays bound to the receptor. A highly rigid chelator, with an open face of exchangeable ligands, will favor the metal coordination of carboxylate oxygen atoms from the aspartate or glutamate side chains of CXCR4, see Figure 1. The inflexibility of the chelator should contribute to both the kinetic stability of the metal-chelator complex by inhibiting the dissociation process9 and the drug-protein complex by providing a preorganized, optimized configuration of the drug.
Figure 1.

(a) Space filling plots showing the access to the binding site at the copper(II) center for the most common cyclam (1) configuration which is expected for the AMD3100 complex (left) and the folded configuration of the cross bridged complex (right) and (b) A ball and stick representation of the X-ray crystal structure of [Cu4(OAc)]PF6. Bond lengths in Å: Cu(1)-O(1) 1.946(2); Cu(1)-N(1) 2.190(3); Cu(1)-N(2) 2.143(3); Cu(1)-N(3) 2.044(3); Cu(1)-N(4) 2.095(3).
Studies were carried out to confirm that the copper(II) complex of 3 binds to the CXCR4 chemokine receptor on Jurkat T-cells (which have ca. 140,000 CXCR4 receptors on the cell surface)10. Competition binding assays showed that the drug was not displaced from the receptor site by anti-CXCR4 monoclonal antibodies. A proliferation assay was then carried out which showed no cytotoxicity and similar growth profiles for [Cu23]4+, AMD3100 and [Cu2AMD3100]4+ at 20 μM concentration. It is proposed that an increased potency of this type of drug will correspond to a longer residence time (slower dissociation rate) at the CXCR4 receptor.
To obtain an indication of how long the drug remains bound to the cell receptor using this type of assay, the inhibition of anti-CXCR4 antibody binding over time in a replicating population of cells was investigated. The cells were exposed to the drug molecule, the excess was washed away, and then the binding was monitored by flow cytometry using antibody competition assays, see Figure 2. After 24 hours the most significant drop in binding inhibition is observed for AMD3100 and by 48 hours inhibition is only observed for [Cu23]4+. These data imply that the copper(II) complex of the configurationally fixed chelator 3 has an increased residence time at the CXCR4 chemokine receptor, which may correlate to an improved potency.
Figure 2.

The inhibition of anti-CXCR4 antibody binding over time after exposure to ca. 1,400 molecules of the drug per receptor. A population of 1 × 105 cells was used for each data point and analyzed by flow cell cytometry using a secondary fluorescein tagged IgG antibody (negative values are not shown).
The anti-HIV properties of the new optimized drug provide an appropriate testing ground as data is available for both AMD3100 and [Cu2AMD3100]4+. The in vitro anti-HIV activity of [Cu23]4+ was investigated in MT-4 cells against a R4 strain of HIV-1 (which gains cell entry using the CXCR4 co-receptor) giving an EC50 value of 4.3 nM. In comparison with the published values for AMD3100 and [Cu2AMD3100]4+, 11 nM and 47 nM respectively,11 [Cu23]4+ shows improved potency which can be related to the receptor residence time as the experiment is carried out over a 5 day period.
The difference in activity between [Cu23]4+ and AMD3100 can be attributed to the formation of coordinate bonds in the copper complex rather than the dominant H-bonding interactions for AMD3100. However, it is clearly not as simple as just incorporating a metal center to improve the drug interaction or improved properties would have been observed for [Cu2AMD3100]4+. The interaction with the metal center must be optimized, and so characterization of the coordination environment of the copper(II) ion is important.
It is most likely that the coordinate bond will form with the side chain from one of the exposed aspartate residues on the surface of the CXCR4 receptor. Therefore acetate bound salts of these copper complexes provide a useful model of the bonding interaction and attempts were made to grow single crystals to gain precise geometric parameters using crystallography. The acetate salt of [Cu23]4+ could not be crystallized and so crystals were grown of the analogous single ring copper(II) complex of 4. In [Cu4(OAc)]PF6, the copper(II) center adopts a distorted square based pyramidal geometry, bound to all four macrocycle nitrogens and one acetate oxygen, with the second acetate O donor at a non-bonded distance, see Figure 1. The tetraaza-macrocycle is forced into a folded arrangement (cis-V)12 by the ethyl bridge and the ring nitrogens occupy three of the equatorial sites and the axial site, which is in contrast to the most stable of the four observed configurations for copper(II) cyclam complexes (trans-III)12 that has the ring N atoms planar and bound in all four of the equatorial sites. On protein binding the most stable configuration of copper(II) AMD3100 would form a weaker axial bond to the aspartate side chain, whereas [Cu23]4+ forms a shorter, stronger equatorial coordinate bond.
In conclusion, coordination chemistry provides strong interactions that can be used to improve receptor binding drugs, offering complementary options to multiple hydrogen bonds. The compound designed in this study has rigid chelator units optimized with an open face and suitable geometry to form strong coordinate bonds with the metal center. This improves the antagonist properties in competition with CXCR4 binding antibodies and its anti-HIV potency over the flexible chelator (AMD3100) and its copper(II) complex. Due to the high stability of radiolabeled bridged macrocyclic complexes13 we are investigating the 64Cu labeling of 3, which could potentially be used for in vivo monitoring of levels of CXCR4 expression by PET imaging.
Supplementary Material
Acknowledgments
We would like to acknowledge Yorkshire Cancer Research, the National Center for Research Resources (a component of the NIH) (P2PRR016478), Research Corporation (CC6505) and The University of Hull for funding.
Footnotes
Supporting Information Available: synthetic details; flow cytometry data; time dependent antibody binding assay and cell viability; anti-HIV assay; crystal data for [Cu4(OAc)]PF6.
References
- 1.Tummino PJ, Copeland RA. Biochemistry. 2008;47(20):5481. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- 2.De Clercq E. Nat Rev Drug Discov. 2003;2(7):581. doi: 10.1038/nrd1134. [DOI] [PubMed] [Google Scholar]
- 3.Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, Liles WC, Li XX, Graham-Evans B, Campbell TB, Calandra G, Bridger G, Dale DC, Srour EF. J Exp Med. 2005;201(8):1307. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Clercq E, Yamamoto N, Pauwels R, Baba M, Schols D, Nakashima H, Balzarini J, Debyser Z, Murrer B, Schwartz D, Thornton D, Bridger G, Fricker S, Henson G, Abrams M, Picker D. Proc Natl Acad Sci U S A. 1992;89(12):5286. doi: 10.1073/pnas.89.12.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hatse S, Princen K, Gerlach LO, Bridger G, Henson G, De Clercq E, Schwartz TW, Schols D. Mol Pharmacol. 2001;60(1):164. doi: 10.1124/mol.60.1.164. [DOI] [PubMed] [Google Scholar]
- 6.Gerlach LO, Jakobsen JS, Jensen KP, Rosenkilde MR, Skerlj RT, Ryde U, Bridger GJ, Schwartz TW. Biochemistry. 2003;42(3):710. doi: 10.1021/bi0264770. [DOI] [PubMed] [Google Scholar]
- 7.Liang XY, Parkinson JA, Weishaupl M, Gould RO, Paisey SJ, Park HS, Hunter TM, Blindauer CA, Parsons S, Sadler PJ. J Am Chem Soc. 2002;124(31):9105. doi: 10.1021/ja0260723. [DOI] [PubMed] [Google Scholar]
- 8.(a) Valks GC, McRobbie G, Lewis EA, Hubin TJ, Hunter TM, Sadler PJ, Pannecouque C, De Clercq E, Archibald SJ. J Med Chem. 2006;49:6162. doi: 10.1021/jm0607810. [DOI] [PubMed] [Google Scholar]; (b) Khan A, Silversides JD, Madden L, Greenman J, Archibald SJ. Chem Commun. 2007:416. doi: 10.1039/b614557d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McRobbie G, Valks GC, Empson CJ, Khan A, Silversides JD, Pannecouque C, De Clercq E, Fiddy SG, Bridgeman AJ, Young NA, Archibald SJ. Dalton Trans. 2007:5008. doi: 10.1039/b705800d. [DOI] [PubMed] [Google Scholar]; (d) Lewis EA, Hubin TJ, Archibald SJ. Patent WO2005121109. 2005
- 9.(a) Weisman GR, Wong EH, Hill DC, Rogers ME, Reed DP, Calabrese JC. Chem Commun. 1996;(8):947. [Google Scholar]; (b) Hubin TJ, McCormick JM, Collinson SR, Alcock NW, Busch DH. Chem Commun. 1998;(16):1675. [Google Scholar]
- 10.Lee B, Sharron M, Montaner LJ, Weissman D, Doms RW. Proc Natl Acad Sci U S A. 1999;96(9):5215. doi: 10.1073/pnas.96.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esté JA, Cabrera C, De Clercq E, Struyf S, Van Damme J, Bridger G, Skerlj RT, Abrams MJ, Henson G, Gutierrez A, Clotet B, Schols D. Mol Pharmacol. 1999;55(1):67. doi: 10.1124/mol.55.1.67. [DOI] [PubMed] [Google Scholar]
- 12.Bosnich B, Poon CK, Tobe ML. Inorg Chem. 1965;4(8):1102. [Google Scholar]
- 13.Lewis EA, Boyle RW, Archibald SJ. Chem Commun. 2004;(19):2212. doi: 10.1039/b406906d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.