

Abstract
The pregnane X receptor (PXR; PXR.1) can be activated by structurally diverse lipophilic ligands. PXR.2, an alternatively spliced form of PXR, lacks 111 nucleotides encoding 37 amino acids in the ligand binding domain. PXR.2 bound a classic CYP3A4 PXR response element (PXRE) in electrophoretic mobility shift assays, but transfected PXR.2 failed to transactivate a CYP3A4-promoter-luciferase reporter plasmid in HepG2 cells treated with various PXR ligands. Cotransfection experiments showed that PXR.2 behaved as a dominant negative, interfering with PXR.1/rifampin activation of CYP3A4-PXRE-LUC. In HepG2 and LS180 cells stably transduced with PXR.1, PXR target genes (CYP3A4, MDR1, CYP2B6, and UGT1A1) were higher than mock-transduced cells in the absence of ligand and were further induced in the presence of rifampin. In contrast, PXR.2 stably introduced into the same host cells failed to induce target genes over levels in mock-transfected cells after drug treatment. Our homology modeling suggests that ligands bind PXR.1 more favorably, probably because of the presence of a key disordered loop region, which is missing in PXR.2. Yeast two-hybrid assays revealed that, even in the presence of ligand, the corepressors remain tightly bound to PXR.2, and coactivators are unable to bind at helix 12. In summary, PXR.2 can bind to PXREs but fails to transactivate target genes because ligands do not bind the ligand binding domain of PXR.2 productively, corepressors remain tightly bound, and coactivators are not recruited to PXR.2.
The pregnane X receptor (PXR; NR1I2 also known as the steroid and xenobiotic receptor or pregnane-activated receptor) (Bertilsson et al., 1998; Blumberg et al., 1998; Kliewer et al., 1998) is a nuclear receptor that is highly expressed in tissues involved in detoxification such as liver and intestine (Lehmann et al., 1998) and other steroid-sensitive tissues such as breast (Fukuen et al., 2002). PXR has been implicated in cholesterol and bile acid synthesis and metabolism, steroid hormone metabolism, and bone homeostasis (Jones et al., 2000; Kliewer and Willson, 2002) as it is activated by a wide array of 5α- and 5β-bile acids and high concentrations of other endogenous ligands (Krasowski et al., 2005a,b). The role of PXR is ever-expanding as ligands are continuing to be added to an already wide range of structurally diverse lipophilic ligands (Schuetz and Strom, 2001; Krasowski et al., 2005b), which include steroids, vitamins, oxysterols, bile acids, and numerous drugs (Yasuda et al., 2008). PXR shows broad ligand binding specificity because its ligand binding domain (LBD) contains a novel insert that expands its size.
On ligand activation, PXR forms a heterodimer with the retinoid X receptor (RXR) to bind to DNA response elements in regulatory regions of target genes. PXR is widely known to be a key regulator of many enzymes (Kliewer and Willson, 2002) and transporters (Synold et al., 2001) involved in drug detoxification and endogenous homeostasis. It is noteworthy that there are striking species differences in PXR activation as a result of sequence differences in the LBD. For example, rifampin activates human PXR but not rat PXR, and residues Leu308 and Phe305 of human and rat PXR, respectively, were found to be important for this difference (Tirona et al., 2004).
To increase diversity of protein isoforms differing in structural or functional properties, human genes can use alternative RNA processing to generate multiple mRNA products. Alternatively spliced PXR mRNAs have been detected in human liver, breast tissue, colon, and small intestine (Dotzlaw et al., 1999; Fukuen et al., 2002; Lamba et al., 2004). The most abundant alternatively spliced PXR mRNA transcripts have deletions compared with the full mRNA transcript. PXR.2 lacks 111 nucleotides, resulting in a deletion of 37 amino acids from the LBD (Fig. 1A). PXR.2 is also the most abundant alternatively spliced transcript in human liver, accounting for nearly 7% of total PXR mRNA transcripts (Lamba et al., 2004).
Fig. 1.

A, comparison of the structures of human PXR.1 and PXR.2. PXR.2 lacks 111 nucleotides, resulting in a deletion of 37 amino acids from the LBD as shown in the gray-shaded region. Three active site residues that line the ligand-binding pocket of PXR.1 and that form contacts with SR12813 are missing from PXR.2. B, amino acid sequence alignment of the LBDs of human PXR.1 and PXR.2. Structural elements (α helical regions and β strands) and several amino acids important for homodimerization (arrows) are indicated.
What is the functionality of PXR.2? One hypothesis is that PXR.2 would be incapable of binding PXR.1 ligands because the amino acids deleted in human PXR.2 reside within the unique ∼50 amino acid insert that expands the PXR ligand binding pocket (Watkins et al., 2001, 2003a,b; Maglich et al., 2003; Moore et al., 2003) allowing it to accommodate structurally diverse ligands. Nevertheless, it is possible that PXR.2 might retain ligand activation because these same amino acids are lacking in other nuclear hormone receptors, e.g., farnesoid X receptor (FXR; NR1H4) and liver X receptor β (NR1H2) (Ekins et al., 2002), that are still activated by some PXR ligands (bile acids and oxysterols) (Reschly et al., 2008). In addition, mouse PXR.3 (lacking the same amino acids as human PXR.2 plus an additional five amino acids) was shown to be activated by dexamethasone, albeit significantly less than mouse PXR.1 (Kliewer et al., 1998). However, human PXR.2 was not capable of inducing UGT1A in transiently transfected HepG2 and Caco-2 cells in response to rifampin (Gardner-Stephen et al., 2004), and it was reported that compared with PXR.1, PXR.2 decreased basal and corticosterone-induced CYP3A4-luciferase activity in LS174T cells (Hustert et al., 2001). However, because PXR activation of target genes shows ligand, promoter, and cell-type specificity, it would be difficult to extrapolate the few studies to date on PXR.2 function to a broader number of substrates and gene targets.
Hence, the primary objectives of this study were as follows: 1) to characterize differences in the functionality of PXR.2 compared with PXR.1 in vitro using cell and biochemical assays; 2) to determine whether PXR.2 could alter PXR.1-regulated gene expression because PXR.2 is coexpressed in human tissues with PXR.1; 3) to characterize differences in the functionality of PXR.2 compared with PXR.1 in silico using homology modeling and intrinsic disorder (ID) analysis; and 4) to evaluate whether the difference in PXR.2 activation by rifampin is caused by altered association with corepressors and coactivators.
Materials and Methods
Reagents. Reagents for polymerase chain reaction (PCR) and quantitative real-time PCR (SYBR Green) were obtained from Invitrogen (Carlsbad, CA) and Roche Palo Alto LLC (Palo Alto, CA), respectively. Dexamethasone butyl-acetate was purchased from Research Plus (Bayonne, NJ); phenobarbital was from Mallinckrodt Baker, Inc. (Phillipsburg, NJ); and all the other drugs, steroids, and sterols from Sigma-Aldrich (St. Louis, MO) were of the highest purity available.
Generation of PXR.1 and PXR.2 Expression Plasmids. pSG5-PXR.1 ΔATG was a gift from Dr. Steven Kliewer. To generate pSG5-PXR.2 ΔATG, PXR.2 was amplified from a human liver sample using primers (forward) TCGTACGAATTCAACATGGAGGTGAGACCC and (reverse) AGAGTCCCTAGGTCAGCTACCTGTGATGCC containing EcoRI and BamHI sites; that changed the initiator methionine from CTG to an ATG, and the product was directionally cloned into pSG5.
MSCV-PXR.1-IRES-GFP and MSCV-PXR.2-IRES-GFP (hereafter referred to as pMSCV-PXR.1 and pMSCV-PXR.2, respectively) were created by amplifying PXR.1 and PXR.2 from pSG5-PXR.1 ΔATG and pSG5-PXR.2 ΔATG using primers containing EcoRI (forward) TCGTACGAATTCAACATGGAGGTGAGACCC and XhoI (reverse) CTCGAGCTCGAGTCAGCTACCTGTGATGCC restriction sites and cloning into MSCV-IRES-GFP, which was derived from MSCV-IRES-neomycin (Clontech, Mountain View, CA), in which neomycin was replaced with green fluorescent protein (GFP) from pEGFP (Clontech).
pcDNA3-PXR.1 ΔATG was created by amplifying PXR from primary human hepatocyte RNA using PXR primer sequences containing EcoRI restriction sites (forward) CCGGAATTCCGGTGGAGGTGAGACCCAAAGAAAGC and (reverse) CCGGAATTCCGGCTCAGCTACCTGTGATGCCG and ligated into the EcoRI site of pcDNA3. The initiator methionine was changed from a CTG to an ATG by site-directed mutagenesis using (forward) GTGCTGGAATTCCGATGGAGGTGAGACCC and (reverse) GGGTCTCACCTCCATCGGAATTCCAGCAC primers. pcDNA3PXR.2 ΔATG, in which amino acids 174 to 210 were deleted, was a gift from Dr. Oliver Burke (Hustert et al., 2001).
Human pCMX-steroid and xenobiotic receptor.1-Gal4DBD (hereafter referred to as pCMX-Gal4-PXR.1) (containing Lys107 to Ser443 of the LBD), pCMX-Gal4 vector plasmids, and TK-(MH100)4-LUC reporter were provided by Dr. Bruce Blumberg (Blumberg et al., 1998). The PXR.2 LBD (position 487-1471 in NM_022002) was amplified from pSG5-PXR.2 using PXR (forward) GGGGAATTCAAGAAGGAGATGATCATGTCC and (reverse) GGGAATTCTCAGCTACCTGTGATGCCGA primers containing EcoRI restriction sites and subcloned into the EcoRI restriction site of the pCMX-Gal4 plasmid to create pCMX-PXR.2-Gal4; these plasmids contain the LBD of PXR.1 or PXR.2 fused to the yeast Gal4 DNA binding domain. Sequence and orientation of PXR in all the plasmids were confirmed by DNA sequencing. The reporter plasmid CYP3A4-PXR response element (PXRE)-LUC (containing the proximal 0/–362 and distal 7208/7797 PXRE regions fused upstream of luciferase) was provided by Dr. Christopher Liddle (University of Sydney, Westmead, Australia).
Full-length human PXR.1 in the pGBT9 vector, pACT-c silencing mediator for retinoid and thyroid receptors (SMRT), pACT-cNcoR, pGAD-RAC3 (1–792), pACT-steroid receptor coactivator-1 (SRC1)-N, and pACT-hADA3 have been described previously (Johnson et al., 2006). Plasmid pGBT-PXR.2 was generated with the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) and confirmed by DNA sequencing. The PCR primers used for PXR.2 were (forward) CATTTCAAGAATTTCCGGGTCTCTCTGCAGCTGCG and (reverse) cgcagctgcagagagacccggaaattcttgaaatg.
Protein Expression Studies and Immunoblot Analysis. PXR expression plasmids were in vitro transcribed and translated (TNT) using the TNTQuick Coupled Rabbit Reticulocyte Lysate Transcription/Translation System according to the manufacturer's protocol (Promega, Madison, WI). Five and 15 μl of the TNT product were resolved by 10% polyacrylamide gel electrophoresis and immunoblotted with either anti-PXR or anti-GAL4 antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), followed by secondary antibodies coupled with peroxidase (Sigma-Aldrich) and developed with the Amersham enhanced chemiluminescence detection system (GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Electrophoretic Mobility Shift Assay. PXR.1 and PXR.2 were synthesized from pcDNA3-PXR expression plasmids using the rabbit reticulocyte TNT kit (Promega). Six hundred thousand counts per minute of double-stranded, 32P-labeled oligonucleotides representing the CYP3A4 PXR DNA binding sequence, an everted repeat with 6 base pair spacer (ER6) 5′-GATCAATATGAACTCAAAGGAGGTCAGTG-3′ or a mutant CYP3A4-ER6 5′-GATCAATATGCCATCAAAGGAATACAGTG-3′ [bolded bases disrupt PXR/RXR binding sites (underlined)], was incubated with PXR.1, PXR.2, or pcDNA3 TNT reaction mixture in the presence or absence of 1- to 500-fold molar excess of unlabeled double-stranded oligonucleotide. Binding reactions contained 10 mM Tris, pH 8.0, 40 mM KCl, 0.05% Nonidet P-40, 6% glycerol, 1 mM dithiothreitol, 0.2 μg of poly(dI-dC), 10 μM ZnCl2, and 4 μl of synthesized PXR or pcDNA3 in a 20-μl reaction. Samples were incubated on ice for 90 min, and complexes were resolved by electrophoresis through a nondenaturing 4% polyacrylamide gel in 0.5× Tris borate-EDTA (45 mM Tris-borate, 1 mM EDTA) at room temperature and analyzed using the Storm 860 PhosphorImager (GE Healthcare).
Transient Transfection and Reporter Gene Assay. HepG2 cells were maintained in minimum Eagle's medium-α and plated in 24-well plates at 3 × 105 cells/well. Twenty-four hours later, they were transfected overnight by calcium phosphate precipitation with 1) 300 ng of pCYP3A4-PXRE2-LUC reporter and 100 ng of MSCV-PXR-GFP expression plasmids, or 2) 1000 ng of TK-(MH100)4-LUC reporter with 400 ng of pCMX-Gal4-PXR expression plasmids and 50 ng of pSV40-LacZ to normalize LUC activity. The next day, the medium was changed to contain 10% charcoal dextran-treated fetal bovine serum (HyClone Laboratories, Logan, UT) with or without drug. Treatments included vehicle [0.1% dimethyl sulfoxide (DMSO)], 10 μM rifampin, 5 μM 20α-hydroxycholesterol, 50 μM dexamethasone butyl-acetate, 50 μM deoxycholic acid, 50 μM chenodeoxycholic acid, 100 μM ursodeoxycholic acid, 100 μM 3,7-diketocholanic acid, 1 mM phenobarbital, 50 μM phenytoin, 10 μM dexamethasone, 10 μM mifepristone, 10 μM estrone, 10 μM estradiol, 10 μM estriol, 50 μM progesterone, 50 μM pregnenolone, 50 μM pregnanolone, 50 μM 5β-pregnane-3,20-dione, and 20 μM 6,16α-dimethyl pregnenolone. Twenty-four hours later, cells were harvested and luciferase activity measured in supernatant according to the manufacturer's instructions (Luciferase Assay System; Promega) using an automated luminometer (Clarity Luminescence Microplate Reader; Bio-Tek Instruments, Winooski, VT). β-Galactosidase activity was measured according to the manufacturer's instructions (β-Galactosidase Enzyme Assay System with Reporter Lysis Buffer; Promega) using a spectrophotometer (μQuant; Bio-Tek Instruments). Protein content was determined by the Bradford reaction (Bio-Rad, Hercules, CA). All the experiments were performed in duplicate or triplicate.
For competition assays, HepG2 cells were cotransfected with either 1) varying amounts (0.1–1000 ng) of pCMV-Gal4-PXR and 750 ng of TK-(MH100)4-luciferase plasmid and 50 ng of pSV40-LacZ or 2) 0.1 to 1000 ng of pMSCV-PXR vectors and 500 ng of CYP3A4-PXRE-LUC plasmid. Cells were transfected overnight, and then media were changed to media containing vehicle or 10 μM rifampin for 24 h. Cells were lysed, and the luciferase reporter and β-galactosidase activities were determined. The IC50 was estimated using KaleidaGraph (Synergy Software, Reading, PA).
Creation and Characterization of HepG2 and LS180 Cell Lines Stably Transduced with pMSCV-PXR.1 and pMSCV-PXR.2 Retroviruses. Human embryonic kidney 293T cells were plated on 100-mm dishes at 45 × 106 cells. Approximately 20 h later, cells were transfected with 5 μg of each recombinant MSCV-PXR retroviral plasmid (mock, PXR.1, or PXR.2) along with 3.3 μg of packaging envelope plasmid (pRD118) and 3.3 μg of gal-Pol expression plasmid (PEQPAM3-E) using GenePorter (Gene Therapy Systems Inc., San Diego, CA). The retroviral supernatant from the cells was collected at 24, 48, and 72 h, stored on ice, pooled, and filtered for subsequent use to transduce cell lines. HepG2 cells or LS180 cells (American Type Culture Collection, Manassas, VA) were plated at a density of 0.1 × 106 cells/60-mm tissue culture dish. The media were replaced with the retroviral supernatant supplemented with 10 μg/ml Polybrene and incubated overnight at 37°C in a 5% CO2 humidified atmosphere. After 3 days, cells were trypsinized, washed, and resuspended in phosphate-buffered saline and analyzed by fluorescence-activated cell sorting analysis on a BD Biosciences (Franklin Lakes, NJ) Vantage flow cytometer. The GFP-positive population of cells was collected, expanded, and repetitively sorted, as above, until nearly 100% of the population displayed a GFP-positive phenotype. These stably transduced cells were used for subsequent experiments.
PXR protein expression was determined in lysates from the cells by immunoblot with an anti-PXR antibody. To determine target PXR gene expression, we harvested the cells, and RNA was extracted using TRIzol (Invitrogen). cDNA was synthesized from 5 μg of total RNA according to manufacturer's instructions using the SuperScript II reverse-transcriptase kit (Invitrogen). Targets of PXR (CYP3A4, CYP2B6, MDR1, UGT1A1, and GAPDH) were amplified by semiquantitative PCR (primers and conditions described in Lamba et al., 2004). CYP3A4 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were also amplified by quantitative real-time PCR using QuantiTech SYBR Green (Roche) with an ABI 7900 (Applied Biosystems, Foster City, CA) as described previously (Lamba et al., 2004). Real-time values were determined for CYP3A4 and GAPDH mRNA using the comparative cycle threshold method (Lamba et al., 2004).
Yeast Two-Hybrid Assay. pGBT9, pGBT-hPXR.1, and pGBT-PXR.2, paired with pACT-SMRTτ (amino acids 2017–2471), and pGAD-RAC3 (SRC3) (amino acids 1–792), pACT-SMARTα, pACT-cNcoR, pACT-SRC1-N, and pACT-hADA3 were cotransformed into yeast Y190 cells according to the manufacturer's instructions for Matchmaker Two-Hybrid System 2 (Clontech). Yeast cells containing the corresponding vectors were grown in –Trp, –Leu selection media for 24 h at 30°C. One hundred-microliter aliquots from each sample were taken and added to the fresh selection medium enriched with DMSO, or 10 μM rifampin. Twenty-four hours later, we harvested the yeast cells and analyzed them for liquid β-galactosidase activity. β-Galactosidase activities were normalized to the cell number.
Protein Modeling: Homology Modeling of PXR.1 versus PXR.2. A random loop of the missing residues for PXR.1 from the PXR crystal structure bound with hyperforin (Protein Databank entry 1M13) (Watkins et al., 2003b) was created with MOE 2006.8 (Chemical Computing Group, Montreal, QC, Canada) and SYBYL 7.2 (Tripos, St. Louis, MO) using the known PXR sequence. Deletion of the 37 amino acid region to produce PXR.2 was also performed with this software.
ID Prediction. The PXR.1 protein sequence was downloaded from the UniProt database. The PXR.1 ID prediction was then performed using the PONDR VL3H algorithm (Peng et al., 2005) available at http://www.ist.temple.edu/disprot/predictor.php. The PXR.2 sequence was also used with the same predictor and the extent of predicted ID compared with PXR.1.
Results
Location of Amino Acids Missing in PXR.2. Compared with many other nuclear hormone receptors, PXR.1 contains a unique insert of ∼50 amino acids. The PXR.1 insert adds a unique helix 2 and two β-strands bordering the PXR ligand-binding cavity (Noble et al., 2006). The 37 residues deleted in PXR.2 are specifically lost from this inserted region in PXR.1 (Fig. 1, A and B). PXR.2 still retains the RXR heterodimerization region (α10 helices) and the activation function-2 surface formed by αAF along with helices α3, α3′, and α4 (Teotico et al., 2008).
PXR.1 and PXR.2 Plasmids Appropriately Direct PXR Protein Expression. To begin testing the functionality of PXR.2, we developed a series of PXR.2 expression plasmids. To confirm that each plasmid produced PXR protein, we performed in vitro transcription and translation in rabbit reticulocyte extracts. PXR.1 and PXR.2 proteins were synthesized from pcDNA3 expression plasmids as assessed by incorporation of 35S-methionine (data not shown) or when probed on immunoblots with anti-PXR IgG (Fig. 2). As expected, translation of PXR.2 resulted in expression of a protein with a lower molecular mass than PXR.1. Additional PXR transcripts were evident with some expression plasmid, and this probably represents use of alternative translation initiation codons. We also confirmed cellular synthesis of these PXR proteins after transient transfection of HepG2 cells (Fig. 2).
Fig. 2.

Expression of PXR.1 and PXR.2 protein. A, left, PXR-Gal4 fusion constructs were TNT in vitro, and products were immunoblotted and reacted with indicated antibodies. Right, pMSCV-PXRs were transiently transfected into HepG2 cells, and lysates were immunoblotted and reacted with anti-PXR and anti-GFP antibodies. B, pcDNA3-PXRs and pcDNA3 were transiently transfected into HepG2 cells (left) or TNT in vitro (right), and lysates and products were immunoblotted with anti-PXR IgG.
PXR.2 Binds to the CYP3A4 Promoter PXRE. To determine whether PXR.2 could bind to the CYP3A4 proximal promoter PXRE, we performed electrophoretic mobility shift experiments. In vitro transcription/translation experiments, based on incorporation of radiolabeled methionine into the proteins, showed that equivalent amounts of PXR.1 and PXR.2 were synthesized (Fig. 3A). When 32P-labeled oligonucleotides containing the consensus PXRE were incubated with synthesized PXR.1 and PXR.2, both proteins formed a specific complex with the PXRE, and unlabeled oligonucleotides competed for formation of the complex (Fig. 3B). The PXR.2 complex migrated with a slightly lower molecular mass (Fig. 3B). Although the relative level of PXR/RXR/PXRE complex formed seemed to be higher for PXR.1 versus PXR.2, despite using equal input of PXR proteins (Fig. 3B), the relative affinities of PXR.1 versus PXR.2 for the PXRE cannot be determined simply by comparing the abundance of the complex formed. To measure DNA affinity more precisely, we performed competition experiments with increasing concentrations of unlabeled probe (Fig. 3C). Titration with 1×, 10×, 50×, and 100× excess of unlabeled oligonucleotides (Fig. 3C) displaced similar amounts of the PXR.1/RXR/PXRE complex (80, 52, 26, and 14%) and the PXR.2/RXR/PXRE complex (80, 51, 21, and 18%), respectively. Thus, PXR.2 can bind to a consensus PXRE with an affinity similar to PXR.1.
Fig. 3.

Electrophoretic mobility shift assay for PXR.1 and PXR.2-PXRE promoter complexes. A, [35S]methionine-labeled PXR.1 and PXR.2 were synthesized in vitro, resolved on polyacrylamide gel, and assessed by phosphorimager. B and C, 32P-labeled oligonucleotides representing the consensus CYP3A4 PXRE were incubated with the unlabeled TNT reaction mixtures containing PXR.1 or PXR.2 with RXR. Reactions were incubated in the absence (no competitor) or presence of 1- to 500-fold molar excess of unlabeled oligonucleotide or anti-PXR IgG. After electrophoresis, complex formation was assessed by a phosphorimager.
Ligand-Treated PXR.2 Fails to Activate the CYP3A4 PXRE. Next, we tested a range of structurally diverse PXR ligands to determine whether PXR.2 had a different ligand activation profile compared with PXR.1. Although two groups had previously tested PXR.2 function (Hustert et al., 2001; Gardner-Stephen et al., 2004), it was in response to rifampin (molecular mass = 822), which is much larger than many of the smaller PXR ligands (e.g., deoxycholic acid, molecular mass = 392) or other steroids and bile acids. We were particularly interested in testing PXR ligands (oxysterols and bile acids), which also bind to the much smaller LBD in FXR and liver X receptor. In transiently transfected HepG2 cells, none of the ligands tested activated either full-length PXR.2 (pMSCV-PXR.2) or the PXR.2 LBD (pCMX-Gal4-PXR.2), whereas PXR.1 and GAL4-PXR.1 readily transactivated the PXRE reporters in the presence of some of these ligands (Fig. 4). To determine whether the results obtained in HepG2 cells were cell type-specific, we performed similar experiments in NIH/3T3 cells using the Gal4-PXR plasmids and found that PXR.2 was incapable of transactivating the LUC reporter in this cell context as well (data not shown).
Fig. 4.

Ligand responsiveness of PXR.1 versus PXR.2. HepG2 cells were transfected with (A) MSCV-PXR plasmids and the CYP3A4-PXRE-LUC reporter and treated 48 h with control vehicle [(CT) 0.1% DMSO], 10 μM rifampin (RIF), 5 μM 20α-hydroxycholesterol (OXS), 50 μM dexamethasone butyl-acetate (DEX-BA), 50 μM deoxycholic acid (DCA), 50 μM chenodeoxycholic acid (CDCA), 100 μM ursodeoxycholic acid (UDCA), and 100 μM 3,7-diketocholanic acid (DKCA). B, pCMX-Gal4-PXR plasmids, the GAL4-responsive reporter TK-(MH100)4-LUC, and pSV40-LacZ were treated with vehicle, 10 μM RIF, 1 mM phenobarbital, 50 μM phenytoin (PHT), 10 μM dexamethasone (DEX), 10 μM mifepristone (RU486), 10 μM estrone (ESR), 10 μM estradiol (ESD), 10 μM estriol (ESL), 50 μM progesterone (PGR), 50 μM pregnenolone (PNL), 50 μM pregnanolone (PNA), 50 μM5 β-pregnane-3,20-dione (5β), and 20 μM 6,16 α-dimethyl pregnenolone (6,16α). Luciferase activities were normalized to either β-galactosidase or total cell protein as indicated. The relative luciferase normalized activity in vehicle-treated cells (for vector, human PXR.1, and human PXR.2) was each set as one, and the -fold change in relative luciferase activity in drug-treated cells was graphed relative to this baseline. Values represent the mean ± S.D. measured in triplicate in at least two independent transfections.
Transiently Cotransfected PXR.2 Represses PXR.1 Transactivation. We tested the effects of increasing the amounts of PXR.1 and PXR.2 on reporter activation. In the presence of rifampin, the GAL4-PXR.1 LBD (Fig. 5A) dose-dependently activated reporter gene activity to a maximum of nearly 4-fold. Conversely, the GAL4-PXR.2 LBD failed to activate the target gene and at high amounts decreased reporter activity to 0.27-fold of vector control (Fig. 5A). Likewise, increasing amounts of full-length MSCV-PXR.1 resulted in a more than 700-fold increase in CYP3A4-PXRE luciferase reporter activity compared with a maximum of 20-fold activation by the highest amount of transfected MSCV-PXR.2 (Fig. 5C).
Fig. 5.

PXR.2 can compete with PXR.1 for rifampin activation of reporter plasmids. HepG2 cells were transfected with expression vectors encoding PXR.1 or PXR.2 (A and B) pCMX-Gal4-PXR or pMSCV-hPXR-IRES-GFP (C and D) with appropriate reporter genes as described under Materials and Methods and treated for 48 h with 10 μM rifampin and relative reporter activity graphed relative to cells without cotransfected PXR. B and D, a constant amount of PXR.1 (10 ng) was cotransfected with increasing amounts of PXR.2 and the reporter plasmids. Values represent the mean ± S.D. measured in triplicate in at least two independent transfections. The ratio of PXR.2 to PXR.1 expression plasmid was graphed relative to activation of reporter genes.
Because PXR.1 and PXR.2 are coexpressed in liver (Lamba et al., 2004), we then examined whether PXR.2 could modulate transcriptional activation by PXR.1. A constant amount of GAL4-PXR.1 LBD (20 ng) was cotransfected with increasing amounts of GAL4-PXR.2 LBD. A 50% decrease in luciferase reporter activity was seen with a 21.1 ± 6.8-fold excess of the GAL4-PXR.2 to GAL4-PXR.1 (Fig. 5B). Likewise, cotransfection of increasing amounts of full-length MSCV-PXR.2 repressed MSCV-PXR.1 reporter activation, with an IC50 of 10.3 ± 2.9 (ng of PXR.2/ng of PXR.1) (Fig. 5D). Thus, the transactivation of PXR.1 can be suppressed by PXR.2 in a dose-dependent manner.
Stably Transfected PXR.2 Fails to Induce Target Genes in Stably Transfected Cells. HepG2 cells and LS180 cells stably transduced with MSCV-PXR retroviruses showed increased amounts of PXR.1 or PXR.2 transcript and protein (Fig. 6A). HepG2 cells stably expressing PXR.1, but not PXR.2, showed increased responsiveness (over vector control) to rifampin, phenobarbital, and simvastatin treatment with increased induction of CYP3A4 mRNA as measured by quantitative real-time PCR (Fig. 6B). Stable transduction of pMSCV-PXR.1 in LS180 cells resulted in a higher basal level of mRNAs of some PXR target genes [CYP3A4, CYP2B6, MDR1 (encoding P-glycoprotein)] and a robust further induction in the presence of rifampin (Fig. 6C). Stable transduction of LS180 cells with pMSCV-PXR.2 resulted in a diminution of basal (CYP3A4, MDR1) and of rifampin-inducible expression of MDR1 compared with pMSCV vector control. It is unclear why CYP2B6 levels were increased in untreated PXR.2 cells but suppressed in the same cells treated with rifampin.
Fig. 6.

Stable transduction of HepG2 and LS180 cells with MSCV-PXR.1 and MSCV-PXR.2 retroviruses. Cells were stably transduced with pMSCV-PXR.1 and pMSCV-PXR.2 and sorted by fluorescence-activated cell sorting for GFP. A, PXR transcript and protein expression were in cells stably transduced with PXR retroviruses as described under Materials and Methods. B, CYP3A4 expression in stably transduced HepG2 and LS180 cells after treatment with 0.1% DMSO, 10 μM rifampin, 2 mM phenobarbital, and 10 μM simvastatin for 48 h. Values represent the mean ± S.D. measured in triplicate in at least two independent transfections. CYP3A4 -fold change is the mean ± S.D. of the ratio of CYP3A4/GAPDH mRNA levels in PXR.1- or PXR.2-transduced cells to CYP3A4/GAPDH mRNA levels in MSCV-transduced cells all measured by quantitative real-time PCR. C, expression of PXR target genes mRNAs [CYP3A4, CYP2B6, MDR1 (encoding P-gp, P-glycoprotein), UGT1A1] in stably transduced HepG2 and LS180 cells in the presence of vehicle (0.1% DMSO) or 10 μM rifampin was assessed by semiquantitative PCR.
In Silico Predictions of PXR.2 Structure and ID. Several methods that have all been individually used to evaluate PXR or other nuclear hormone receptors were used to compare PXR.1 and PXR.2.
Protein Modeling. We took advantage of the extensive information on various PXR ligand bound structures (Watkins et al., 2001, 2003a,b) and used this to model the PXR.2 interaction with hyperforin. The deletion of 37 amino acids (Figs. 1 and 7, A and B) causes a wide opening in the binding pocket of PXR.2 (Fig. 7C), and it is likely that hyperforin or other similar-sized molecules cannot be kept inside the flexible binding site when this loop is missing. The white hairline seen next to the red tube (Fig. 7B) is the Leu209 residue that is important for interaction with hyperforin in PXR.1. Once this residue is removed in PXR.2, it is likely that hyperforin will have a weaker interaction with the LBD. Both the loop and specific interactions with amino acids on it are suggested to be critical for holding molecules in a productive orientation in the LBD. As PXR.2 is missing this important region and Leu209 (Fig. 7C), this could contribute significantly to its lack of function.
Fig. 7.

A, homology modeling of PXR.2. The trace of PXR.1 full-length in magenta. The 37 amino acids missing from PXR.2 are depicted in red. The green surface of hyperforin is shown behind. B, close-up view of Leu209 involved in the interaction with hyperforin in PXR.1. C, the PXR.2 homology model surface is displayed and colored in green, whereas the helix 12 is colored in magenta. The ligand hyperforin is displayed in ball-and-stick representation and colored by atom type (carbon gray, oxygen red).
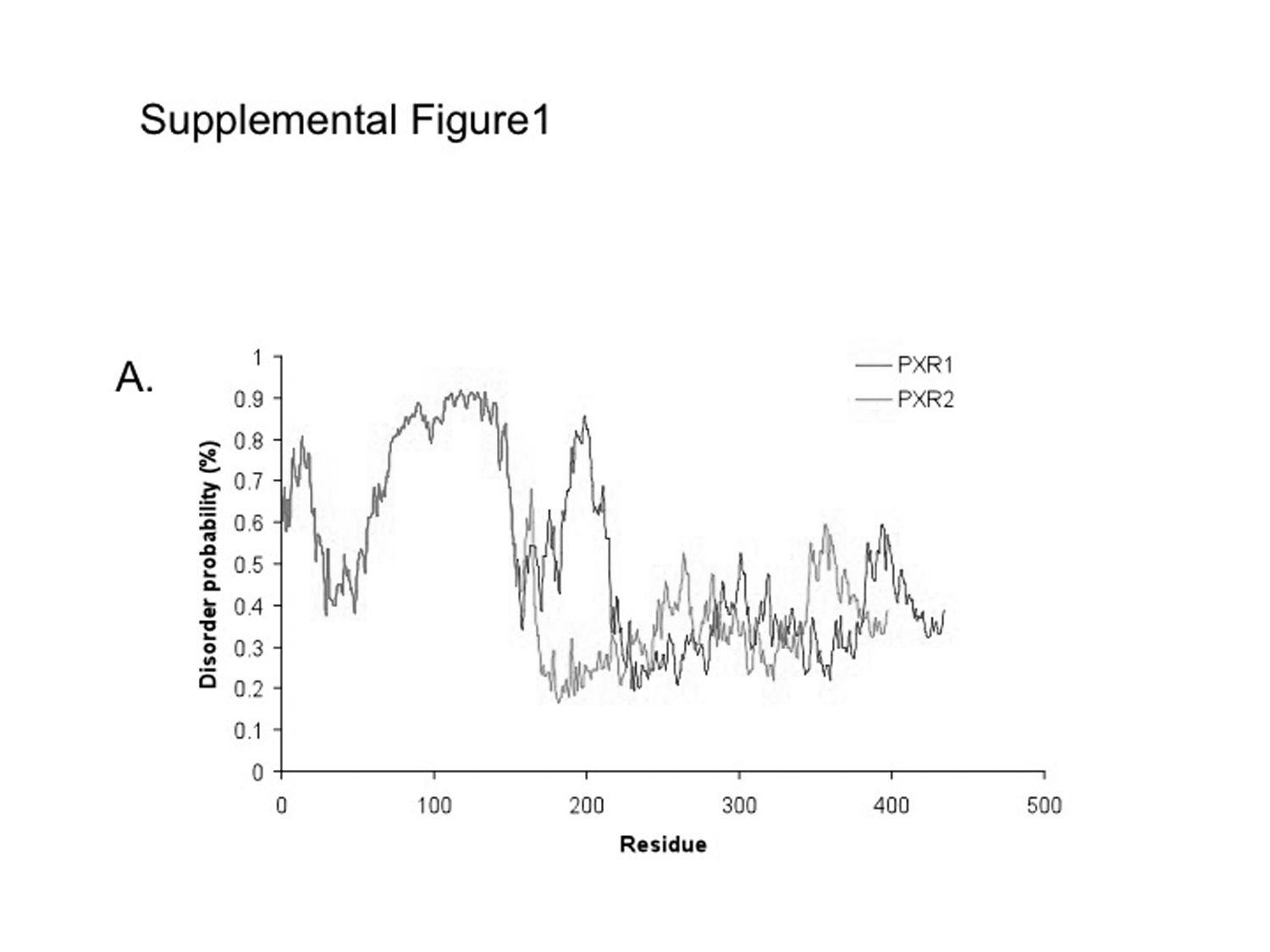
ID Prediction. The 37 amino acids removed from PXR.1 to form PXR.2 are in an area predicted to be highly disordered (Supplemental Fig. 1). The predicted ID for PXR.2 (38.5% residues with predicted ID greater than 50%) would be expected to be lower than PXR.1 (44.5% residues with predicted ID greater than 50%). This decrease in level of overall predicted ID might also be a contributing factor to the altered PXR.2 function observed.
Ligand-Treated PXR.2 Retains Corepressors and Fails to Recruit Coactivators. To explore the mechanism accounting for PXR.2 repression of target genes in vitro, a yeast two-hybrid assay was used to probe the association of PXR.2 with nuclear receptor corepressors and coactivators. The results of the yeast two-hybrid studies indicated that in wild-type PXR.1 with all three coactivators, binding was higher in the presence of rifampin compared with DMSO. The splice variant PXR.2 showed low or no interaction with the three coactivators in either the presence or absence of rifampin (Fig. 8A). PXR.1 was found to have relatively weak interactions with the corepressors SMRTτ and NcoR in the presence or absence of rifampin, whereas PXR.2 had stronger interactions with SMRTτ and NcoR (Fig. 8B). Both PXRs had the strongest interaction with SMRTα in the presence or absence of rifampin. These results indicate that PXR.2 is unable to bind to coactivators, a necessary step as part of ligand activation. In addition, it seems that corepressors are bound more tightly, independent of ligand addition. Taken together, the present results suggest that PXR.2 is not functional (at least with rifampin) as corepressors are not released and coactivators cannot bind.
Fig. 8.

Interaction between PXRs and coactivators (CoA) or corepressors (CoR) in the yeast two-hybrid assay. A, human PXR.1 and PXR.2 interactions with CoAs SRC1, SRC3, and ADA3 in the yeast two-hybrid assay incubated with either DMSO or 10 μM rifampin (Rif) as described under Materials and Methods. The β-galactosidase unit reflects the activity determined using O-nitrophenyl-β-galactopyranoside and normalized by cell number and incubation time. B, human PXR.1 and PXR.2 interactions with CoRs SMRTτ, NcoR, and SMRTα in the yeast two-hybrid assay incubated with either DMSO or Rif. All of the values represent the mean ± S.D. (n = 3) from a representative experiment performed at least two times.
Discussion
We present in vitro and in silico evidence that loss of amino acids from the LBD of PXR.2 does not affect the overall ability of PXR to form a complex with DNA, but nevertheless reduces the activity of the receptor in transient transfection experiments and eliminates receptor recruitment of transcriptional coactivators. The data suggest PXR.2 still heterodimerizes with RXR because PXR.2 retains the α10 helices that heterodimerize with RXR, and PXR.2/RXR, but not PXR.2 alone, formed a DNA complex in electrophoretic mobility shift assays. In addition, PXR.2 behaved as a dominant negative receptor in transient cotransfection experiments, repressing activation of PXRE sequences by rifampin-activated PXR.1. The combination of yeast two-hybrid and computational modeling indicates that mechanistically the PXR.2 variant is missing the disordered loop region. This effectively prevents ligands such as rifampin (data not shown) used in this study from binding productively in the LBD; the corepressors also remain tightly bound to PXR.2 such that coactivators would be unable to bind at helix 12.
Therefore, we suggest PXR.2 can join other alternatively spliced nuclear hormone receptor mRNAs that produce a protein product that is functional and/or can regulate the protein product of the major spliced mRNA. For example, a truncated isoform of the peroxisome proliferator activated receptor-γ (NR1C3), peroxisome proliferator activated receptor-γ1tr, exhibits dominant negative activity (Kim et al., 2006). FXR also produces multiple alternative mRNAs that differentially activate gene expression in human tissues (Zhang et al., 2003).
Can PXR.2 regulate PXR.1 target expression in human liver and therefore contribute to variable expression of its targets genes (e.g., CYP3A4, MDR1) in vivo? Our previous quantitation of PXR.1 and PXR.2 transcripts in donor human livers (Lamba et al., 2004) found that 1) the amounts of PXR.1 and PXR.2 are highly correlated (r2 = 0.82), and 2) the ratio of PXR.1 to PXR.2 is fairly constant in human livers. Although PXR.2 is the most abundant alternative PXR mRNA transcript in human liver, on average it represented only 7% of total PXR mRNA transcripts (Lamba et al., 2004). However, our cotransfection experiments showed that a greater than 10-fold excess of PXR.2 to PXR.1 was needed to attenuate PXR.1 transcriptional activation of CYP3A4-PXRE-LUC by 50%, with no significant attenuation of PXR.1 transactivation when PXR.2 represented <10% of the PXR pool. This suggests the relative ratio of PXR.2 to PXR.1 in human liver is typically insufficient for PXR.2 to inhibit PXR.1 target gene activation, e.g., CYP3A4, because the system needs to exceed 2.5 times more PXR.2 relative to PXR.1 to achieve dominant negative effects. However, it remains plausible that in some individuals or pathophysiological states where levels of PXR.2 might be higher than PXR.1 this could result in impaired expression of PXR target genes.
Although our study found that human PXR.2 was unable to be significantly activated by any PXR.1 ligands tested, this contrasts with work by Kliewer et al. (1998), who showed that both mouse PXR.1 and mouse PXR.2 (similar to human PXR.2) were activated by dexamethasone t-butylacetate and 6,16α-dimethyl pregnenolone. This difference may be because of the differences in the human versus mouse PXR LBD (Ekins et al., 2008) because neither of these steroids activated human PXR.1 well (Fig. 4).
Sequence alignment indicated that helix 2, which normally forms the bottom of the ligand binding pocket in PXR.1, is missing in the PXR.2 splice variant. With this deletion one would expect it to lead to weak or complete loss of binding of ligands for the PXR.2 receptor. In the PXR.1 crystal structure used in this study, hydrophobic residues are within 5 Å of the PXR-cocrystallized ligand hyperforin. Although Val211 is present in PXR.2, it or other favorable hydrophobic contacts are probably not in close contact with the ligands based on our homology model. Therefore, hyperforin or other similar-sized molecules would not be expected to be held inside PXR.2 (Fig. 6C). In addition, if a ligand could bind to PXR.2, it would probably be solvent-exposed, resulting in reduced entropic contributions to the binding. The deleted helix 2 region is far away from helix 12 and so cannot directly disrupt coactivator SRC1 binding in PXR.2 (Fig. 6C, magenta region).
Xenopus PXR is also missing the insertion domain between H1 and H3 compared with human PXR.1. Homology modeling of Xenopus PXR can illustrate how the missing domain limits the accessibility of the ligands to the Xenopus PXR ligand binding pocket (volume 850 Å3) (data not shown). Although the missing residues in human PXR.2 are not identical to those absent in Xenopus PXR, we might expect a similar effect on ligand accessibility and specificity for PXR.2 versus PXR.1. We have tested the Xenopus PXR ligand n-butyl 4-aminobenzoate and found it did not activate human PXR.1 or PXR.2 (E. Schuetz, unpublished observation), further indicative of important species differences in PXR (Ekins et al., 2008).
Proteins may be functional in different states such as ordered, random coil, and molten globule, which accommodate those proteins possessing ID in some part of their sequence. This property can be readily determined in silico (Dunker et al., 2001). ID prediction was recently performed for nearly 400 nuclear hormone receptors (NHRs) (Krasowski et al., 2008). We found that the in silico predictions correctly identified regions in 20 of 23 NHRs known to be disordered experimentally. ID in the D-domain and LBD was also significantly higher in well connected or “hub” human NHRs (Krasowski et al., 2008). Others have shown regions affected by alternative splicing in human genes are most often intrinsically disordered enabling regulatory and functional diversity, facilitating more rapid evolution (Romero et al., 2006).
Consistent with this model, the 37 amino acids removed from PXR.1 to form the splice variant PXR.2 are in the same area predicted to be highly disordered (Supplemental Fig. 1). Therefore, the predicted sequence ID for PXR.2 is lower than PXR.1 and might be considered a contributing factor to the activation observed experimentally. Further assessment of protein disorder in other PXR variants across species may be warranted. The PXR.2 sequence moves its LBD closer in size to that of the constitutive androstane receptor (NR1I3). The amino acids deleted in PXR.2 are part of the “H1–H3” insertion found in PXR, FXR (NR1H4), and the vitamin D receptor (VDR; NR1I1). X-ray crystallography of the human PXR LBD (Carnahan and Redinbo, 2005) includes regions that cannot be resolved in this insertion and others (e.g., residues 178–197) (Watkins et al., 2003a). It is interesting to note that the binding of the agonist rifampin was suggested to induce disorder in human PXR (residues 178–209, 229–235, and 310–317; Chrencik et al., 2005). Using the PONDR predictor, residues 178 to 209 (in closest proximity to the ligand) are predominantly predicted as disordered, whereas residues 229 to 235 and 310 to 317 are predicted as ordered. The human FXR and fish VDR crystal structures also show disorder between the H1 and H3 region (Downes et al., 2003; Ciesielski et al., 2007), whereas human VDR was crystallized without the disordered H1 to H3 insert (Rochel et al., 2001).
VDR is the proposed original NR1I gene, and the evolutionary history of PXR and VDR has been studied using the invertebrate ortholog to these receptors from Ciona intestinalis (sea squirt) (Reschly et al., 2007; Ekins et al., 2008). In terms of ID in the LBD, the Ciona VDR/PXR was closer to PXRs than to VDRs (Ekins et al., 2008). Homology modeling studies indicated that the smaller size of the human VDR LBD, relative to human FXR and human PXR, renders human VDR responsive only to small α/β cis bile acids (Reschly et al., 2008). The evolution of PXRs has also been suggested to be driven by adaptation to changes in bile salt structures as increasing size and altered topology of the human PXR LBD, compared with nonmammalian PXRs, permit recognition of the evolutionary recent 5β-bile acids while retaining sensitivity to early 5α-bile salts (Krasowski et al., 2005a). It remains unclear whether we will be able to find any ligands that can activate PXR.2 or what is the evolutionary significance of this splice variant.
Finally, although it remains to be formally tested, the data in this report also support a model in which PXR.2 may lose its ability to homodimerize, and this contributes to PXR.2's loss of functionality. All the PXR LBD crystal structures have revealed PXR forms a homodimer that uses the unique 50 residue insert in PXR that form β1 and β1′ strands that associate in an antiparallel fashion. The LBD insert creates a unique PXR/PXR tryptophan zipper-mediated homodimer interface that is involved in receptor function (Noble et al., 2006). Moreover, PXR.1 homodimerization requires interlocking of Trp223 and Tyr225 residues in the terminal β-strands of the LBD. Important homodimer oligomerization interactions include van der Waals contacts between Pro175 (missing in PXR.2) (Fig. 1B) and Trp223 and Tyr225, as well as hydrogen bonding interactions between Pro175's main carbonyl oxygen and the indole nitrogen on Trp223 (Noble et al., 2006). The authors also further showed that PXR homodimerization is critical for directing long-range interactions of the activation function-2 domain with coactivators (Noble et al., 2006; Teotico et al., 2008). An updated model of a PXR/RXR heterotetramer has been proposed (Noble et al., 2006) in which all the oligomerization interfaces are important for PXR function. It is noteworthy that our computational results in this report agree perfectly with those of a recent study [published by Teotico et al. (2008) while the current work was ongoing] in which the contacts in the 37 amino acids missing in PXR.2 are required for motions essential to coactivator binding by PXR.1. This probably explains, in part, why PXR.2 is incapable of recruiting coactivators.
In conclusion, although it would be difficult to predict the functionality of PXR.2 toward the multitude of structurally different PXR ligands, in this study we used computational modeling alongside various experimental methods to rationalize the functional differences between PXR.1 and PXR.2. We have shown that PXR.2 1) is unable to transactivate CYP3A4 in HepG2 cells treated with PXR ligands, although it is capable of binding to a consensus CYP3A4 PXRE; 2) behaves as a dominant negative interfering with PXR.1-mediated activation of the CYP3A4 promoter; and 3) is unable to bind coactivators because corepressors are bound more tightly. The complementary computational and in vitro approaches could be applied to splice variants of other nuclear receptors.
Supplementary Material
Acknowledgments
We thank Jie Zheng and Jufang Shan (Department of Structural Biology, St. Jude Children's Research Hospital, Memphis, TN) for discussions on docking of ligands to PXR.1 versus PXR.2. We also thank Dr. David Lawson (Takeda San Diego Inc., San Diego, CA) for providing information on the disorder analysis approach and Drs. Matthew D. Krasowski and Erica J. Reschly (University of Pittsburgh, Pittsburgh, PA) for earlier extensive discussions on NHR disorder and evolution.
This work was supported in part by the National Institutes of Health National Institute of General Medical Sciences [Grant GM60346]; the National Institutes of Health National Cancer Institute [Grant P30-CA21765]; the National Institutes of Health National Center for Research Resources [Grant KL2-RR025015]; the American Lebanese Syrian Associated Charities; and the U.S. Environmental Protection Agency-funded Environmental Bioinformatics and Computational Toxicology Center [STAR Grant GAD R 832721-010].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.108.025213.
ABBREVIATIONS: PXR, pregnane X receptor; LBD, ligand binding domain; RXR, retinoid X receptor; FXR, farnesoid X receptor; ID, intrinsic disorder; PCR, polymerase chain reaction; GFP, green fluorescent protein; PXRE, pregnane X receptor response element; SMRT, silencing mediator for retinoid and thyroid receptors; SRC1, steroid receptor coactivator-1; TNT, transcribed and translated; DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NHR, nuclear hormone receptor; VDR, vitamin D receptor.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
References
- Bertilsson G, Heidrich J, Svensson K, Asman M, Jendeberg L, Sydow-Bäckman M, Ohlsson R, Postlind H, Blomquist P, and Berkenstam A (1998) Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc Natl Acad Sci U S A 95 12208–12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg B, Sabbagh W Jr, Juguilon H, Bolado J Jr, van Meter CM, Ong ES, and Evans RM (1998) SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev 12 3195–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnahan VE and Redinbo MR (2005) Structure and function of the human nuclear xenobiotic receptor PXR. Curr Drug Metab 6 357–367. [DOI] [PubMed] [Google Scholar]
- Chrencik JE, Orans J, Moore LB, Xue Y, Peng L, Collins JL, Wisely GB, Lambert MH, Kliewer SA, and Redinbo MR (2005) Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol Endocrinol 19 1125–1134. [DOI] [PubMed] [Google Scholar]
- Ciesielski F, Rochel N, and Moras D (2007) Adaptability of the vitamin D nuclear receptor to the synthetic ligand gemini: remodelling the LBP with one side chain rotation. J Steroid Biochem Mol Biol 103 235–242. [DOI] [PubMed] [Google Scholar]
- Dotzlaw H, Leygue E, Watson P, and Murphy LC (1999) The human orphan receptor PXR messenger RNA is expressed in both normal and neoplastic breast tissue. Clin Cancer Res 5 2103–2107. [PubMed] [Google Scholar]
- Downes M, Verdecia MA, Roecker AJ, Hughes R, Hogenesch JB, Kast-Woelbern HR, Bowman ME, Ferrer JL, Anisfeld AM, Edwards PA, et al. (2003) A chemical, genetic, and structural analysis of the nuclear bile acid receptor FXR. Mol Cell 11 1079–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, et al. (2001) Intrinsically disordered protein. J Mol Graph Model 19 26–59. [DOI] [PubMed] [Google Scholar]
- Ekins S, Mirny L, and Schuetz EG (2002) A ligand-based approach to understanding selectivity of nuclear hormone receptors PXR, CAR, FXR, LXRa and LXRb. Pharm Res 19 1788–1800. [DOI] [PubMed] [Google Scholar]
- Ekins S, Reschly EJ, Hagey LR, and Krasowski MD (2008) Evolution of pharmacologic specificity in the pregnane X receptor. BMC Evol Biol 8 103–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman MC and Kirby ML (1998) The human orphan receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest 102 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuen S, Fukuda T, Matsuda H, Sumida A, Yamamoto I, Inaba T, and Azuma J (2002) Identification of the novel splicing variants for the hPXR in human livers. Biochem Biophys Res Commun 298 433–438. [DOI] [PubMed] [Google Scholar]
- Gardner-Stephen D, Heydel JM, Goyal A, Lu Y, Xie W, Lindblom T, Mackenzie P, and Radominska-Pandya A (2004) Human PXR variants and their differential effects on the regulation of human UDP-glucuronosyltransferase gene expression. Drug Metab Dispos 32 340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustert E, Zibat A, Presecan-Siedel E, Eiselt R, Mueller R, Fuss C, Brehm I, Brinkmann U, Eichelbaum M, Wojnowski L, et al. (2001) Natural protein variants of pregnane X receptor with altered transactivation activity toward CYP3A4. Drug Metab Dispos 29 1454–1459. [PubMed] [Google Scholar]
- Johnson DR, Li CW, Chen LY, Ghosh JC, and Chen JD (2006) Regulation and binding of pregnane X receptor by nuclear receptor corepressor silencing mediator of retinoid and thyroid hormone receptors (SMRT). Mol Pharmacol 69 99–108. [DOI] [PubMed] [Google Scholar]
- Jones SA, Moore LB, Shenk JL, Wisely GB, Hamilton GA, McKee DD, Tomkinson NC, LeCluyse EL, Lambert MH, Willson TM, et al. (2000) The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol Endocrinol 14 27–39. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Woo IS, Kang ES, Eun SY, Kim HJ, Lee JH, Chang KC, Kim JH, and Seo HG (2006) Identification of a truncated alternative splicing variant of human PPARgamma1 that exhibits dominant negative activity. Biochem Biophys Res Commun 347 698–706. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterström RH, et al. (1998) An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 92 73–82. [DOI] [PubMed] [Google Scholar]
- Kliewer SA and Willson TM (2002) Regulation of xenobiotic and bile acid metabolism by the nuclear pregnane X receptor. J Lipid Res 43 359–364. [PubMed] [Google Scholar]
- Krasowski MD, Reschly EJ, and Ekins S (2008) Intrinsic disorder in nuclear hormone receptors. J Proteome Res 7 4359–4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasowski MD, Yasuda K, Hagey LR, and Schuetz EG (2005a) Evolution of the pregnane X receptor: adaptation to cross-species differences in biliary bile salts. Mol Endocrinol 19 1720–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasowski MD, Yasuda K, Hagey LR, and Schuetz EG (2005b) Evolutionary selection across the nuclear hormone receptor superfamily with a focus on the NR1I subfamily (vitamin D, pregnane X, and constitutive androstane receptors). Nucl Recept 3 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamba V, Yasuda K, Lamba JK, Assem M, Davila J, Strom S, and Schuetz EG (2004) PXR (NR1I2): splice variants in human tissues, including brain, and identification of neurosteroids and nicotine as PXR activators. Toxicol Appl Pharmacol 199 251–265. [DOI] [PubMed] [Google Scholar]
- Maglich JM, Parks DJ, Moore LB, Collins JL, Goodwin B, Billin AN, Stoltz CA, Kliewer SA, Lambert MH, Willson TM, et al. (2003) Identification of a novel human CAR agonist and its use in the identification of CAR target genes. J Biol Chem 278 17277–17283. [DOI] [PubMed] [Google Scholar]
- Moore JT, Moore LB, Maglich JM, and Kliewer SA (2003) Functional and structural comparison of PXR and CAR. Biochim Biophys Acta 1619 235–238. [DOI] [PubMed] [Google Scholar]
- Noble SM, Carnahan VE, Moore LB, Luntz T, Wang H, Ittoop OR, Stimmel JB, Davis-Searles PR, Watkins RE, Wisely GB, et al. (2006) Human PXR forms a tryptophan zipper-mediated homodimer. Biochemistry 45 8579–8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK, and Obradovic Z (2005) Optimizing long intrinsic disorder predictors with protein evolutionary information. J Bioinform Comput Biol 3 35–60. [DOI] [PubMed] [Google Scholar]
- Reschly EJ, Ai N, Ekins S, Welsh WJ, Hagey LR, Hofmann AF, and Krasowski MD (2008) Evolution of the bile salt nuclear receptor FXR in vertebrates. J Lipid Res 49 1577–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reschly EJ, Bainy AC, Mattos JJ, Hagey LR, Bahary N, Mada SR, Ou J, Venkataramanan R, and Krasowski MD (2007) Functional evolution of the vitamin D and pregnane X receptors. BMC Evol Biol 7 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochel N, Tocchini-Valentini G, Egea PF, Juntunen K, Garnier JM, Vihko P, and Moras D (2001) Functional and structural characterization of the insertion region in the ligand binding domain of the vitamin D nuclear receptor. Eur J Biochem 268 971–979. [DOI] [PubMed] [Google Scholar]
- Romero PR, Zaidi S, Fang YY, Uversky VN, Radivojac P, Oldfield CJ, Cortese MS, Sickmeier M, LeGall T, Obradovic Z, et al. (2006) Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc Natl Acad Sci USA 103 8390–8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz E and Strom S (2001) Promiscuous regulator of xenobiotic removal. Nat Med 7 536–537. [DOI] [PubMed] [Google Scholar]
- Synold TW, Dussault I, and Forman BM (2001) The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Med 7 584–590. [DOI] [PubMed] [Google Scholar]
- Teotico DG, Frazier ML, Ding F, Dokholyan NV, Temple BR, and Redinbo MR (2008) Active nuclear receptors exhibit highly correlated AF-2 domain motions. PLoS Comput Biol 4 e1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirona RG, Leake BF, Podust LM, and Kim RB (2004) Identification of amino acids in rat pregnane X receptor that determine species-specific activation. Mol Pharmacol 65 36–44. [DOI] [PubMed] [Google Scholar]
- Watkins RE, Davis-Searles PR, Lambert MH, and Redinbo MR (2003a) Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J Mol Biol 331 815–828. [DOI] [PubMed] [Google Scholar]
- Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, Lambert MH, Kliewer SA, and Redinbo MR (2003b) 2.1A crystal structure of human PXR in complex with the St. John's wort compound hyperforin. Biochemistry 42 1430–1438. [DOI] [PubMed] [Google Scholar]
- Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, and Redinbo MR (2001) The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science 292 2329–2333. [DOI] [PubMed] [Google Scholar]
- Yasuda K, Ranade A, Venkataramanan R, Strom S, Chupka J, Ekins S, Schuetz E, and Bachmann K (2008) A comprehensive in vitro and in silico analysis of antibiotics that activate PXR and induce CYP3A4 in liver and intestine. Drug Metab Dispos 36 1689–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kast-Woelbern HR, and Edwards PA (2003) Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation. J Biol Chem 278 104–110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}