

Abstract
A complex program of cell intrinsic and extrinsic signals guide cortical development. Although genetic studies in mice have uncovered roles for numerous genes and gene families in multiple aspects of corticogenesis, determining their cell autonomous functions is often complicated by pleiotropic defects. Here we describe a novel lentiviral-based method to analyze cell autonomy by generating two-color chimeric mouse embryos. Ena/VASP-deficient mutant and control embryonic stem (ES) cells were labeled with different fluorescent chimeric proteins (EGFP and mCherry) that were modified to bind to the plasma membrane. These labeled ES cells were used to generate two-color chimeric embryos possessing two genetically distinct populations of cortical cells, permitting multiple aspects of neuronal morphogenesis to be analyzed and compared between the two cell populations. We observed little difference between the ability of control and Ena/VASP-deficient cells to contribute to cortical organization during development. In contrast, we observed axon fiber tracts originating from control neurons but not Ena/VASP-deficient neurons, indicating that loss of Ena/VASP causes a cell autonomous defect in cortical axon formation. This technique could be applied to determine other cell autonomous functions in different stages of cortical development.
Introduction
Cortex formation requires the movement and guidance of neurons and their processes to specific targets. Neurons are born in internal germinal regions and migrate tangentially and radially to occupy more superficial layers and establish the layered pattern of the forebrain (Kriegstein and Noctor, 2004; Marin and Rubenstein, 2003). During migration, neurons initiate axon formation (Noctor et al., 2004), and subsequent outgrowth and guidance of axons is critical to cortical organization and function (Sur and Rubenstein, 2005).
Multiple proteins are required for axon formation including the Ena/VASP proteins, a family of proteins that regulate actin dynamics in multiple cell types (Drees and Gertler, 2008; Krause et al., 2003). We showed recently that loss of all three murine Ena/VASP proteins blocks cortical axon fiber tract formation during development, and that the underlying cause is a failure in neurite initiation, a prerequisite to axon formation (Dent et al., 2007; Kwiatkowski et al., 2007). To determine if the in vivo defect in axon formation was cell autonomous, we generated chimeric embryos contain unlabeled control cells and Ena/VASP-null mutant cells marked with cytoplasmic EGFP. We were unable to detect axons arising from EGFP-positive mutant cortical neurons, but neuronal processes were difficult to visualize unequivocally with cytoplasmic EGFP (Kwiatkowski et al., 2007). Therefore, we sought a method to label neuronal processes – specifically axons – and directly compare processes from mutant and control neurons within the same cortex.
Here we describe a lentiviral-based method to generate ES cells with a marker for neuronal processes and two-color chimeric embryos composed of EGFP-labeled Ena/VASP-null cells and mCherry-labeled control cells. Cortices isolated from two-color chimeric embryos contained two discrete populations of labeled cells, which allowed us to assess cortical organization and, notably, axon fiber tract formation. Targeting EGFP and mCherry to the plasma membrane served as a superior marker of neuronal processes compared to standard cytoplasmic fluorescent proteins, permitting detailed analysis of axonal fibers. We could show unequivocally that Ena/VASP-deficient neurons, unlike control neurons, did not contribute to axon formation, indicating that loss of Ena/VASP causes a cell autonomous defect in cortical axon formation. Thus, this system offered new and refined insights into the involvement of Ena/VASP in cortical organization. This technique may be applied to study the involvement of genetically distinct populations of cells in multiples aspects of cortical development.
Results and Discussion
Lentiviruses enable effective and efficient gene delivery in both mitotic and post-mitotic cells. Furthermore, lentiviral-expressed transgenes are not silenced during development, permitting production of transgenic animals from infected ES cells (Rubinson et al., 2003). However, commonly used promoters for mammalian expression, notably the cytomegalovirus (CMV) promoter, do not provide adequate levels of expression in certain cell types, such as neurons (Boulos et al., 2006). Therefore, a lentiviral expression vector in which transgene expression is driven by the CMV-enhancer/chicken beta-actin/beta-globin intron (CAG) promoter was designed (Kwiatkowski et al., 2007). The CAG promoter (Niwa et al., 1991) provides high levels of expression in most tissues (Okabe et al., 1997). The lentiviral expression vector, a modified version of pLentiLox3.7 (pLL3.7, (Rubinson et al., 2003)) and designated pLL4.4, was constructed to express either EGFP or mCherry downstream of a CAG promoter (pLL4.4-E/C, Figure 1A).
Figure 1. Lentivirus expression system for labeling cellular processes and generation of two-color chimeric embryos.

(A) Diagram of pLentiLox 4.4 and pLentiLox 4.4 gap. Expression in pLentiLox 4.4 (pLL 4.4) is driven by the CAG promoter. In pLL4.4, EGFP or mCherry (pLL4.4 E/C) is expressed downstream of the CAG promoter; fusions proteins can be created by cloning into the BamHI and EcoRI sites at the 3′ end of EGFP. The expression cassette is flanked by LoxP sites, allowing EGFP or EGFP-tagged proteins to be excised by Cre recombinase expression post-infection. To improve labeling of cellular processes in neurons, pLL4.4 was reengineered to express the first 19 amino acids of GAP-43 (shown at bottom) fused to EGFP, mCherry or DsRed2 (pLL4.4 gap-E/C/D). The N-terminus of GAP-43, referred to as a “gap” tag, contains palmitoylation sequences that target the plasma membrane.
(B) Cartoon schematic of two-color chimeric embryo generation. Wild type and Ena/VASP-null (mmvvee) ES cells were infected with lentiviruses expressing gap-EGFP and gap-mCherry, respectively, and sorted for fluorescent protein (FP) expression. FP-positive wild type and mmvvee ES cell clones were co-injected into wild type blastocysts. Brains isolated from two-color chimeric embryos contain mCherry-labeled cells derived from control cells and EGFP-labeled cells derived from mmvvee cells.
(C–E) Coronal vibratome sections from an E16.5 two-color chimeric cortex.
(C) In high percentage chimeras, most cells were labeled with either EGFP or mCherry, indicating they were derived from ES cells. Membrane-targeted FPs clearly labeled cell bodies and processes throughout the cortex. Cortical plate (CP) and subventricular zone (SVZ) are noted.
(D–E) Cortical organization in two-color chimeras. Labeling two populations of founder cells revealed the striking columnar organization of the cortex, stretching from the ventricular zone (VZ) shown in (E) to the marginal zone (MZ, in D). White dashed line in (E) marks the VZ boundary.
Scale bars for (C), (D) and (E) are 50 microns.
Neurons infected with lentivirus prepared with pLL4.4-EGFP express high levels of cytoplasmic EGFP 48–72 hours post-infection, and expression persists for 28 days in culture (Supplemental Figure 1). pLL4.4-EGFP was used previously to infect Ena/VASP-null (mmvvee) ES cells, and EGFP-expressing ES cells injected into blastocysts to generate chimeric mouse embryos composed of unlabeled control cells and EGFP-positive Ena/VASP-null cells (Kwiatkowski et al., 2007). However, while Ena/VASP-null cell somas in chimeric embryos were clearly labeled with cytoplasmic EGFP, we observed limited diffusion of EGFP into neuronal extensions, making migratory and axonal processes from labeled cells difficult to distinguish. Therefore, we reengineered pLL4.4 to express membrane-targeted fluorescent proteins (FPs) by adding the 19 N-terminal amino acids of gap-43, an axonal-targeted protein, upstream of EGFP, mCherry and DsRed2 (referred to as a “gap” tag, pLL4.4 gap-E/C/D, Figure 1A). This sequence contains a palmitolaytion site that targets the plasma membrane, and when added to EGFP successfully labels neuronal processes in vitro and in vivo (Okada et al., 1999). When expressed in early stage neurons in vitro, pLL4.4 gap-tagged FPs effectively target and label all neuronal processes or neurites (Supplementary Figure 1B); in later stage cultures, gap-tagged FPs favor axonal projections (Supplementary Figure 1C–F). These results suggested that lentiviral-expressed membrane-targeted proteins could be used in vivo to analyze neuronal process formation, in particular axon fiber tract formation, in distinct cell populations.
Since we wanted to compare neuronal processes between control and Ena/VASP-deficient cells within the same cortex, we generated two-color chimeric embryos using gap-tagged FPs (outlined in Figure 1B). Employing this technique, control ES cells were infected with gap-mCherry-expressing lentivirus and Ena/VASP-null ES cells were infected with gap-EGFP-expressing lentivirus. Isolated, stable gap-mCherry and gap-EGFP expressing ES clones were then co-injected into wild type blastocysts. The resulting chimeric embryos were composed of three individual cell types: unlabeled wild type cells, gap-mCherry labeled control cells, and gap-EGFP labeled Ena/VASP-null cells (Figure 1B). Respective contributions of control and Ena/VASP-null cell types in the developing embryo were controlled by varying both the total number and ratio of injected gap-EGFP Ena/VASP-null ES cells to gap-mCherry control ES cells.
Chimeric embryos were collected at E16.5, the stage at which Ena/VASP-deficient embryos survive yet demonstrate striking cortical defects (Kwiatkowski et al., 2007). The percentage of labeled cells in embryos varied greatly, with some embryos containing only a small percentage (<20%) of a single color and others possessing a high percentage (>80%) of mCherry and EGFP-labeled cells (Figures 1–3). Embryos composed of both mCherry- and EGFP-labeled cells are referred to as two-color chimeric embryos.
Figure 3. Cortical fiber tracts in dual-color chimeras originate from mCherry control neurons.
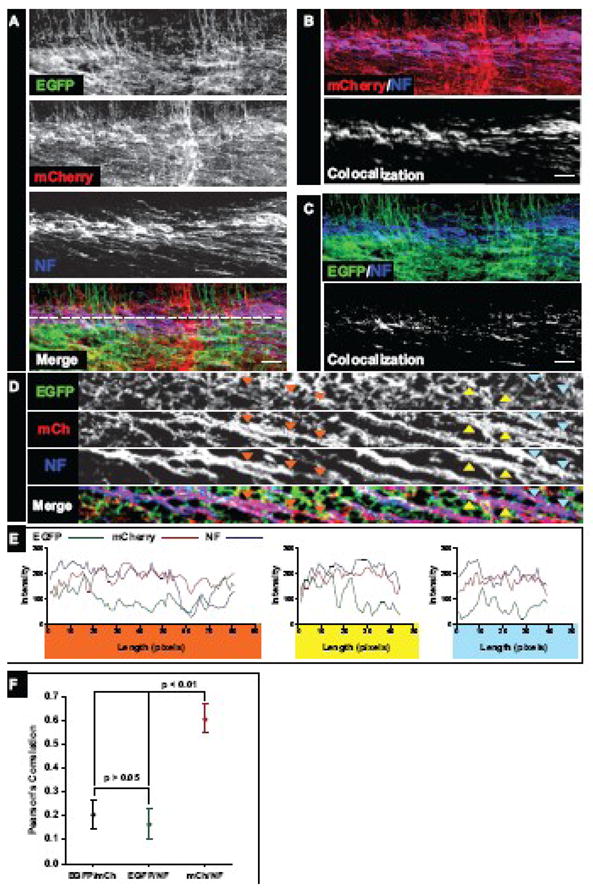
(A) Coronal vibratome sections (100 μm) from two-color chimeric E16.5 embryo cortices stained for Neurofilament-M (NF). Individual mmvvee EGFP (green), control mCherry (red), and NF (blue) channels as well as the Merge are shown. NF-positive fibers in the intermediate zone (IZ) of chimeric cortices showed strong mCherry expression but largely lacked EGFP, indicating they arose from control, not mutant mmvvee, neurons (see Merge, Panels B, C).
(B, C) Two channel merge and colocalization. NF channel was merged with individual mCherry (B, top panel) or EGFP (C, top panel) signals. Prominent colocalization is observed in the mCherry/NF overlay (B, top panel, note the strong purple-colored fibers) compared to the EGFP/NF overlay (C, top panel). Colocalization is demonstrated visually in the bottom panels of B and C, where the white signal reflects overlap between NF and mCherry (B) or NF and EGFP (C) channels.
(D) To visualize individual axon fibers in high resolution, optical slices through the IZ of NF-labeled cortical sections were generated (the source of the optical slice is represented by the dashed white line in Figure 3A, Merge). Individual EGFP (green), mCherry (red), and NF (blue) slices as well as the merge are shown. Colored arrows mark NF-positive axons and corresponding regions in the EGFP and mCherry channels.
(E) Linescans along NF-positive axon fibers reveal strong overlap with mCherry signal. Linescans were drawn along NF-positive axons marked by the colored triangles, and pixel intensities measured across the linescans in all channels and graphed. Individual graphs show changes in signal intensity over length (in pixels) and are colored coded to match triangles in (D).
(F) Pearson’s correlation was measured between pairings of all channels to determine if colocalization was significant. Pearson’s correlation was measured between mCherry and EGFP signals to obtain a “background” value, as these two channels should be mutually exclusive. The correlation between EGFP and NF signals was low and similar to that observed between EGFP and mCherry; in contrast, the correlation between mCherry and NF signals was high and statistically significant (p < 0.01).
Scale bars for (A), (B) and (C) are 50 microns.
Coronal sections through the cortex of a high percentage E16.5 two-color chimeric embryo (Figure 1C-E) revealed a striking columnar organization of labeled cells, stretching from the ventricular zone (VZ, Figure 1D) to the cortical plate (CP, Figure 1E). This distribution is consistent with previous studies of cortical organization (Rakic, 2007) that have relied largely on in utero techniques to label cortical cells. However, analysis of two-color chimeras offered unique advantages compared with established in utero manipulations. The first cells of the developing cortex – founder cells – are labeled in chimeric embryos, in contrast to in utero-labeling that marks cortical cells at later stages of development (E10 versus E12–E15, see Supplemental Figure 2B, C). Thus, the entire lineage of cells derived from a parent germinal cell is labeled, rather than just a subset of cells. In addition, labeling two genetically distinct populations of cells within the same cortical region allows a direct comparison of the contribution of each population to cortical organization. Interestingly, cortical columns tended to be dominated by neurons of one origin (based on color), suggesting limited tangential migration of cortical neurons between columns and limited entry of tangentially migrating interneurons at this stage of development. Previous studies using an X-linked lacZ transgene to label roughly half of the cortical progenitor cells revealed similar patterns of dispersion (Tan and Breen, 1993; Tan et al., 1995).
Using these chimeric embryos, we first determined whether there were any gross differences in the pattern or distribution of control mCherry-labeled cells and processes versus Ena/VASP-deficient EGFP-labeled cells and processes in embryonic cortices. We focused analysis on two-color chimeric embryos with large populations of both mCherry and EGFP-expressing cortical cells, as these embryos were best suited for comparative analysis. We first measured the percentage of EGFP versus mCherry-labeled tissues in individual cortical sections; results from one representative two-color chimeric embryo are shown (Figure 2A). In this embryo, the percentage of EGFP-labeled structures was more than double that of mCherry-labeled structures, suggesting a larger contribution from Ena/VASP-deficient ES cells than mCherry control ES cells to the developing cortex.
Figure 2. Cortical column size and uniformity in two-color chimeric cortices.
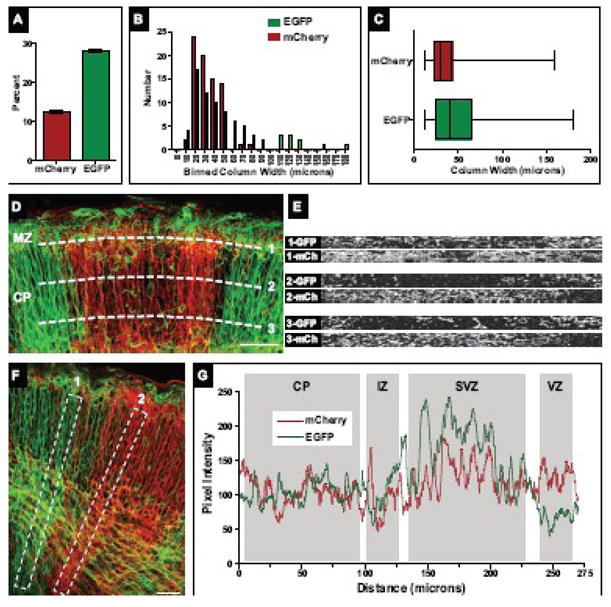
(A) Percentage of mCherry and EGFP-labeled cellular structures in a two-color chimeric cortex. The number of mCherry or EGFP-positive pixels was measured in individual confocal sections over multiple regions of the cortex. Shown is the average number of labeled pixels over total pixel area from all confocal images measured (n=135).
(B) Histogram of binned column width data. Both mCherry and EGFP-labeled columns showed a similar distribution in widths, though more large (>50 micron) EGFP-labeled columns were observed.
(C) Cortical column width measurements were compiled and are presented in a horizontal box and whisker format: the left and right ends of the box mark the lower and upper quartiles, respectively; the vertical line in the box is the median; and the whiskers outside the box extend to the highest and lowest value. The range in column size is similar between mCherry and EGFP-labeled populations; however, the increased number of large EGFP-labeled columns noted in (C) is reflected in the raised median and upper quartile compared to mCherry-labeled columns.
(D) Column uniformity throughout the CP. Optical slices (dashed white lines) were taken at various layers of the CP: just above the IZ (CP layer 5–6, bottom line), halfway through the CP (layer 2–4, middle line), and through the MZ (layer 1, top line). Corresponding optical slices are shown in right panels. Pronounced mixing of cell populations was largely restricted to the MZ (top right panel). (E, F) Distribution of mCherry and EGFP-labeled structures along the cortical radial axis. Linescans along predominately EGFP-positive cortical columns (left box in E, signal intensity plotted as green line in F) or mCherry-positive columns (right box in E, signal intensity plotted as red line in F) revealed increased signal variability in EGFP-dominated columns, most notably in the SVZ and VZ.
Scale bars for (D) and (F) are 50 microns.
To determine if loss of Ena/VASP affected cortical column formation or distribution, we measured the width of individual cortical columns in two-color chimeric embryos. To do this, 20 to 40 micron-thick confocal Z-stacks were collected and assembled from cortical vibratome sections. From these, optical slices through the CP were generated and column width measured (Supplemental Figure 3). Results are shown as a histogram of binned column width data (Figure 2B) and compiled measurements in a horizontal box and whisker format (Figure 2C). A similar range in cortical column widths was observed in both populations (Figure 2C), with the smallest unit size (10 microns, approximately 1 cell width) shared. However, a difference in distribution was evident, with mCherry-labeled columns tending to be smaller relative to EGFP-labeled columns (Figure 2B and 2C). We suggest that this difference in column width likely reflects an increased number of EGFP-positive founder cells during early stages of corticogenesis (Figure 2A), rather than a unique phenotypic property associated with the loss of Ena/VASP.
To determine the level of population mixing within cortical columns, we assessed column uniformity throughout the CP. Optical slices were taken at various layers of the CP: just above the intermediate zone (IZ) (CP layers 5–6), halfway through the CP (layers 2–4), and through the marginal zone (MZ, layer 1). Representative optical slices are shown in Figure 2D. Columns of either control (mCherry) or Ena/VASP-deficient (EGFP) origin tended to be relatively homogenous at deeper layers (bottom two optical slices, Figure 2D). However, increased mixing was noted in the MZ (top optical slice, Figure 2D). We speculate that increase heterogeneity in the MZ arises from tangential migration of Cajul-Retzius cells into this layer (Marin and Rubenstein, 2003; Soriano and Del Rio, 2005), as opposed to radial migration of cortical neurons that predominates deeper cortical layer development. These observations are consistent with current models of cortical development (Rakic, 2007), and highlight the clonal organization of cortical layer development.
Since loss of Ena/VASP alters cortical neuron migration (Goh et al., 2002; Kwiatkowski et al., 2007), we determined if there was any difference in distribution of mCherry versus EGFP-labeled cells along the path of cortical neuron migration, the cortical radial axis. We measured signal intensity along individual columns (Figure 2E, F) and observed slight differences between the distribution of mCherry and EGFP signals (linescans in Figure 2F). Whereas the distribution of mCherry was relatively uniform along the length of the column, more variability was detected in EGFP-expressing processes. In particular, signal increase in the subventricular zone (SVZ) and slight decrease in the VZ was observed in many EGFP-positive columns. Such an increase in signal could result from an increase in the number of cells and their processes in the SVZ, where cortical neurons are believed to pause their migration during polarization (Kriegstein and Noctor, 2004; LoTurco and Bai, 2006). This interpretation is consistent with our earlier work showing that loss of Ena/VASP alters migratory potential in vivo (Goh et al., 2002; Kwiatkowski et al., 2007) and inhibits neuronal polarization in vitro (Dent et al., 2007; Kwiatkowski et al., 2007).
Finally, we examined the potential of EGFP-positive Ena/VASP-deficient neurons to form cortical axon fiber tracts. Loss of Ena/VASP blocks cortical fiber tract formation in Ena/VASP-null embryos (Kwiatkowski et al., 2007); however, a direct comparison of the ability Ena/VASP-deficient neurons to make axons relative to control neurons was not tested. To assess fiber tract formation in two-color chimeric cortices, we stained for Neurofilament-M (NF), a marker for cortical axon fibers. Stained coronal vibratome sections from cortices with a high percentage of EGFP and mCherry-labeled cells (Figure 3A) revealed a lack of EGFP-positive, NF-positive axon fibers, as highlighted in two-channel merges and respective colocalization filters (Figure 3B, C). To obtain better resolution of individual axon fibers amidst the background of migrating neurons and processes, we generated optical slices through the IZ of NF-stained sections (Figure 3D, Supplemental Figure 4). In these optical slices, individual NF-positive axon fibers could be discerned easily (Figure 3D, Supplemental Figure 4). Linescans along NF-positive axon bundles (colored arrowheads, Figure 3D; plots, Figure 3E) revealed a striking correlation in signal intensity between NF (blue) and mCherry (red) signals but no or limited correlation between NF and EGFP (green) signals. To measure colocalization, we applied Pearson’s correlation to these images. We found strong colocalization between the NF and mCherry channels (Pearson’s Correlation = 0.6), which was significantly higher than background colocalization between mCherry and EGFP channels, two channels that should exhibit no overlap (Figure 3F). In contrast, the level of colocalization between NF and EGFP channels (Pearson’s Correlation = 0.15) was not significantly different from background (Figure 3F). Overall, these findings demonstrate that loss of Ena/VASP causes a cell autonomous defect in axon fiber tract formation.
In this study, we generated chimeric embryos using two populations of labeled ES cells as an alternative to traditional electroporation and retroviral-mediated lineage analysis of cortical development. While potentially more labor intensive, this technique offers certain advantages, including the ability to label, and thus identify and study, the earliest cells of the cortex and their involvement in cortical development. The lentiviral system could also be easily modified to express fluorescently-tagged fusions to proteins of interest or altered to express labeled proteins in a cell or temporal-specific manner by exchanging the CAG promoter for a neuronal-specific promoter.
Using this novel lentiviral-mediated technique to effectively label two genetically distinct populations of cells, we show that loss of Ena/VASP causes a cell autonomous defect in axon fiber tract formation during cortical development. Together, our findings demonstrate the utility of this system in analyzing the role of Ena/VASP in cortical development, and expect that it will serve as a useful tool in future studies of neuronal development.
Experimental Methods
Lentiviral Vectors
pLentiLox 4.4 (pLL 4.4) is modified version of pLentiLox 3.7 (Rubinson et al., 2003) first described in (Kwiatkowski et al., 2007). Fluorescent protein (EGFP, mCherry or DsRed2) expression is driven from a CAG (CMV-enhancer, chicken beta-actin, beta-globin intron) promoter. pLL4.4 gap-tagged constructs were made by PCR amplifying a region containing the 19 N-terminal amino acids of gap-43 from plasmid pCA-GAP-EGFP (gift of Ami Okada, (Okada et al., 1999)) and cloning them upstream of EGFP, mCherry and DsRed2.
Lentivirus Production
Lentivirus was produced essentially as described in (Rubinson et al., 2003).
Labeled ES Cell and Chimera Generation
All ES cell and mouse work in this study adhered to guidelines established by the MIT Committee on Animal Care. Control R1 and Ena/VASP-deficient ES cells (Kwiatkowski et al., 2007) were cultured following standard protocols. ES cells were infected with gap-mCherry or gap-EGFP lentiviruses as previously described (Rubinson et al., 2003). Following infection, ES cells were sorted for FP expression by FACS and plated onto feeder cells at low density to permit individual colony formation. After one week, colonies with strong, homogenous FP expression were picked and expanded. Control gap-mCherry and gap-EGFP Ena/VASP-null ES cells were injected into E3.5 C57/B6 blastocysts and implanted into pseudo-pregnant females. Chimeric embryos were dissected from E12.5 to E16.5, scored for FP protein expression on a dissection scope outfitted with a fluorescent lamp, and prepped for vibratome sectioning as described (Kwiatkowski et al., 2007).
Immunostaining and Confocal Imaging
100 micron vibratome sections were cut from the embryonic brains and stored in PBS. For two-channel imaging, sections were mounted directly in Fluoromount G (EMS) on slides. For staining, sections were blocked for 3 hours at room temperature in blocking solution (PBS plus 5% normal goat serum, 5 % BSA and 0.2% Triton X-100). Primary antibodies were diluted in blocking solution, and sections incubated overnight with agitation in 4 degrees. Sections were washed 4 times, 15 minutes/wash, in PBS. Secondary antibodies were diluted in blocking solution, and sections incubated with agitation for 3 hours at room temperature. Sections were washed as before and mounted in Fluoromount G for imaging. All confocal imaging was performed on a Leica SP2 AOBS confocal laser scanning microscope. 30 to 40 micron thick z-stacks were collected from both unstained and stained tissue sections to ensure uniform signal intensity through the stack.
Neuronal Culture, Immunostaining and Imaging
Low density hippocampal cultures were prepared from embryonic rats as previously described (Leal-Ortiz et al., 2008). Concentrated lentivirus was added to cultured neurons on the day of plating (DIV0) or the next (DIV1). Neurons at either DIV4/5 or DIV14/15 were fixed in PFA/PHEM (Strasser et al., 2004) and immunostained as described (Dent et al., 2007). Stained neurons were imaged on an Applied Precision Deltavision wide-field deconvolution system, and images deconvolved post-capture.
Confocal Image Processing and Analysis
Confocal image stacks were compiled in Volocity (Improvision) to examine tissue organization and staining and to generate projections for illustrative purposes. All image analysis was performed on unmodified image stacks using Metamorph or ImageJ software. For all linescans, the average pixel intensity along the width of the line was calculated. For all optical slices, the maximum pixel value across a 4 pixel wide swath along the slice was calculated.
Supplementary Material
Supplemental Figure 1. Expression and localization of gap-tagged FPs in cultured neurons
(A, B) Early stage (DIV 4) cultured neurons infected with pLL4.4 (A) and pLL4.4 gap-E (B). Neuron were infected with lentivirus at DIV 0, fixed at DIV 4 and stained for F-actin and EGFP. Similar to EGFP, gap-EGFP labeled the cell body and neurites (axons and dendrites). However, gap-EGFP labeled small protrusions (filopodia) and growth cones better than EGFP alone (note the prominent yellow colocalization at neurite growth cones in B compared A). (C-F) gap-mCherry localization in later stage (DIV 14) neuronal cultures. In more mature cultures, gap-mCherrry localization became more restricted to axons, evident in fixed cells stained for F-actin and MAP2, a marker for dendrites (C). Higher magnification images revealed that gap-mCherry was an excellent marker of axonal membranes, labeling synaptic contacts and synaptic buotons (white arrows, D and E) and F-actin-rich axonal growth cones (F).
Scale bars for (A) and (B) are 10 microns.
Supplemental Figure 2. Early cortical migration events
(A, B) gap-EGFP-expressing mmvvee cells during early stages of corticogenesis in an E12.5 chimeric embryo. The dashed red line marks the approximate location of the pial membrane and the dashed white line marks the ventricular surface in (A). Many labeled cells were morphologically identical to cortical neurons undergoing somal translocation, with long, branched pial-directed leading processes (white asterisks in A and right panel of B). During somal translocation, believed to be the most common form of radial migration in early corticogenesis, the leading process becomes thicker and shorter as the cell advances (white arrows in right panel of B).
Scale bars for (A) and (B) are 10 microns.
Supplemental Figure 3. Cortical column width
The width of individual mCherry (wild type) and EGFP (mmvvee) columns of neurons in the CP was measured. 20 to 40 micron-thick confocal Z-stacks from coronal vibratome sections of high percentage two-color cortices were gathered and assembled (left panel). From these, optical slices through the CP (right panel, white dashed line in left panel represents site of optical slice) were generated and columns measured (black lines below the optical slice represent the width of mCherry-labeled columns in shown optical slice).
Scale bar in left panel is 50 microns.
Supplemental Figure 4. Optical slices through NF-stained cortical sections
(A–C) Optical slices through the intermediate zone of multiple NF-labeled cortical sections. Individual EGFP (green), mCherry (red), and NF (blue) slices as well as the merge are shown.
Acknowledgments
Doug Rubinson designed and constructed the original pLL4.4 vector. We thank Ulrike Philippar and Elizabeth Alcamo for technical assistance, Clarissa Waites and Sergio Leal-Ortiz for cultured neuron preparation and Aurora Burds and the MIT Rippel Facility for chimera generation. We appreciate Sue McConnell, Carla Shatz and Carlos Lois for their helpful comments and advice. A.V.K is supported by NRSA grant 5T32 CA09302. C.C.G is supported by NIH grants NS39471 and NS353862. W.J.N. is supported by NIH grants GM 78270 and 35527. F.B.G. is supported by NIH grant NM068678.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Boulos S, Meloni BP, Arthur PG, Bojarski C, Knuckey NW. Assessment of CMV, RSV and SYN1 promoters and the woodchuck post-transcriptional regulatory element in adenovirus vectors for transgene expression in cortical neuronal cultures. Brain Res. 2006;1102:27–38. doi: 10.1016/j.brainres.2006.04.089. [DOI] [PubMed] [Google Scholar]
- Dent EW, Kwiatkowski AV, Mebane LM, Philippar U, Barzik M, Rubinson DA, Gupton S, Van Veen JE, Furman C, Zhang J, Alberts AS, Mori S, Gertler FB. Filopodia are required for cortical neurite initiation. Nat Cell Biol. 2007;9:1347–1359. doi: 10.1038/ncb1654. [DOI] [PubMed] [Google Scholar]
- Drees F, Gertler FB. Ena/VASP: proteins at the tip of the nervous system. Curr Opin Neurobiol. 2008;18:53–59. doi: 10.1016/j.conb.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh KL, Cai L, Cepko CL, Gertler FB. Ena/VASP proteins regulate cortical neuronal positioning. Curr Biol. 2002;12:565–569. doi: 10.1016/s0960-9822(02)00725-x. [DOI] [PubMed] [Google Scholar]
- Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003;19:541–564. doi: 10.1146/annurev.cellbio.19.050103.103356. [DOI] [PubMed] [Google Scholar]
- Kriegstein AR, Noctor SC. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27:392–399. doi: 10.1016/j.tins.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski AV, Rubinson DA, Dent EW, Edward van Veen J, Leslie JD, Zhang J, Mebane LM, Philippar U, Pinheiro EM, Burds AA, Bronson RT, Mori S, Fassler R, Gertler FB. Ena/VASP Is Required for neuritogenesis in the developing cortex. Neuron. 2007;56:441–455. doi: 10.1016/j.neuron.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Leal-Ortiz S, Waites CL, Terry-Lorenzo R, Zamorano P, Gundelfinger ED, Garner CC. Piccolo modulation of Synapsin1a dynamics regulates synaptic vesicle exocytosis. J Cell Biol. 2008;181:831–846. doi: 10.1083/jcb.200711167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoTurco JJ, Bai J. The multipolar stage and disruptions in neuronal migration. Trends Neurosci. 2006;29:407–413. doi: 10.1016/j.tins.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL. Cell migration in the forebrain. Annu Rev Neurosci. 2003;26:441–483. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- Okada A, Lansford R, Weimann JM, Fraser SE, McConnell SK. Imaging cells in the developing nervous system with retrovirus expressing modified green fluorescent protein. Exp Neurol. 1999;156:394–406. doi: 10.1006/exnr.1999.7033. [DOI] [PubMed] [Google Scholar]
- Rakic P. The radial edifice of cortical architecture: from neuronal silhouettes to genetic engineering. Brain Res Rev. 2007;55:204–219. doi: 10.1016/j.brainresrev.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Ihrig MM, McManus MT, Gertler FB, Scott ML, Van Parijs L. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- Soriano E, Del Rio JA. The cells of cajal-retzius: still a mystery one century after. Neuron. 2005;46:389–394. doi: 10.1016/j.neuron.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Strasser GA, Rahim NA, VanderWaal KE, Gertler FB, Lanier LM. Arp2/3 is a negative regulator of growth cone translocation. Neuron. 2004;43:81–94. doi: 10.1016/j.neuron.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Sur M, Rubenstein JL. Patterning and plasticity of the cerebral cortex. Science. 2005;310:805–810. doi: 10.1126/science.1112070. [DOI] [PubMed] [Google Scholar]
- Tan SS, Breen S. Radial mosaicism and tangential cell dispersion both contribute to mouse neocortical development. Nature. 1993;362:638–640. doi: 10.1038/362638a0. [DOI] [PubMed] [Google Scholar]
- Tan SS, Faulkner-Jones B, Breen SJ, Walsh M, Bertram JF, Reese BE. Cell dispersion patterns in different cortical regions studied with an X-inactivated transgenic marker. Development. 1995;121:1029–1039. doi: 10.1242/dev.121.4.1029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Expression and localization of gap-tagged FPs in cultured neurons
(A, B) Early stage (DIV 4) cultured neurons infected with pLL4.4 (A) and pLL4.4 gap-E (B). Neuron were infected with lentivirus at DIV 0, fixed at DIV 4 and stained for F-actin and EGFP. Similar to EGFP, gap-EGFP labeled the cell body and neurites (axons and dendrites). However, gap-EGFP labeled small protrusions (filopodia) and growth cones better than EGFP alone (note the prominent yellow colocalization at neurite growth cones in B compared A). (C-F) gap-mCherry localization in later stage (DIV 14) neuronal cultures. In more mature cultures, gap-mCherrry localization became more restricted to axons, evident in fixed cells stained for F-actin and MAP2, a marker for dendrites (C). Higher magnification images revealed that gap-mCherry was an excellent marker of axonal membranes, labeling synaptic contacts and synaptic buotons (white arrows, D and E) and F-actin-rich axonal growth cones (F).
Scale bars for (A) and (B) are 10 microns.
Supplemental Figure 2. Early cortical migration events
(A, B) gap-EGFP-expressing mmvvee cells during early stages of corticogenesis in an E12.5 chimeric embryo. The dashed red line marks the approximate location of the pial membrane and the dashed white line marks the ventricular surface in (A). Many labeled cells were morphologically identical to cortical neurons undergoing somal translocation, with long, branched pial-directed leading processes (white asterisks in A and right panel of B). During somal translocation, believed to be the most common form of radial migration in early corticogenesis, the leading process becomes thicker and shorter as the cell advances (white arrows in right panel of B).
Scale bars for (A) and (B) are 10 microns.
Supplemental Figure 3. Cortical column width
The width of individual mCherry (wild type) and EGFP (mmvvee) columns of neurons in the CP was measured. 20 to 40 micron-thick confocal Z-stacks from coronal vibratome sections of high percentage two-color cortices were gathered and assembled (left panel). From these, optical slices through the CP (right panel, white dashed line in left panel represents site of optical slice) were generated and columns measured (black lines below the optical slice represent the width of mCherry-labeled columns in shown optical slice).
Scale bar in left panel is 50 microns.
Supplemental Figure 4. Optical slices through NF-stained cortical sections
(A–C) Optical slices through the intermediate zone of multiple NF-labeled cortical sections. Individual EGFP (green), mCherry (red), and NF (blue) slices as well as the merge are shown.