

Conditions are reported for the crystallization of the flexible nuclear import receptor importin-β. Preliminary X-ray diffraction data indicate that the structure can be solved using molecular replacement.
Keywords: nucleocytoplasmic transport, importins, karyopherins
Abstract
The transport of macromolecules across the nuclear envelope is an essential eukaryotic process that enables proteins such as transcription factors, polymerases and histones to gain access to the genetic material contained within the nucleus. Importin-β plays a central role in the nucleocytoplasmic transport process, mediating nuclear import through a range of interactions with cytoplasmic, nuclear and nuclear pore proteins such as importin-α, Ran, nucleoporins and various cargo molecules. The unliganded form of the full-length yeast importin-β has been expressed and crystallized. The crystals were obtained by vapour diffusion at pH 6.5 and 290 K. The crystals belonged to space group P21 (unit-cell parameters a = 58.17, b = 127.25, c = 68.52 Å, β = 102.23). One molecule is expected in the asymmetric unit. The crystals diffracted to 2.4 Å resolution using a laboratory X-ray source and were suitable for crystal structure determination.
1. Introduction
Nucleocytoplasmic transport is a vital eukaryotic process that enables the specific targeting of proteins such as transcription factors, polymerases and histones to the nucleus. Importantly, regulation of nucleocytoplasmic transport provides the cell with an additional level of control in regulating processes such as development, cell division and other cellular processes that are important in higher eukaryotes. Aberrations in the transport process have been shown to lead to cancer and other diseases such as genetic sex reversal (Cook et al., 2007 ▶).
The mechanism by which proteins are actively shuttled across the nuclear envelope into the nucleus is dependent on a superfamily of carrier proteins known as importins (or karyopherins; Bednenko et al., 2003 ▶; Forwood et al., 2008 ▶; Lee et al., 2005 ▶). Importins recognize a diverse range of proteins through their interaction with nuclear localization signals (NLSs) contained within proteins that are destined for the nucleus. Importin–cargo binding is the first step of the nuclear import cycle and is followed by docking and translocation through the nuclear pore complex (Cingolani et al., 1999 ▶; Forwood et al., 2008 ▶; Fries et al., 2007 ▶; Stewart, 2006 ▶). Once in the nucleus, the importin–cargo complex is dissociated through a high-affinity interaction with RanGTP, which displaces the cargo protein and allows the recycling of importins back to the nucleus (Bischoff & Ponstingl, 1991 ▶; Forwood et al., 2001 ▶; Jans, 1995 ▶).
Importin-β plays a central role in the nucleocytoplasmic transport process. It mediates nuclear import through a range of interactions with cytoplasmic and nuclear pore-containing proteins such as importin-α, Ran, nucleoporins and many cargo proteins (Bednenko et al., 2003 ▶; Cingolani et al., 1999 ▶). Details of these interactions and their importance in mediating nuclear transport have been elucidated in structural studies, predominantly by X-ray crystallography (Lee et al., 2005 ▶; Pyhtila & Rexach, 2003 ▶; Ryan et al., 2007 ▶; Stewart, 2006 ▶). An emerging theme is that the flexibility of importin-β plays an important role in mediating the binding to different partners and thereby the nucleocytoplasmic transport cycle (Lee et al., 2005 ▶; Saric et al., 2007 ▶; Stewart, 2006 ▶). Despite the range of importin-β–protein complexes that have been solved over the last decade, a high-resolution structure of unliganded importin-β has yet to be determined, possibly owing to the size and flexibility of the molecule. To obtain a better understanding of the function of this molecule, we have expressed, purified and determined conditions that induce the crystallization of importin-β from Saccharomyces cerevisiae (often referred to as Kap95p or karyopherin-β1; we will use the term ‘yeast importin-β’ hereafter throughout this paper). Here, we describe the crystallization and preliminary X-ray diffraction analysis of full-length yeast importin-β.
2. Experimental methods
2.1. Expression and purification
Yeast importin-β was recombinantly expressed as a GST-fusion protein as described previously (Forwood et al., 2001 ▶). Briefly, the pGEX2T-yImportin-β expression vector was transformed into chemically competent Escherichia coli strain BL21 (DE3). Using a single colony, 5 ml Luria–Bertani (LB) medium was inoculated and incubated overnight at 310 K and 225 rev min−1 in the presence of ampicillin (100 µg ml−1). This culture was used to inoculate 500 ml auto-induction media (Studier, 2005 ▶) with ampicillin and incubated until an OD600nm of 6–10 was reached. The cells were harvested, with the typical yield of importin-β being 30–50 mg per litre of culture. The GST-tagged protein was purified utilizing affinity chromatography (Glutathione-Sepharose 4B column, GE Healthcare). Following elution of the protein, 100 µl thrombin protease (1 U µl−1) was added to cleave the GST tag from approximately 40 mg yeast importin-β-GST fusion protein in 10 ml and incubated for 12 h at room temperature. The protein mixture was then further purified by size-exclusion chromatography (S-200 column, GE Healthcare) and collected in 20 mM Tris pH 7.4, 50 mM NaCl. The elution profile of yeast importin-β from the size-exclusion column was indicative of a monomeric species. Fractions containing the purified recombinant yeast importin-β were passed through a glutathione-Sepharose affinity matrix to remove residual traces of GST and uncleaved GST-yeast importin-β. The protein was concentrated to 40 mg ml−1 and stored in 20 mM Tris pH 7.4 and 50 mM NaCl at 193 K. The protein was >95% pure as assessed by SDS–PAGE.
2.2. Crystallization
Crystallization conditions were screened by utilizing the sparse-matrix and hanging-drop vapour-diffusion techniques across a range of commercially available crystal screens (Crystal Screen and Crystal Screen 2 from Hampton Research and PACT Premier from Molecular Dimensions). 1 µl protein sample was combined with 1 µl reservoir solution and suspended above 500 µl reservoir solution and incubated at 290 K. Small needle-shaped crystals were obtained using conditions from the PACT Premier crystallization screen (E11, F11, G11 and H11). To obtain single and large diffraction-quality crystals, the protein was diluted to 2 mg ml−1 for crystallization using 20 mM Tris pH 7.4 and 50 mM NaCl and the crystallization conditions were iteratively optimized around different molecular weights and concentrations of PEG and by varying the buffers, salts and additives. Diffraction-quality crystals were formed in 20% PEG 4K, 0.1 M 2-(N-morpholino)ethanesulfonic acid (MES) pH 6.5, 20 mM MgCl2, 125 mM NaCl, 12% MPD with 0.1 mg ml−1 of the peptide HREKYPNYKYRPRRKAKMLGC. The peptide was synthetically manufactured by Auspep and was solubilized to a final stock concentration of 10 mg ml−1 in 20 mM Tris, 50 mM NaCl. The peptide is a small region of the human sex-determining region Y (hSRY) protein and was originally included in the crystallization conditions with the aim of identifying the interaction interface with yeast importin-β as a basis for the interaction between hSRY and importin-β. However, the peptide did not bind specifically to yeast importin-β as shown by mass-spectrometric analysis of the crystals. It also did not alter the shape of the molecule in solution as shown by small-angle X-ray scattering experiments (Fig. 1 ▶). However, the peptide serendipitously improved the diffraction (crystals lacking the peptide were small and diffracted poorly).
Figure 1.

(a) SAXS profiles of yeast importin-β in the absence (open circles) and presence (grey triangles) of hSRY peptide plotted as a logarithm of scattering intensity as a function of the scattering vector q (Å−1). (b) The P(r) profiles of the scattering data as determined using the program GNOM (Svergun, 1992 ▶), showing a maximum particle dimension of 120 Å for both SAXS conditions.
2.3. Diffraction data collection
Translucent rod-shaped crystals (100 × 100 × 300 µm) grew after 2 d at 290 K. For data collection, these were transiently soaked in reservoir solution and flash-cooled under a 100 K nitrogen stream (Cryocool, Cryo Industries, New Hampshire, USA). Diffraction data were collected from a single crystal on an R-AXIS IV++ image-plate detector using Cu Kα radiation produced by a Rigaku FR-E rotating-anode generator (Rigaku/MSC, Texas, USA). Raw data were auto-indexed, integrated and scaled using the HKL-2000 package (data-collection statistics are summarized in Table 1 ▶). The structure of Kap95p will be solved by molecular replacement.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Reservoir solution | 20% PEG 4000, 0.1 M MES pH 6.5, 20 mM MgCl2, 125 mM NaCl, 12% MPD |
| Crystal system | Monoclinic |
| Space group | P21 |
| Wavelength (Å) | 1.54 |
| Unit-cell parameters (Å, °) | a = 58.17, b = 127.25, c = 68.52, β = 102.23 |
| Resolution (Å) | 30–2.4 (2.49–2.40) |
| Observations | 251525 (24460) |
| Unique reflections | 37902 (3727) |
| Completeness (%) | 97.1 (95.5) |
| Mosaicity | 0.3 |
| Multiplicity | 6.6 (6.6) |
| Rmerge† (%) | 7.5 (52.9) |
| 〈I/σ(I)〉 | 11.4 (2.6) |
| Wilson B factor (Å2) | 31.8 |
| Matthews coefficient (Å3 Da−1) | 2.68 [for one molecule in the ASU] |
| Solvent content (%) | 54.06 |
R
merge = 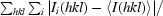
 , where I
i(hkl) is the intensity of an individual measurement of the reflection with Miller indices hkl and 〈I(hkl)〉 is the mean intensity of that reflection. Calculated for I > −3σ(I).
, where I
i(hkl) is the intensity of an individual measurement of the reflection with Miller indices hkl and 〈I(hkl)〉 is the mean intensity of that reflection. Calculated for I > −3σ(I).
2.4. Small-angle X-ray scattering
Small-angle X-ray scattering (SAXS) data were collected at the Centre of Microscopy and Microanalysis at the University of Queensland using an Anton Paar/Panalytical SAXSess system. The instrument uses Cu Kα radiation (1.54 Å) from a sealed X-ray tube, line collimation and a CCD detector (Princeton Instruments). Purified yeast importin-β in the presence and absence of peptide (0.1 mg ml−1) was dialysed overnight in 20 mM Tris pH 7.4, 125 mM NaCl, 10 mM DTT at 277 K. SAXS measurements were collected at 293 K at a concentration of 2–5 mg ml−1 in a 1 mm silica capillary. To monitor for radiation damage, 2–6 successive 15 min exposures were compared and no differences in scattering intensity were seen for either sample, suggesting that no structural changes had occurred. The scattering data were collected from q = 0.005 to 0.20 Å−1 and were reduced to remove the contributions from the dark current of the detector, the scattering of the dialysis buffer and the empty capillary. Data were further normalized to absolute intensity using pure water as a calibration standard using SAXS-Quant software from Anton Paar. The indirect transform program GNOM (Svergun, 1992 ▶) was used to evaluate the pair distance distribution function P(r), the radius of gyration R g and the maximum size D max of the scattering particle. P(r) is a real-space representation of the scattering profile and represents the frequency of vector lengths connecting small-volume elements within the entire volume of the scattering particle.
3. Results and discussion
Crystals of unliganded full-length yeast importin-β (residues 1–861) were obtained by vapour diffusion using 20% PEG 4000, 0.1 M MES pH 6.5, 20 mM MgCl2, 125 mM NaCl, 12% MPD as the reservoir solution in the presence of 0.1 mg ml−1 peptide HREKYPNYKYRPRRKAKMLGC. This peptide did not bind specifically to the protein as shown by mass-spectrometric analysis of the crystals. It also did not alter the shape of the molecule in solution as shown by SAXS experiments when added at the same concentration used for crystallization. The scattering profiles are shown in Fig. 1 ▶(a). The indirect Fourier transform program GNOM (Svergun, 1992 ▶) was used to calculate the radius of gyration (R g), maximum particle dimension (D max) and pair distance distribution function [P(r)] from the scattering patterns of the protein. The calculated P(r) curves display similar distributions for both peptide-free and peptide-supplemented samples of yeast importin-β (Fig. 1 ▶ b), with a D max value of 120 Å for both samples, suggesting that the peptide does not change the shape of the protein in solution. Consistently, the calculated R g values are in close agreement for the peptide-free and peptide-supplemented conditions (39.7 ± 0.5 and 39.9 ± 0.4 Å, respectively).
MPD was incorporated into the crystallization conditions at 12% primarily as a cryoprotectant and was not required for crystal growth. The crystals were rod-shaped (dimensions of 0.3 × 0.1 × 0.1 mm) and belonged to the monoclinic space group P21. The crystals appeared after 2 d and grew to maximum dimensions within one week. Using the laboratory source, the crystals diffracted X-rays to 2.4 Å resolution. A representative diffraction image is shown in Fig. 2 ▶ and the data-collection statistics are shown in Table 1 ▶.
Figure 2.

Diffraction image of the crystal described in Table 1 ▶. The crystal-to-detector distance was 170 mm; the resolution at the edge of the image is 2.2 Å.
Because of the availability of the crystal structures of several ligand-bound forms of yeast importin-β (Liu & Stewart, 2005 ▶), we believe it is likely that the structure will be solved using molecular replacement. Determination of the crystal structure of the nuclear import receptor will provide a greater understanding of the flexibility of importin-β in nuclear transport. Importin-β is a solenoid protein consisting of 19 tandem HEAT (huntingtin, EF3, A subunit of PP2A, TOR1) repeats. Solenoid proteins generally lack long-range intramolecular contacts and are therefore often very flexible (Kobe & Kajava, 2000 ▶). This flexibility is often an important feature in the specific function of these proteins. The structure of the unliganded molecule will provide insights into the flexibility of the molecule and the role of this flexibility in nuclear transport (Suel et al., 2006 ▶).
Acknowledgments
We acknowledge the use of the University of Queensland ROCX Diffraction Facility. BK is an Australian Research Council (ARC) Federation Fellow and a National Health and Medical Research Council (NHMRC) Honorary Research Fellow and JKF was an NHMRC C. J. Martin Fellow.
References
- Bednenko, J., Cingolani, G. & Gerace, L. (2003). Traffic, 4, 127–135. [DOI] [PubMed]
- Bischoff, F. R. & Ponstingl, H. (1991). Nature (London), 354, 80–82. [DOI] [PubMed]
- Cingolani, G., Petosa, C., Weis, K. & Muller, C. W. (1999). Nature (London), 399, 221–229. [DOI] [PubMed]
- Cook, A., Bono, F., Jinek, M. & Conti, E. (2007). Annu. Rev. Biochem.76, 647–671. [DOI] [PubMed]
- Forwood, J. K., Harley, V. & Jans, D. A. (2001). J. Biol. Chem.276, 46575–46582. [DOI] [PubMed]
- Forwood, J. K., Lonhienne, T. G., Marfori, M., Robin, G., Meng, W., Guncar, G., Liu, S. M., Stewart, M., Carroll, B. J. & Kobe, B. (2008). J. Mol. Biol.383, 772–782. [DOI] [PubMed]
- Fries, T., Betz, C., Sohn, K., Caesar, S., Schlenstedt, G. & Bailer, S. M. (2007). J. Biol. Chem.282, 19292–19301. [DOI] [PubMed]
- Jans, D. A. (1995). Biochem. J.311, 705–716. [DOI] [PMC free article] [PubMed]
- Kobe, B. & Kajava, A. V. (2000). Trends Biochem. Sci.25, 509–515. [DOI] [PubMed]
- Lee, S. J., Matsuura, Y., Liu, S. M. & Stewart, M. (2005). Nature (London), 435, 693–696. [DOI] [PubMed]
- Liu, S. M. & Stewart, M. (2005). J. Mol. Biol.349, 515–525. [DOI] [PubMed]
- Pyhtila, B. & Rexach, M. (2003). J. Biol. Chem.278, 42699–42709. [DOI] [PubMed]
- Ryan, K. J., Zhou, Y. & Wente, S. R. (2007). Mol. Biol. Cell, 18, 886–898. [DOI] [PMC free article] [PubMed]
- Saric, M., Zhao, X., Korner, C., Nowak, C. & Kuhlmann, J. (2007). FEBS Lett.581, 1369–1376. [DOI] [PubMed]
- Stewart, M. (2006). Biochem. Soc. Trans.34, 701–704. [DOI] [PubMed]
- Studier, F. W. (2005). Protein Expr. Purif.41, 207–234. [DOI] [PubMed]
- Suel, K. E., Cansizoglu, A. E. & Chook, Y. M. (2006). Methods, 39, 342–355. [DOI] [PMC free article] [PubMed]
- Svergun, D. I. (1992). J. Appl. Cryst.25, 495–503.