

The peptide hormone hepcidin exerts its function by binding to the transmembrane cellular iron exporter ferroportin and inducing its internalization and degradation, resulting in decreased intestinal iron uptake and iron retention in the reticulo-endothelial (RE) macrophages. Inflammatory cytokines and iron loading increase hepcidin production, while increased bone marrow activity, and anemia suppress hepcidin synthesis.1,2 However, most of the evidence of these regulatory processes is obtained by molecular in vitro work and mice models, and much is still unknown about how these different stimuli interact in man.
Sickle cell disease (SCD) patients are characterized by chronic hemolytic anemia, increased erythropoiesis and a chronic inflammatory state with endothelial activation and enhanced red cell and leukocyte adhesion. Sickle cell patients have iron overload due to chronic blood transfusions in the treatment or prevention of the severe sickle cell-related complications such as stroke.3 SCD has been associated with low urinary hepcidin levels in children.4 However, serum hepcidin 25-amino acid isoform (hepcidin-25) levels, which are directly responsible for the biological effect, have not been documented and factors that contribute to hepcidin regulation in this disease have not been assessed.
Samples were collected from adult steady state SCD patients5 with various hemoglobin (Hb) genotypes (9 HbSS, 3 HbSβ0-thalassemia and 4 HbSC, Table 1) between February 2005 and February 2006, and stored in polypropylene tubes at –80°C until analysis. Patients received no transfusions or chelation therapy for two months prior to sampling. Race matched controls were heterozygous for HbS or C.5 Serum and urinary hepcidin-25 measurements were performed in November and December 2007 by use of surface enhanced laser desorption ionization-time of flight mass spectrometry (SELDI-TOF MS) as previously described.6,7 The hepcidin regulators’ inflammation, iron store and erythropoiesis (reflected in the serum markers C-reactive protein (CRP), ferritin and soluble transferrin receptor (sTfR), respectively) were assessed to delineate the regulatory pathways of hepcidin.8 Approval for the study was obtained from the Medical Ethics Committee of the Academic Medical Center in Amsterdam.
Table 1.
Characteristics of study populations of adult sickle cell disease patients in steady state of their disease.

We found the various serum parameters to vary widely within this population (Table 1). Of note is the pattern of the serum iron parameters, which shows highly variable ferritin levels, not simply related to the transfusion history and in the presence of normal transferrin saturation (TS). This suggests an iron distribution pattern of the anemia of chronic disease, with relatively more iron in the RE system.
Serum hepcidin-25 levels were below the lower limit of detection (LLOD <0.5 nM) in 5 SCD patients, while in the rest, the levels were between 1 and 10 nM, which is considered to be the normal range (Table 1). The median serum and urine hepcidin-25 levels were similar for patients and controls (p>0.2), but hepcidin-25/ferritin ratio’s as a measure of appropriateness of hepatocyte-produced hepcidin for the iron burden,4 were significantly lower for patients (p<0.01) (Table 1). However, as ferritin in SCD might be increased by inflammation and iron loading of RE cells by transfusions, this ratio might not be suitable in the evaluation of the adequacy of hepcidin in response to hepatocyte iron loading.9
Results confirm that erythropoiesis down-regulates hepcidin-25, i.e. when only sTfR is increased, serum hepcidin-25 levels are in the lower normal range or even not detectable (<LLOD-3.6 nM; patients 2, 4, 5, 13–16). In cases where next to a substantially increased sTfR inflammation and/or high iron stores are also present, serum hepcidin-25 levels are in the normal range (1.2 −9.5 nM; patients 1, 3, 6, 8–12) confirming the induction of hepcidin by inflammation and elevated iron stores in sickle cell patients. Interestingly, in patient 7 the low hepcidin-25 level due to increased erythropoiesis (highly elevated sTfR) is not compensated by low grade inflammation (CRP of 10 mg/L) and a slightly elevated iron store (ferritin of 210 μg/L), resulting in undetectable serum hepcidin-25 levels.
While this is a small study, the results only describe the qualitative contribution of the various parameters to hepcidin-25 levels. Nevertheless, Spearman’s correlation analysis showed that serum hepcidin-25 levels were significantly correlated with urine hepcidin-25, log ferritin, Body Mass Index (BMI)10 (Figure 1A–C) and age, but not with CRP, sTfR, TS (Figure 1D–F) and hemoglobin.
Figure 1.
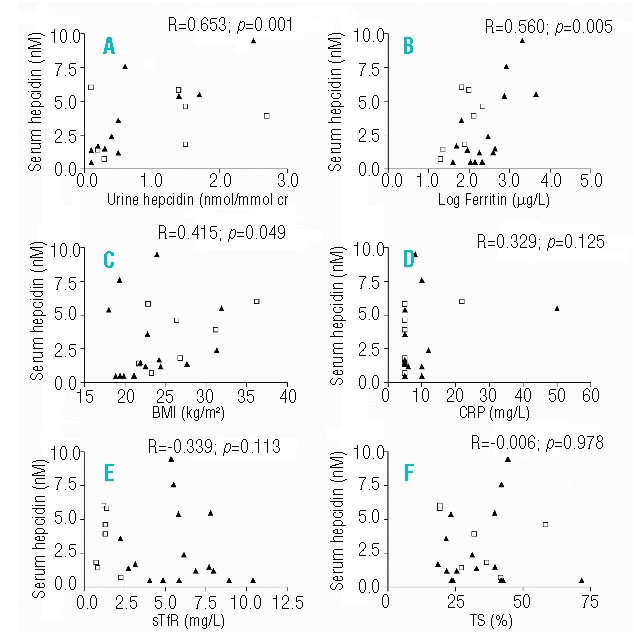
Spearman’s correlation analysis of serum hepcidin-25 with (A) urine hepcidin-25 (B) Log Ferritin (C) BMI (D) CRP (E) sTfR, F) TS. Data represent the whole study population of 16 SCD patients (HbSS, HbSβ0-thalassemia and HbSC) (triangle) and 7 controls (square). BMI: body mass index; CRP: C-reactive protein; sTfR: soluble transferrin receptor; TS: transferrin saturation.
In conclusion, this proof of principle study in a heterogeneous group of SCD patients indicates that: (i) previous results obtained in vitro and mice studies of hepcidin-25 suppression by increased erythropoietic activity that is counterbalanced by iron stores and (low grade) inflammation are also valid in man; (ii) larger studies are needed to determine the quantitative contribution of various factors to hepcidin-25 regulation in this disease.
The insights gained in this study could be clinically beneficial in the identification and treatment of patients most at risk of iron mediated tissue damage.
References
- 1.Nemeth E, Ganz T. Hepcidin and iron-loading anemias. Haematologica. 2006;91:727–32. [PubMed] [Google Scholar]
- 2.Kemna EH, Tjalsma H, Willems HL, Swinkels DW. Hepcidin: from discovery to differential diagnosis. Haematologica. 2008;93:90–7. doi: 10.3324/haematol.11705. [DOI] [PubMed] [Google Scholar]
- 3.Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004;364:1343–60. doi: 10.1016/S0140-6736(04)17192-4. [DOI] [PubMed] [Google Scholar]
- 4.Kearney SL, Nemeth E, Neufeld EJ, Thapa D, Ganz T, Weinstein DA, Cunningham MJ. Urinary hepcidin in congenital chronic anemias. Pediatr Blood Cancer. 2007;48:57–63. doi: 10.1002/pbc.20616. [DOI] [PubMed] [Google Scholar]
- 5.van Beers EJ, Nieuwdorp M, Duits AJ, Evers LM, Schnog JJ, Biemond BJ. Sickle cell patients are characterized by a reduced glycocalyx volume. Haematologica. 2008;93:307–8. doi: 10.3324/haematol.12027. [DOI] [PubMed] [Google Scholar]
- 6.Kemna EH, Tjalsma H, Podust VN, Swinkels DW. Mass spectrometry-based hepcidin measurements in serum and urine: analytical aspects and clinical implications. Clin Chem. 2007;53:620–8. doi: 10.1373/clinchem.2006.079186. [DOI] [PubMed] [Google Scholar]
- 7.Swinkels DW, Girelli D, Laarakkers C, Kroot J, Campostrini N, Kemna EH, et al. Advances in quantitative hepcidin measurements by time-of-flight mass spectrometry. PLoS ONE. 2008;3:e2706. doi: 10.1371/journal.pone.0002706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kemna EH, Kartikasari AE, van Tits LJ, Pickkers P, Tjalsma H, Swinkels DW. Regulation of hepcidin: insights from biochemical analyses on human serum samples. Blood Cells Mol Dis. 2008;40:339–46. doi: 10.1016/j.bcmd.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Swinkels DW, Drenth JP. Hepcidin in the management of patients with mild non-hemochromatotic iron overload: Fact or fiction? J Hepatol. 2008;49:680–5. doi: 10.1016/j.jhep.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 10.Bekri S, Gual P, Anty R, Luciani N, Dahman M, Ramesh B, et al. Increased adipose tissue expression of hepcidin in severe obesity is independent from diabetes and NASH. Gastroenterology. 2006;131:788–96. doi: 10.1053/j.gastro.2006.07.007. [DOI] [PubMed] [Google Scholar]