

Abstract
The insertion and removal of N-methyl D-aspartate (NMDA) receptors from the synapse are critical events that modulate synaptic plasticity. While a great deal of progress has been made on understanding the mechanisms that modulate trafficking of NMDA receptors, we do not currently understand the molecular events required for the fusion of receptor containing vesicles with the plasma membrane. Here we show that sphingomyelin phosphodiesterase3 (also known as neutral sphingomyelinase-2; nSMase2) is critical for TNFα-induced trafficking of NMDA receptors and synaptic plasticity. TNFα initiated a rapid increase in ceramide that was associated with increased surface localization of NMDA receptor NR1 subunits and a specific clustering of NR1 phosphorylated on serines 896 and 897 into lipid rafts. Brief applications of TNFα increased the rate and amplitude of NMDA-evoked calcium bursts and enhanced excitatory postsynaptic currents (EPSCs). Pharmacological inhibition or genetic mutation of nSMase2 prevented TNFα-induced generation of ceramide, phosphorylation of NR1 subuints, clustering of NR1, enhancement of NMDA-evoked calcium flux and EPSCs.
Introduction
Modifications in the number and complement of glutamate-sensing receptors in the postsynaptic membrane are key mechanisms used to adjust synaptic strength. There is abundant evidence that the trafficking of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors is critical for long-term potentiation (LTP) and long-term depression (LTD) of synaptic strength (see (Malinow & Malenka 2002, Song & Huganir 2002, Bredt & Nicoll 2003) for reviews), and emerging evidence suggests that trafficking of N-methyl-D-asparated (NMDA) receptors is also important for synaptic plasticity (Lan et al. 2001, Roche et al. 2001, Nong et al. 2003, Scott et al. 2004, Lavezzari et al. 2004, Washbourne et al. 2004, Barria & Malinow 2002, Rao & Craig 1997, Quinlan et al. 1999, Watt et al. 2000). Although the identification and characterization of protein components involved in the trafficking of glutamate receptors has been an active and productive area of research, there has been little progress in understanding how changes in membrane lipid components affect the function and trafficking of glutamate receptors. Recent experimental evidence suggests that up to 60% of NMDA receptors are located in lipid rafts (Besshoh et al. 2005, Fullekrug & Simons 2004). These highly specialized membrane domains enriched in sphingomyelin, ceramide, cholesterol and the ganglioside GM1 are thought to play important roles in signal transduction by compartmentalizing membrane proteins into focal signaling units (Bollinger et al. 2005). When the structure of lipid platforms was disrupted by the removal of cholesterol, NMDA receptor currents and associated calcium flux were reduced and neurons protected from ischemic and excitotoxic insults (Frank et al. 2004, Abulrob et al. 2005). It has also been reported that cholesterol depletion increased the basal internalization rate of AMPA receptors (Hering et al. 2003), suggesting that one way in which lipid platforms may modulate receptor trafficking is by stabilizing surface expression. However, it is not known if rapid changes in the metabolism of raft-associated lipids modulate receptor trafficking under physiological conditions, nor whether lipid metabolism is critically involved in synaptic plasticity.
The traffic of transmembrane receptors requires focal changes in the biophysical properties of cellular membranes. For example, receptor internalization requires a focal invagination of the plasma membrane, while the traffic of receptors to the cell surface requires a fusion of receptor-laden vesicles with the plasma membrane. For these events to occur there must be rapid changes in local lipid content that allow changes in shape and fusigenic properties of membranes. Because ceramide has a high aggregation index, we hypothesized that a rapid generation of ceramide may be involved in synaptic plasticity by modulating the fusion of NMDA receptor containing vesicles with the plasma membrane. There are several lines of evidence that suggest ceramide could be involved in the regulation of receptor trafficking and synaptic plasticity. The cytokine TNF is a potent activator of sphingomyelin phosphodiesterase 3 (SMPD3, also known as neutral sphingomyelinase 2; nSMase2) and can modulate plastic events at the synapse. For example, an acute exposure of hippocampal slices to TNF resulted in a rapid insertion of AMPA receptors and enhanced synaptic transmission, whereas long-term exposure to TNF inhibited LTP (Tancredi et al. 1992, Beattie et al. 2002). Additional evidence suggests TNF mediates homeostatic synaptic scaling in hippocampal neurons, an activity-dependent refinement of neural circuitry that involves, at-least in part, alterations in receptor content at synapses (Stellwagen & Malenka 2006). We therefore sought to determine if TNFα could modulate synaptic plasticity by actions that involve nSMase2. The findings from our experiments suggest that TNF-induced activation of nSMase2 and generation of ceramide are critical for NMDA receptor clustering and synaptic plasticity in the hippocampus.
Methods
Cell Culture and Experimental Treatments
Hippocampal neuronal cultures were prepared from embryonic day 18 Sprague Dawley rats using methods similar to those described previously (Haughey et al. 2004). Tissues were dissociated by gentle tituration in a calcium-free Hank's balanced salt solution and centrifuged at 1000 × g. Cells were resuspended in MEM media containing 10 % heat-inactivated fetal bovine serum and 1% antibiotic solution (104 U of penicillin G/ml, 10 mg streptomycin/ml and 25 μg amphotericin B/ml) in 0.9 % NaCl (Sigma). Neurons were plated at a density of 200,000 cells/ml on 15 mm diameter poly-D-lysine coated glass coverslips. Three hours after plating the media for hippocampal cultures was replaced with serum-free Neurobasal medium containing 1% B-27 supplement (Gibco). Immunofluorescent staining for MAP-2 (neurons) showed that hippocampal cultures were > 98 % neurons; the remainder of cells were predominantly astrocytes. Hippocampal cultures were used between 14 and 21 days in vitro.
Lipid extraction and measurement of sphingolipids, phospholipids and sterols
Total lipids from samples will be prepared according to a modified Bligh and Dyer procedure (Shaikh 1994). Each sample was homogenized at room temperature in 10 volumes of deionized water, then in 3 volumes of 100% methanol containing 30 mM ammonium acetate, and vortexed. Four volumes of chloroform were added and the mixture vortexed and then centrifuged at 1,000g for 10 min. The bottom (chloroform) layer was removed and analyzed by direct injection into an electrospray ionization tandem mass spectrometer. Lipid extractions were performed using borosilicate-coated glass tubes, pipettes, and injectors.
Electrospray ionization tandem mass spectrometry (ESI/MS/MS) analyses were performed using methods similar to those used in previous studies (Haughey et al. 2004). Samples were injected using a Harvard Apparatus pump at 15 μl/min into a Sciex API 3,000 triple stage quadrupole tandem mass spectrometer from Sciex Inc. (San Francisco, CA) operated in the positive mode. The ion spray voltage (V) was 5,500 at a temperature of 80°C with a nebulizer gas of 8 psi, curtain gas of 8 psi, and the collision gas set at 4 psi. The declustering potential was 80V, the focusing potential 400V, the entrance potential −10V, the collision energy 30V, and a collision cell exit potential of 18V. The MS/MS was set to scan from 300 to 2,000 atomic mass units (amu) per second at a step of 0.1amu. Each species of sphingolipids, phospholipids, and sterols was identified by a Q1 mass scan, then by precursor ion scanning or neutral loss scanning of a purified standard. Samples were injected into the ES/MS/MS for 3 min, where the mass counts accumulated and the sum of the total counts under each peak were used to quantify each species. Sphingomyelins, ceramides, cholesterol, and cholesterol ester standards C16:0, C18:0, C18:1, and cholesteryl-arachidonate (C20:0) were purchased from Sigma. Ceramides C20:0, C24:0, C24:1, phosphatidylcholine C16:0-C18:1, C18:0-C18:1, phospatidylethanolamine C16:0-C18:1, phosphatidylglycerol C16:0-C18:1, phosphatidylserine C16:0-C18:1, phosphatidylinositol C16:0-C18:1, and phosphatidic acid C16:0-C18:1 were purchased from Avanti Polar Lipids (Alabaster, AL). Palmitoyl-lactosyl ceramide C16:0-C16:0, stearoyl-lactosyl-ceramide C16:0-C18:0, lignoceryl-glucosyl-ceramide C16:0-C24:0, lignoceryl-galactosyl-ceramide C16:0-C24:0, and stearoyl-galactosyl-ceramide-sulfate C18:1-C24:0 were purchased from Matreya Inc. (Pleasant Gap, PA).
Immunofluorescence and confocal microscopy
Labeling of surface located NR1
was accomplished using procedures similar to those published by other groups (Washbourne et al. 2004). In brief, neurons were exposed to TNFα, IL-1, or IL-6 (50 − 100 ng/ml each) and the glass coverslips containing neurons were incubated for 30 min at 15 °C in 5% CO2with a mouse monoclonal antibody against the NR1 subunit of the NMDA receptor that recognizes an extracellular epitope in the region of the amino-terminal amino acids 341−561 (R1JHL, 1:100. Affinity Bioreagents, Golden, CO). During the last 10 min of incubation, a Cholera toxin subunit B (CT-B) conjugated with Alexa Fluor 555 that binds the ganglioside GM1 was added (1 ng/ml; Invitrogen/Molecular Probes Inc, Carlsbad, California). CTX-555 co-localizes with flotillin, a protein that is known to be located in lipid rafts and ceramide, a sphingolipid that is enriched in lipid rafts (Supplemental Fig 1A, B). Cells were then washed 3 times with ice cold PBS and fixed with ice-cold 4% paraformaldehyde in PBS. Non-specific binding was blocked with 5% normal goat serum in PBS, followed by a 2 h incubation at room temperature in PBS containing 2% normal goat serum with AlexaFluor 488 (1:2000; Invitrogen /Molecular Probes, Carlsbad, CA).
For standard immunofluorescence
cell were fixed with ice-cold 4% paraformaldehyde in PBS and membranes were permeabilized by incubation for 10 min in a solution of 0.1% Triton X-100 in PBS then incubated for 1 h in blocking solution (2% normal goat serum and 2% normal horse serum in PBS). Primary antibodies were added overnight at 4°C and included: mouse monoclonal antibodies NR1-CT (Upstate, Charlottesville, VA) and a rabbit polyclonal antibodies that recognize NR1S896, NR1S897 (1:500; Upstate, Charlottesville, VA), flotillin-1 (1:1000; Beckton Dickinson, Franklin Lakes, NJ), ceramide (1:250; Sigma-Aldrech, St. Louis, MO). Cultures were washed with PBS and then incubated for 2 h in the presence of appropriate fluorescently tagged anti-mouse and anti-rabbit secondary antibodies (AlexaFluor 546 or 488; 1:2000 dilution; Invitrogen/Molecular Probes, Carlsbad, CA). In some experiments, cultures were further stained with propidium iodide or Hoechst 33342. For quantification of immunopositive puncta on dendritic branches, images were acquired with a 100× objective lens using a Zeiss axiovert 200 microscope equipped with an Orca CCD camera and Improvision imaging software (Lexington, MA). Immunopositive puncta were counted along dendritic branches and standardized to area. The criteria for a positive identification were that the puncta must be clear and distinguishable in a single plane of focus. Co-localization was considered as any amount of overlap (yellow) between AlexaFluor 555 (red) and AlexaFluor 488 (green). To confirm co-localization, confocal images of serial z-stacks (0.38 μm optical slices) were acquired using a Zeiss 510 CSLM microscope. Co-localization was confirmed in the orthogonal views and quantified after 3D deconvolution of z-stacks using Zeiss's LSM Image Examiner software.
Calcium imaging
Cytosolic calcium levels ([Ca2+]c) were measured using the Ca2+-specific fluorescent probe Fura-2FF. Rat hippocampal neurons were incubated for 25 min at 37°C in Locke's buffer (154 mM NaCl, 3.6 mM NaHCO3, 5.6 mM KCl, 1 mM MgCl2, 5 mM HEPES, 2.3 mM CaCl2, 10 mM glucose) 2 μM Fura-2FF; pH 7.4. Neurons were washed with Locke's to remove extracellular Fura-2 and incubated at 37°C for an additional 10 min to allow complete de-esterfication of the probe. Coverslips containing Fura-2 loaded cells were mounted in an imaging chamber and maintained at 37°C during perfusion (RC-26 chamber and V8 channel controller; Warner Instruments, Hamden CT). Cells were excited at 340 and 380 nm, and emission was recorded at 510 nm with a video-based intrtacellular imaging system (Intracellular Imaging Inc.). Image pairs were acquired at the rate of 3 images/sec using a digital intensified CCD imaging system (Cooke Corp.). Rmax/Rmin were converted to nM [Ca2+]c as described previously(Grynkiewicz et al. 1985) using reference standards (Molecular Probes). N-methyl-D-aspartic acid (10 μM) + glycine (100 nM) was applied by rapid perfusion in the presence of nifedipine (10 μM) to prevent depolarization-induced activation of voltage sensitive calcium channels.
Slice Preparation and Electrophysiology
Hippocampal slices were prepared using procedures described previously (Wang et al. 2004). Briefly, transverse slices of whole brain were cut at a thickness of 350 μm and were allowed to recover for at least 1 h in a holding chamber in artificial cerebral spinal fluid (ACSF), bubbled with 95/5% (O2/CO2) at room temperature up to 6 h. Field potentials were recorded from CA1 stratum radiatum using pipettes (1−3 MΩ) filled with bubbled ACSF, placed in stratum radiatum in response to stimulation of Schaffer collateral/commissural afferents. The stimuli (30 μs duration at 0.033 Hz) were delivered through fine bipolar tungsten electrodes. A stimulation intensity was used that evoked a response that was approximately 30−40% of the maximum fEPSP and LTP was induced by high frequency stimulation (HFS, 100 Hz 1 s). Whole-cell excitatory postsynaptic currents (EPSC) were recorded from CA1 pyramidal neurons. Whole-cell patch clamp recordings of excitatory postsynaptic currents (EPSCs) from CA1 pyramidal cells were performed by visualization of the cells with a 40× water immersion lens (2 mm working distance). Voltage steps from −60 to +40 mV were applied for recording NMDAR component of EPSCs. All recording solutions for LTP studies contained 50 uM picrotoxin to block GABAA activity with or without CNQX (10 μM) or DL-AP5 (100 μM). The series resistance was 8 to 14 MΩ, as measured directly from the amplifier, the mean input resistance was 118 ± 10 MΩ and the mean resting membrane potential was −66 ± 5 mV. Patch-clamp electrodes, with a typical resistance of 3−5 MΩ, were filled with a solution containing (mM): potassium gluconate, 130; KCl, 10; EGTA, 10; CaCl2, 1; MgCl2, 3; Hepes, 20; Mg-ATP, 5; Na-GTP, 0.5; QX 314, 10; pH 7.2 (the osmolality was adjusted to 280 mmol/kg). The input resistance was monitored continuously, and the recording terminated if it varied by more than 10%. Data were considered valid only when the following criteria were met: a resting membrane potential of at least −60 mV, a high input resistance (at least 100 MΩ), and a steep input-output curve obtained with a low stimulation intensity. Data were collected and analyzed using an Axopatch 200B and pCLAMP 8 software (Axon Instruments). All signals were recorded and filtered at 2 kHz and digitized at 10 kHz. All slices were pre-selected; only those with a steep input–output curve were included in the study. During recording, slices were maintained at 30−32 °C.
Results
TNFα rapidly increases the ceramide content in neurons
To investigate if nSMase2 can trigger the insertion of NMDA receptors into the plasma membrane by modification of sphingolipid content, we first stimulated primary hippocampal neurons with TNFα, a cytokine that is known to rapidly increase nSMase2 activity in a variety of cell types including neurons (Sortino et al. 1999b, Kronke 1999b, Oral et al. 1997, Chatterjee 1994). Initial dose response experiments ranging from 10 − 100 ng/ml TNFα showed that 50 ng/ml of TNFα was the minimal dose that consistently increased ceramide levels in our cultured neurons (data not shown). We therefore used a dose of 50 ng/ml TNFα in subsequent experiments. TNFα induced a rapid and transient increase of long-chain ceramides that peaked at 2 min and began to return to baseline values within 5 min, consistent with previous reports that TNFα can activate nSMase2 in seconds with peak activity at 1.5 min (Fig. 1A) (Wiegmann et al. 1994). IL-1 and IL-6 did not alter ceramide levels in neurons (data not shown) and TNFα did not alter cholesterol, phosphoinositol, phosphatidylcholine or phosphatidylethanolamine levels within the 5 min test period (Supplemental Fig. 2A-E). TNF-α-induced increases of ceramide were reduced by pre-treating neurons with the nSMase2 inhibitor GW4869 (10 μM), but not by inhibiting the de novo synthesis of ceramide with ISP-1 (10 μM; Fig. 1B). GW4869 attenuated TNFα-induced increases of ceramide by 84%-C16:0, 95%-C18:0, 89%-C20:0, 52%-C22:0 and 90%-C24:0.
Figure 1. TNFα rapidly increases sphingomyelin and ceramide levels in hippocampal neurons.
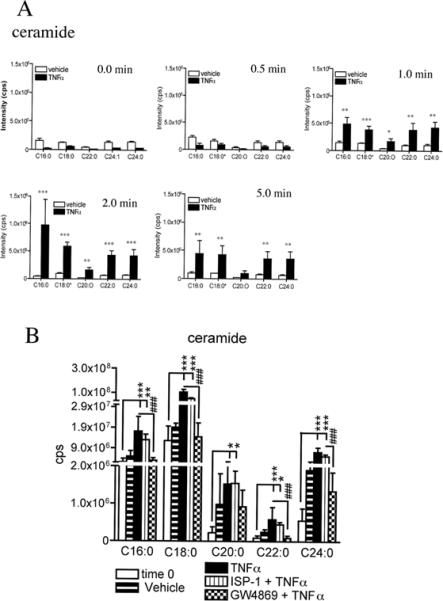
A Hippocampal neurons were treated with vehicle or TNFα (50 ng/ml) and lysed with ddH2O at the indicated times. Lipids were extracted and analytes were detected and quantified by ESI/MS/MS. Sphingomyelin C16:0, C18:0, C20:0, C22:0 and C24:0 steadily increased from 1 to 5 min. Ceramide C16:0, C18:0, C20:0, C22:0 and C24:0 increased from 1 to 2 min and began to decrease toward baseline values within 5 min. B. A pre-incubation of neurons with the nSMase2 inhibitor GW4869 (10 μM), but not the serine palmitoyltransferase inhibitor ISP-1 (10 μM), prevented TNFα from increasing cellular ceramide content. GW4869 attenuated TNFα-induced increases of ceramide by 84%-C16:0, 95%-C18:0, 89%-C20:0, 52%-C22:0 and 90%-C24:0. Only the 2 min TNFα-treatment timepoint is shown. Data are mean ± S.D. * = p < 0.05, ** = p < 0.01, *** = p < 0.001. Two-way ANOVA with Tukey post hoc comparisons.
TNFα induces NR1 trafficking into lipid rafts
Using a fluorophore-conjugated peptide that recognizes an extracellular domain of NR1 (Luo et al. 1997) we first determined that a 2 min treatment with TNFα doubled the number of surface located NR1 from 0.97 ± 0.16/μm2 to 1.8 ± 0.29/μm2. These newly integrated receptors appeared as clusters along the length of dendrites (Fig. 2A, B).
Figure 2. TNFα increases surface expression of NR1 and promotes the clustering of NR1S896 into lipid rafts.
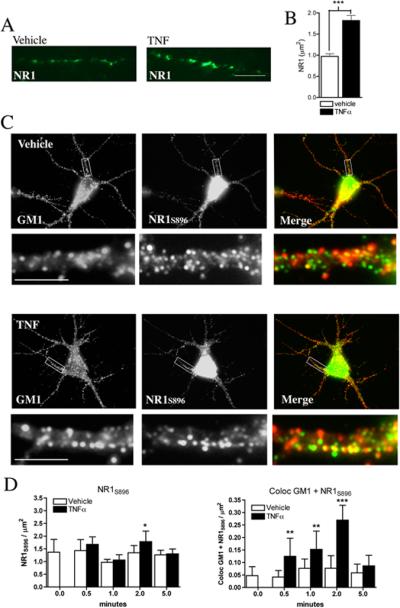
Hippocampal neurons were treated with vehicle or TNFα (50 ng/ml) for 2 min and endogenous surface expression of NR1 was detected using the peptide R1JHL (recognizes an extracellular domain of NR1) conjugated to AlexaFluor 488. A. TNFα increased the surface expression of NR1, which appeared in clusters along dendritic branches. B. Summary data are mean ± S.D./area from a minimum of 21 dendrites derived from three separate experiments. ***p < 0.001. Two-way ANOVA with Tukey post hoc comparisons. C. Hippocamapal neurons were treated with vehicle or TNFα (50 ng/ml) for 0 to 5 min and NR1 were visualized using a phosphospecific antibody that recognizes NR1 phosphorylated on serine 896 (NR1S896). Lipid platforms were identified using AlexaFluor 555-cholera toxin that binds the gangloside GM1. Shown are images of hippocampal dendrites immunopositive for NR1S896 (green) and GM1 (red). Co-localized immunopositive puncta are yellow in the merged images. D. TNFα did not alter the number of GM1+ domains or total number of NR1S896 during the 5 min test period, but increased the number of GM1+/NR1S896+ dual positive sites from 0.5 to 2 min. Data are mean ± S.D./area of a minimum 15 dendrites/condition derived from 3−4 separate experiments. Scale bar = 10 μm. ** = p < 0.01, *** = p < 0.001. Two-way ANOVA with Tukey post hoc comparisons.
We next sought to determine if the clustering of NR1 occurred in the highly ordered gangloside-, sphingomyelin- and ceramide-rich membrane domains known as lipid rafts. Although we found no increase in the fraction of total NR1 that associated with GM1+ lipid rafts, a 2 min pulse with TNFα did increase the fraction of NR1 phosphorylated on serine 896 (NR1S896) that located to GM1+ lipid rafts by four-fold (Fig 2C, D), consistent with evidence that phosphorylation is associated with a redistribution of NR1 to lipid rafts (Besshoh et al. 2005) and evidence that NR1S896 is important for clustering and surface localization of NR1 (Tingley et al. 1997). Similar results were obtained when TNFα-stimulated cells were immunostained for NR1 phosphorylated on serine 897 (data not shown), a residue that is also known to be important for the surface localization of NR1 (Tingley et al. 1997). However, TNFα-stimulation did not alter the fraction of NR1 subunits phosphorylated on serine 890 that co-localized with GM1 (Supplementary 3A), consistent with data that suggest phosphorylation of this residue is not critical for surface localization of NR1 (Tingley et al. 1997). The ability of TNFα to promote the surface localization and clustering of NR1 was specific to this cytokine as IL-1β and IL-6 did not alter the fraction of NR1S896 that co-localized with GM1 at any time during the 5 min test period (Supplemental Fig 3B, C). TNFα-induced clustering of NR1S896 into lipid rafts was confirmed using 3D deconvolution of z-stack fluorescence images quantified using Pearson's correlation of the colocalization coefficient (Fig. 3A-C) and by density gradient centrifugation coupled with immunoblot analysis to track the cellular location of NR1S896 (Fig. 3D).
Figure 3. NR1S896 is colocalized to lipid rafts following a 2 min treatment with TNFα.

Cultures of hippocamapal neurons were treated with vehicle or TNFα (50 ng/ml) for 2 min. Surface localized NR1 were visualized using a phosphospecific antibody that recognizes NR1S896. Lipid platform regions were identified by AlexaFluor 555-CTX that binds the ganglioside GM1. A. Confocal images of dendritic branches showing staining of GM1+ lipid rafts (red) and NR1S896 (green) and the orthogonal views. The images are maximal projection stacks of confocal z-series from fixed hippocamal neurons at rest (0 min), after treatment with vehicle for 2 min and after treatment with TNFα (50 ng/ml) for 2 min. B. Quantification of fluorescence showing that TNFα increases the amount of NR1S896 that colocalized with GM1; note that the correlation coefficient analysis was done on 3D reconstructed images. * = p < 0.05 Students T-test. C. Neuronal cultures were treated with vehicle or TNFα (50 ng/ml) for 2 min and Triton-X-100 soluble membrane rafts were isolated and separated by density centrifugation with OptiPrep™. NR1S896 and flotillin-1 (a protein known to be enriched in lipid rafts) were then detected by Western blot. The location of flotillin to fractions 3 and 4 was similar in vehicle and TNFα-treated cultures. The location of NR1S896 shifted from fraction 7 in vehicle-treated cultures to fraction 4 in cultures exposed to TNFα for 2 min.
We next determined if TNFα promoted the clustering of NR1S896 into lipid rafts by a mechanism dependent on nSMase2. Neurons were pre-treated with GW4869 (a nSMase2 inhibitor), or ISP-1 (an inhibitor of de novo ceramide synthesis) and pulsed with TNFα for 2 min. GW4869, but not ISP-1 reduced the number NR1S896 that clustered into GM1+ lipid rafts following TNFα stimulation (Fig. 4A). To confirm the involvement of nSMase2 we cultured neurons from mice with a deletion in the gene coding for nSMase2 (fro/fro mice)(Aubin et al. 2005). TNFα- induced clustering of NR1S896 with GM1 was slightly reduced in +/fro mice and absent in fro/fro mice compared with cultures from wild-type mice (Fig 4B). These data suggest that TNFα-induced clustering of NR1S896 requires nSMase2.
Figure 4. TNFα-induced clustering of NR1S896 into lipid rafts is dependent on nSMase2 activity.
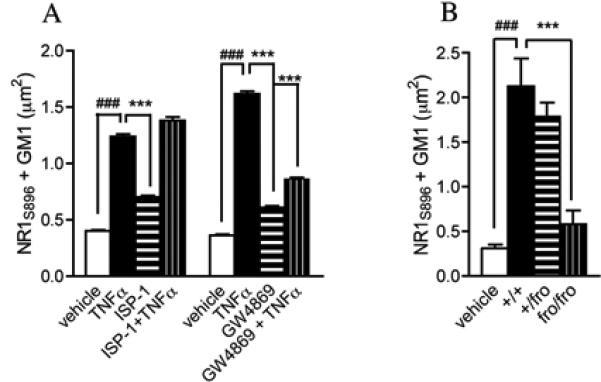
A. Hippocampal neurons were pre-treated with the nSMase2 inhibitor GW4869 (10 μM) or the serine palmitoyltransferase inhibitor ISP-1 (10 μM) then exposed to TNFα for 2 min. GW4869, but not ISP-1 prevented TNFα from increasing the clustering of NR1S896 into GM1+ lipid rafts. Data are mean ± S.D. from a minimum 21 dendrites/condition derived from 3 separate experiments. B. TNFα (50 ng/ml) induced clustering of NR1S896 into GM1+ domains in wt/wt mouse hippocampal cultures was slightly reduced in cultures from wt/fro mice and was not different from vehicle controls in neurons cultured from fro/fro mice. ### = p < 0.001 compared with vehicle and *** = p < 0.001 compared with TNFα or +/+. Two-way ANOVA with Tukey post hoc comparisons.
Based on findings that TNFα increased the area of lipid rafts (Fig 5A, B) we though it possible that simply increasing the ceramide content of neuronal membranes could increase raft area and promote clustering of NR1S896. To test this hypothesis we increased the area of lipid rafts by enhancing de novo ceramide synthesis with the ceramide precursor palmitoyl CoA. It is important to note that increasing de novo ceramide increases ceramide content in multiple cellular membranes over several hours. This is in contrast to nSMase which can be induced to translocate to the inner surface of the plasma membrane where ceramide can be generated at focal points with peak activity in 1.5 minutes (Levy et al. 2006, Visnjic et al. 1999, Clarke et al. 2008). Palmitoyl CoA increased the size of lipid rafts from 169 ± 19 to 209 ± 35 nm2, similar to the increase induced by TNFα, but the fraction of NR1S896 that was associated with GM1+ lipid rafts was 3.6 ± 1.6%, similar to the basal amount of 3.5 ± 2.5 % (Fig. 5B, C). Thus, increasing the ceramide content of cells was sufficient to increase lipid raft size, it was not sufficient to enhance the insertion of NR1S896 into lipid rafts. However, we did observe a synergistic effect on NR1S896 clustering when cultures were pre-conditioned with palmitoyl CoA and then stimulated for 2 min with TNFα. Under these conditions, the fraction of NR1S896 that was associated with lipid rafts doubled from 6.6 ± 3.8% in non-conditioned cultures treated with TNFα, to 11.3 ± 6.8% in cultures pre-treated with palmitoyl CoA before TNFα (Fig. 5C).
Figure 5. Increasing ceramide content by de novo synthesis was not sufficient to increase clustering of NR1S896 with lipid rafts.
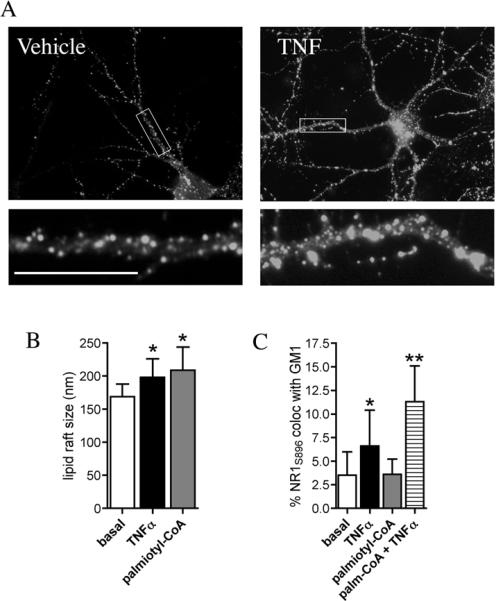
A. Hippocamapal neurons were treated with vehicle or TNFα (50 ng/ml) for 0 to 5 min (the 2 min time point is shown) and lipid raft regions were visualized using AlexaFluor 555 conjugated cholera toxin that binds the ganglioside GM1. B. Quantitative summary of GM1+ lipid rafts showing that a 2 min treatment with TNFα or a 24 h treatment with the ceramide precursor palmitoyl-CoA increase the average size of lipid rafts. C. Quantitative summary of NR1S896 colocalized with GM1+ lipid rafts shows that a 2 min treatment with TNFα, but not a 24 h treatment with palmitoyl-CoA increased the percent of NR1S896 that was found in lipid rafts. Palimtoyl-CoA pre-treatment followed by a 2 min TNFα exposure further enhanced the percent of NR1S896 that colocalized with GM1. Scale bar = 10 μm. * = p < 0.05, ** = p < 0.001 compared with basal. Two-way ANOVA with Tukey post hoc comparisons.
Diacylglycerol and nSMase2 are required for membrane fusion
Our findings suggest that TNFα promotes the insertion of NR1S896 into lipid rafts by a mechanism this is dependent on nSMase2-mediated generation of ceramide. Since this event would require the fusion of NMDA receptor containing vesicles with the plasma membrane we next determined whether ceramide could directly mediate membrane fusion. Vesicles were created from crude rat cortical lipid extract, or a 1:1:1:1 ratio of phosphatidylcholine, sphingomyelin (C24:0), phosphatidylethanolamine and cholesterol. Fluorescence resonance energy transfer (FRET) was used to detect when NBD-PE containing vesicles fused with rhodamine DHPE-labeled vesicles. Surprisingly, direct additions of nSMase2 only resulted in a small amount of vesicle fusion (Fig 6A), suggesting that the generation of ceramide was not sufficient to promote membrane fusion. Based on findings that TNFα can increase diacylglycerol (DAG) via the phospholipase C pathway(Schutze et al. 1995, Schutze et al. 1991), we hypothesized that DAG may be required to modify the membrane and create a fusion point. Consistent with this notion, we found that TNFα increased DAG levels in a time frame similar to ceramide (data not shown). Moreover, while the addition of PLC (to hydrolyze phosphatidylcholine to DAG) induced only a small amount of fusion, the simultaneous addition of PLC and nSMase2 robustly increased fusion by 135.7 ± 2.0% at 5 min, and 151.9 ± 1.8% at 10 min, compared with the vehicle control. TritonX-100 (0.1%) was added at the end of each experiment as a positive control for fusion and heat-inactivated nSMase2 and PLC were used as controls (Fig. 6A-B). We interpret these findings to suggest that a dual generation of DAG and ceramide are required for efficient membrane fusion.
Figure 6. Dual actions of nSMase2 and phospholipase C are required to promote membrane fusion.
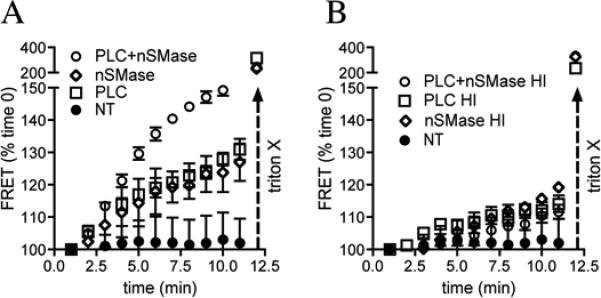
Liposomes were created from 1:1:1:1 ratio of phosphatidylcholine, phosphatidylethanolamine, sphingomeylin and cholesterol using an extruder (0.8 μm pore size) and labeled with either NBD-PE or rhodamine-DHPE. Vesicle fusion was monitored using fluorescence resonance energy transfer (FRET). A. The addition of vehicle did not alter fusion during the 5 min test period. The addition phospholipase C (PLC), or nSMase slightly increased FRET, while the combined addition of PLC + nSMase resulted in a robust increase in FRET. Triton X (0.1 %) is known to rapidly induce the fusion of membranes and is shown as a positive control at the end of experiments. B. Heat inactivated PLC, nSMase or PLC + nSMase had no effect on FRET. Summary data are mean ± S.D. (normalized to time 0) from 5 experiments per condition. **p < 0.01, ***p < 0.001. Two-way ANOVA with Tukey post hoc comparisons.
Enhanced NMDA receptor function by nSMase2-dependent trafficking
We next measured how the kinetics of NMDA receptor function are modified when NR1S896 is clustered into lipid rafts by treating neuronal cultures with TNFα for 2 min and measuring calcium and electrophysiological responses evoked by NMDA applications. Calcium flux was measured in ∼ 1.0 μm2 regions along dendritic branches at the rate of 3 images/sec. We categorized NMDA-evoked calcium responses based on amplitude into high (greater than 1000 nM), medium (500−100 nM) and low (less than 500 nM). Following a 2-minute treatment with vehicle 4.0 ± 4.7 % of NMDA-evoked calcium responses were of high amplitude, 4.2 ± 1.6 % were in the medium range and the remaining 91.0 ± 7.3% were low amplitude (Fig. 7A, C). A 2 minute treatment with TNFα increased the fraction of high amplitude NMDA-evoked calcium responses to 20.2 ± 7.6 %, with 19.4 ± 4.4 % in the medium range and 60.5 ± 7.7% low amplitude, (Fig. 7B, C). TNFα also increased the frequency of NMDA-evoked calcium bursts from 0.67 ± 0.03 peaks/sec to 1.60 ± 0/05 peaks/sec (Fig 7D). Inhibition of nSMase2 with GW4869 prevented TNFα from enhancing the rate of NMDA-evoked calcium bursts and increasing the relative number of high calcium response regions on neurites (Fig. 7C, D).
Figure 7. TNFα increases the amplitude and frequency of focal calcium bursts by a nSMase2-dependent mechanism.
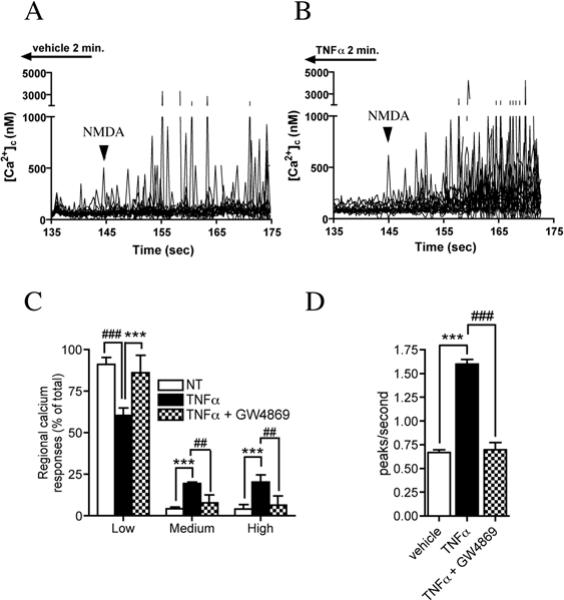
Calcium flux was measured in ∼1.0 μm2 regions along the dendrites of hippocampal neurons in culture using the calcium binding dye Fura-2FF. A. Tracings showing enhancement of focal NMDA evoked (100 μM) calcium bursts in cultures perfused for 2 min with TNFα (50 ng/ml) compared to cultures perfused with vehicle. Arrowheads indicate initiation point of NMDA infusion. B. Summary figure showing the amplitude of regional calcium responses organized into percent of Low (0−500 nM), Medium (500−1000 nM) and High (> 1000 nM) responses. TNFα reduced the relative abundance of low amplitude calcium responses while increasing the fraction of medium and high responses. Pre-incubating cultures with GW4869 (10 μM) prevented TNFα from shifting NMDA-evoked calcium bursts to higher relative response amplitudes. C. Summary data showing that a 2 min perfusion of neurons with TNFα (50 ng/ml) increased the number of NMDA-evoked calcium peaks/second. Pre-treating neurons with GW4869 (10 μM) prevented TNFα from increasing the number of NMDA-evoked focal bursts in calcium. Data are the mean ± S.D of recordings from 47−73 dendritic microdomains in 3−4 separate experiments per condition. *** = p < 0.001 compared with vehicle, ## = p < 0.01 and ### = p < 0.001 compared with TNFα-treated cultures. Two-way ANOVA with Tukey post-hoc comparisons.
As a final functional measure of the requirement for nSMase2 in TNFα-induced clustering and surface localization of NR1, we recorded EPSCs in acute rodent hippocampal acute slice preparations. A 2 min perfusion with TNFα enhanced the NMDA-evoked current recorded from CA1 pyramidal cells to 115 ± 8.89% at +40mV (Fig 8A, B). A1 h pre-incubation with the nSMase2 inhibitor GW4869 blocked the ability of TNFα to enhance NMDA (103 ± 5.64%), while ISP-1, a potent inhibitor of de novo sphingolipid biosynthesis, did not prevent TNFα-induced enhancement of NMDA-evoked EPSCs (117 ± 5.18%; Fig. 8A, B). BSA (0.1 %), used to assist in the solubility of GW4869 and ISP-1, did not alter NMDA currents (101 ± 8.15%; data not shown). To confirm that TNFα-induced modification of NMDA currents involves nSMase2, we recorded currents in acute hippocampal slice preparations from mice containing a deletion in the gene encoding nSMase2 (fro/fro) and their wild type (+/+) littermates. In +/+ mice a 2-minute perfusion with TNFα enhanced the NMDA-evoked current recorded from CA1 pyramidal cells to 118 ± 6.8% at 40 mV (Fig 8C). In +/fro mice, the NMDA-currents were 108 ± 6.8% and in fro/fro mice TNFα did not alter NMDA-currents (101 ± 4.3%; Fig. 8C). Thus, TNFα-induced modification of whole-cell NMDA receptor currents required nSMase2.
Figure 8. NMDA currents are modulated by short exposures to TNFα by a mechanism that requires nSMase2.
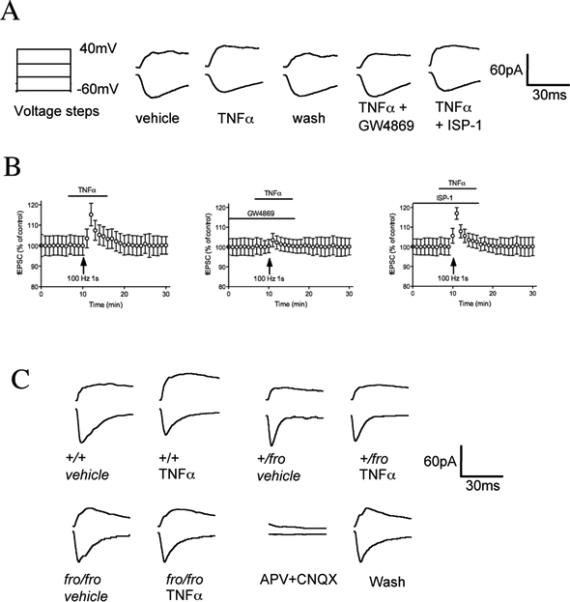
Whole-cell patch clamp recordings of excitatory postsynaptic currents (EPSCs) from CA1 pyramidal cells in hippocamal slice. A. On the left is the voltage protocol. Cells were held at −60 mV during a series voltage steps (150 ms) from −60 mV to +40 mV. To avoid capacitance transients generated by the step effect on the current, there was a delay of 100 ms between start of step and pre-synaptic stimulation. Original EPSCs recorded at CA1 pyramidal cells show currents generated by the voltage steps after a 2 min perfusion with vehicle, TNFα (50 ng/ml), after wash-out of TNFα and in slice cultures pre-treated with GW4869 (10 μM) or ISP-1 (10 μM) for 1 hr before TNFα. Enhancement of NMDA-evoked current was attenuated by GW4869, but not by ISP-1. B. Pooled group data showing that the modulation of NMDA current by TNF-α exhibited a rapid onset and offset. Data are mean ± S.D. from 8 different slices/condition. C. In wt/wt mice, a 2 min perfusion with TNFα enhanced the NMDA-evoked current recorded from CA1 pyramidal −60mV (n =10, in 4 mice). In fro/wt mice, TNFα-induced enhancement of NMDA-evoked current was partially attenuated (n =12, from 4 mice), and in fro/fro mice TNFα did not alter NMDA current (98.8 ± 4.8, n =15, from 3 mice).
Discussion
Recent work has demonstrated that NMDA receptors undergo rapid insertion and removal from neuronal membranes (Barria & Malinow 2002, Roche et al. 2001, Nong et al. 2003, Scott et al. 2004, Lavezzari et al. 2004, Washbourne et al. 2004). Our understanding of the mechanisms that regulate these endocytotic and exocytotic events have come primarily from the discovery and functional analysis of complex protein-protein interactions involved in the removal and insertion of receptors at the plasma membrane (see Perez-Otano & Ehlers 2005 for a review). While a great deal of progress has been made in understanding the molecular mechanisms that regulate NMDA receptor trafficking, it is currently unknown how receptor-laden vesicles become fused with the plasma membrane to allow for receptor insertion. Our findings suggest that a rapid and specific reorganization of the lipid content at focal points in the plasma membrane may be necessary for the phosphorylation and trafficking of NR1 to the surface of plasma membranes.
We found that stimulation of neurons with TNFα increased the phosphorylation of NR1 on serine 896 and 897 that clustered into lipid rafts. Phosphorylation of NMDA receptor subunits is one mechanism that can regulate receptor trafficking (see Swope et al. 1999 for a review). The C-terminal of NR1 contains three serines (890, 896, 897) that when phosphorylated can differentially regulate NMDA receptor trafficking. Protein kinase C (PKC) phosphorylates NR1 serine residues 890 and 896, while cAMP-dependent protein kinase (PKA) phosphorylates serine residue 897 of the NR1 subunit (Tingley et al. 1997). Phosphorylation of serine 890 by PKC results in the dispersion of surface-associated clusters of the NR1 subunit. This PKC-induced redistribution of the NR1 subunit occurs within minutes of serine 890 phosphorylation and is reversed upon dephosphorylation (Tingley et al. 1997). Dual mutations in serine 896 and 897 that mimic the phosphorylated state increase the surface localization of NR1 (Tingley et al. 1997) suggesting that the coordinated activity of PKC and PKA are required for surface location of NR1. Interestingly, we found that pharmacological inhibition or genetic mutation in nSMase2 inhibited the ability of TNFα to increase the phosphorylation of NR1 on serine 896 or to promote the clustering of these modified subunits into lipid rafts. These findings suggest that a rapid and focal generation of ceramide may shift the composition of membrane lipids to bring PKC and PKA into close proximity with NR1. However, it is not clear at this time if these events are the result of lateral diffusion of kinases and membrane docked receptors, or a translocation of one or both components to the plasma membrane following TNFα stimulation. Adding to the complexity of these events are recent data that suggest PKC also controls the translocation and activation of nSMase2 (Visnjic et al. 1999, Clarke et al. 2008). Thus, PKC may serve dual roles that promote the translocation of nSMase2 to the plasmamembrane while mediating phosphorylation of NR1. There may also be a biochemical preference for the localization of NR1 phosphorylarted on serines 896 and 897 to lipid rafts where the lipid composition can be rapidly modified to favor the fusion of receptor-laden vessicles with the plasma membrane. Under basal conditions, when two membranes are in opposition, the polar head groups of the component lipids exert a repulsive force and are not likely to spontaneously fuse. Our findings demonstrate that a rapid and focal generation of DAG and ceramide can trigger the fusion of vesicles and insertion of NR1 phosphorylated on serines 896 and 897 into lipid rafts. Based on the biophysical properties of DAG, it is likely that that the generation of this lipid component serves to destabilize the membranes and creates a fusion-point. A rapid generation of ceramide would then mediate fusion by increasing the relative volume of carbon chains over hydrophilic head-groups thus enhancing the hexagonal II phase propensity of the membranes (Kronke 1999a). In addition to enhancing membrane fusion, the protrusion of ceramide alkyl chains may interact with the hydrophobic pocket on NR1 to promote receptor clustering (Kronke 1997).
Our functional studies demonstrate that nSMase2 is critical for TNFα-induced enhancement of NMDA-evoked focal calcium bursts and EPSCs, suggesting that a rapid and focal generation of ceramide may be critical for certain forms of synaptic plasticity. Thus, stimuli that increase the activity of nSMase2 in neurons including TNFα, Fas ligand and NGF (Sortino et al. 1999a, Castiglione et al. 2004, Sanchez-Alavez et al. 2006, Brann et al. 1999) may modulate synaptic plasticity by controlling the insertion of NR1 into the postsynaptic membrane. Indeed, recent findings have shown that TNFα is involved in the homeostatic activity-dependent regulation of synaptic connectivity (Stellwagen & Malenka 2006), and NGF is known as modulator of synaptic strength (Kang & Schuman 1995a, Kang & Schuman 1995b). There is however, evidence that TNFα may impair synaptic plasticity in models of Alzheimer's disease, in which the length of TNFα stimulation is considerably longer, mimicking pathological conditions (see (Rowan et al. 2007). The rapid versus long-term effects of TNFα on synaptic plasticity may have important consequences for the development of TNF receptor antagonists as neuroprotective therapeutics (Tracey et al. 2008).
In addition to critical roles for nSMase2 and ceramide in some types of synaptic plasticity, there are potential implications for disruptions in sphingolipid metabolism in the pathogenesis of neurodegenerative diseases. For example, dysfunctions in sphingolipid metabolism with accumulations of ceramide have been recently reported in Alzheimer's disease, amyotrophic lateral sclerosis and HIV-associated dementia (Cutler et al. 2002, Cutler et al. 2004, Haughey et al. 2004). A prolonged increase in the ceramide content of neuronal membranes may perturb NMDA receptor trafficking and could increase the susceptibility of neurons to excitotoxic death by locking NMDA receptors at the plasmamembrane for prolonged periods of time. Thus, therapeutics designed to attenuate the activity nSMase2 may preserve neuronal function by stabilizing NMDA receptor trafficking under pathological conditions.
Supplementary Material
Supplementary Figure 1. The protein flotillin and the sphingolipid ceramide are found in GM1+ lipid rafts. Hippocampal neurons were stained with a fluorescent-bound cholera toxin that interacts with the gangloside GM1 (red) and antibodies to flotillin-1 or ceramide (green). A. A hippocampal dendrite showing immunostaining for flotillin-1, GM1 and the overlay of the two images showing a near perfect colocalization of flotillin-1 with GM1. B. An example of a hippocampal dendrite showing immunostaining for ceramide, GM1 and the overlay of the two images showing high degree of association between ceramide and GM1. Scale bar = 10 μm.
Supplementary Figure 2. TNFα did not alter cholesterol or phospholipids content of hippocampal neurons. Hippocampal neurons were treated with vehicle or TNFα (50 ng/ml) and lysed at the indicated times. Lipids were extracted and analytes were detected and quantified by ESI/MS/MS. TNFα did not alter A, the cholesterol, B, phosphatidylinositol, C, phosphatidylethanolamine, D, phosphatidylserine, or C, phosphatidylcholine content during the 5 min test period.
Supplementary Figure 3. Specificity of TNFα to promote the clustering of NR1S896 into lipid rafts. A. TNFα did not alter the total number of NR1 phosphorylated on serine 890 (NR1S890) that colocalized with GM1 at any time during the 5 min stimulation period. Numbers of GM1, NR1S890 and dual positive regions are shown. B, C. Quantitative summaries of the number of GM1+, NR1S896+ and dual positive regions present on dendritic branches following a 0 − 5 min treatment with vehicle, IL-1β or IL-6. IL-1β or IL-6 did not alter the number of GM1, NR1S896 or colocalized NR1S896 and GM1 during the 5 min test period.
Acknowledgements
This research was supported by NIH grants AG023471, MH077542 and AA017408 to NJH and the Intramural Research Program of the National Institute of Aging.
References
- Abulrob A, Tauskela JS, Mealing G, Brunette E, Faid K, Stanimirovic D. Protection by cholesterol-extracting cyclodextrins: a role for N-methyl-D-aspartate receptor redistribution. J Neurochem. 2005;92:1477–1486. doi: 10.1111/j.1471-4159.2005.03001.x. [DOI] [PubMed] [Google Scholar]
- Aubin I, Adams CP, Opsahl S, et al. A deletion in the gene encoding sphingomyelin phosphodiesterase 3 (Smpd3) results in osteogenesis and dentinogenesis imperfecta in the mouse. Nat Genet. 2005;37:803–805. doi: 10.1038/ng1603. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–353. doi: 10.1016/s0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Besshoh S, Bawa D, Teves L, Wallace MC, Gurd JW. Increased phosphorylation and redistribution of NMDA receptors between synaptic lipid rafts and post-synaptic densities following transient global ischemia in the rat brain. J Neurochem. 2005;93:186–194. doi: 10.1111/j.1471-4159.2004.03009.x. [DOI] [PubMed] [Google Scholar]
- Bollinger CR, Teichgraber V, Gulbins E. Ceramide-enriched membrane domains. Biochim Biophys Acta. 2005;1746:284–294. doi: 10.1016/j.bbamcr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Brann AB, Scott R, Neuberger Y, Abulafia D, Boldin S, Fainzilber M, Futerman AH. Ceramide signaling downstream of the p75 neurotrophin receptor mediates the effects of nerve growth factor on outgrowth of cultured hippocampal neurons. J Neurosci. 1999;19:8199–8206. doi: 10.1523/JNEUROSCI.19-19-08199.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Castiglione M, Spinsanti P, Iacovelli L, et al. Activation of Fas receptor is required for the increased formation of the disialoganglioside GD3 in cultured cerebellar granule cells committed to apoptotic death. Neuroscience. 2004;126:889–898. doi: 10.1016/j.neuroscience.2004.04.038. [DOI] [PubMed] [Google Scholar]
- Chatterjee S. Neutral sphingomyelinase action stimulates signal transduction of tumor necrosis factor-alpha in the synthesis of cholesteryl esters in human fibroblasts. J Biol Chem. 1994;269:879–882. [PubMed] [Google Scholar]
- Clarke CJ, Guthrie JM, Hannun YA. Regulation of neutral sphingomyelinase-2 (nSMase2) by tumor necrosis factor-alpha involves protein kinase C-delta in lung epithelial cells. Mol Pharmacol. 2008;74:1022–1032. doi: 10.1124/mol.108.046250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler RG, Pedersen WA, Camandola S, Rothstein JD, Mattson MP. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann Neurol. 2002;52:448–457. doi: 10.1002/ana.10312. [DOI] [PubMed] [Google Scholar]
- Frank C, Giammarioli AM, Pepponi R, Fiorentini C, Rufini S. Cholesterol perturbing agents inhibit NMDA-dependent calcium influx in rat hippocampal primary culture. FEBS Lett. 2004;566:25–29. doi: 10.1016/j.febslet.2004.03.113. [DOI] [PubMed] [Google Scholar]
- Fullekrug J, Simons K. Lipid rafts and apical membrane traffic. Ann N Y Acad Sci. 2004;1014:164–169. doi: 10.1196/annals.1294.017. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Haughey NJ, Cutler RG, Tamara A, McArthur JC, Vargas DL, Pardo CA, Turchan J, Nath A, Mattson MP. Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann Neurol. 2004;55:257–267. doi: 10.1002/ana.10828. [DOI] [PubMed] [Google Scholar]
- Hering H, Lin CC, Sheng M. Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J Neurosci. 2003;23:3262–3271. doi: 10.1523/JNEUROSCI.23-08-03262.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995a;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Schuman EM. Neurotrophin-induced modulation of synaptic transmission in the adult hippocampus. J Physiol Paris. 1995b;89:11–22. doi: 10.1016/0928-4257(96)80547-x. [DOI] [PubMed] [Google Scholar]
- Kronke M. The mode of ceramide action: the alkyl chain protrusion model. Cytokine Growth Factor Rev. 1997;8:103–107. doi: 10.1016/s1359-6101(97)00006-3. [DOI] [PubMed] [Google Scholar]
- Kronke M. Biophysics of ceramide signaling: interaction with proteins and phase transition of membranes. Chem Phys Lipids. 1999a;101:109–121. doi: 10.1016/s0009-3084(99)00059-6. [DOI] [PubMed] [Google Scholar]
- Kronke M. Involvement of sphingomyelinases in TNF signaling pathways. Chem Phys Lipids. 1999b;102:157–166. doi: 10.1016/s0009-3084(99)00084-5. [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS. Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci. 2001;4:382–390. doi: 10.1038/86028. [DOI] [PubMed] [Google Scholar]
- Lavezzari G, McCallum J, Dewey CM, Roche KW. Subunit-specific regulation of NMDA receptor endocytosis. J Neurosci. 2004;24:6383–6391. doi: 10.1523/JNEUROSCI.1890-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M, Castillo SS, Goldkorn T. nSMase2 activation and trafficking are modulated by oxidative stress to induce apoptosis. Biochem Biophys Res Commun. 2006;344:900–905. doi: 10.1016/j.bbrc.2006.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Wang Y, Yasuda RP, Dunah AW, Wolfe BB. The majority of N-methyl-D-aspartate receptor complexes in adult rat cerebral cortex contain at least three different subunits (NR1/NR2A/NR2B). Mol Pharmacol. 1997;51:79–86. doi: 10.1124/mol.51.1.79. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Nong Y, Huang YQ, Ju W, Kalia LV, Ahmadian G, Wang YT, Salter MW. Glycine binding primes NMDA receptor internalization. Nature. 2003;422:302–307. doi: 10.1038/nature01497. [DOI] [PubMed] [Google Scholar]
- Oral H, Dorn GW, 2nd, Mann DL. Sphingosine mediates the immediate negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian cardiac myocyte. J Biol Chem. 1997;272:4836–4842. doi: 10.1074/jbc.272.8.4836. [DOI] [PubMed] [Google Scholar]
- Perez-Otano I, Ehlers MD. Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci. 2005;28:229–238. doi: 10.1016/j.tins.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci. 1999;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron. 1997;19:801–812. doi: 10.1016/s0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- Rowan MJ, Klyubin I, Wang Q, Hu NW, Anwyl R. Synaptic memory mechanisms: Alzheimer's disease amyloid beta-peptide-induced dysfunction. Biochem Soc Trans. 2007;35:1219–1223. doi: 10.1042/BST0351219. [DOI] [PubMed] [Google Scholar]
- Sanchez-Alavez M, Tabarean IV, Behrens MM, Bartfai T. Ceramide mediates the rapid phase of febrile response to IL-1beta. Proc Natl Acad Sci U S A. 2006;103:2904–2908. doi: 10.1073/pnas.0510960103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutze S, Berkovic D, Tomsing O, Unger C, Kronke M. Tumor necrosis factor induces rapid production of 1'2'diacylglycerol by a phosphatidylcholine-specific phospholipase C. J Exp Med. 1991;174:975–988. doi: 10.1084/jem.174.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutze S, Wiegmann K, Machleidt T, Kronke M. TNF-induced activation of NF-kappa B. Immunobiology. 1995;193:193–203. doi: 10.1016/s0171-2985(11)80543-7. [DOI] [PubMed] [Google Scholar]
- Scott DB, Michailidis I, Mu Y, Logothetis D, Ehlers MD. Endocytosis and degradative sorting of NMDA receptors by conserved membrane-proximal signals. J Neurosci. 2004;24:7096–7109. doi: 10.1523/JNEUROSCI.0780-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh NA. Assessment of various techniques for the quantitative extraction of lysophospholipids from myocardial tissues. Anal Biochem. 1994;216:313–321. doi: 10.1006/abio.1994.1047. [DOI] [PubMed] [Google Scholar]
- Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- Sortino MA, Condorelli F, Vancheri C, Canonico PL. Tumor necrosis factor-alpha induces apoptosis in immortalized hypothalamic neurons: involvement of ceramide-generating pathways. Endocrinology. 1999a;140:4841–4849. doi: 10.1210/endo.140.10.7062. [DOI] [PubMed] [Google Scholar]
- Sortino MA, Condorelli F, Vancheri C, Canonico PL. Tumor necrosis factor-alpha induces apoptosis in immortalized hypothalamic neurons: involvement of ceramide-generating pathways. Endocrinology. 1999b;140:4841–4849. doi: 10.1210/endo.140.10.7062. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Swope SL, Moss SJ, Raymond LA, Huganir RL. Regulation of ligand-gated ion channels by protein phosphorylation. Adv Second Messenger Phosphoprotein Res. 1999;33:49–78. doi: 10.1016/s1040-7952(99)80005-6. [DOI] [PubMed] [Google Scholar]
- Tancredi V, D'Arcangelo G, Grassi F, Tarroni P, Palmieri G, Santoni A, Eusebi F. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci Lett. 1992;146:176–178. doi: 10.1016/0304-3940(92)90071-e. [DOI] [PubMed] [Google Scholar]
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem. 1997;272:5157–5166. doi: 10.1074/jbc.272.8.5157. [DOI] [PubMed] [Google Scholar]
- Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244–279. doi: 10.1016/j.pharmthera.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Visnjic D, Batinic D, Banfic H. Different roles of protein kinase C alpha and delta isoforms in the regulation of neutral sphingomyelinase activity in HL-60 cells. Biochem J. 1999;344(Pt 3):921–928. doi: 10.1042/0264-6021:3440921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Haughey NJ, Mattson MP, Furukawa K. Dual effects of ATP on rat hippocampal synaptic plasticity. Neuroreport. 2004;15:633–636. doi: 10.1097/00001756-200403220-00012. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Liu XB, Jones EG, McAllister AK. Cycling of NMDA receptors during trafficking in neurons before synapse formation. J Neurosci. 2004;24:8253–8264. doi: 10.1523/JNEUROSCI.2555-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt AJ, van Rossum MC, MacLeod KM, Nelson SB, Turrigiano GG. Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron. 2000;26:659–670. doi: 10.1016/s0896-6273(00)81202-7. [DOI] [PubMed] [Google Scholar]
- Wiegmann K, Schutze S, Machleidt T, Witte D, Kronke M. Functional dichotomy of neutral and acidic sphingomyelinases in tumor necrosis factor signaling. Cell. 1994;78:1005–1015. doi: 10.1016/0092-8674(94)90275-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. The protein flotillin and the sphingolipid ceramide are found in GM1+ lipid rafts. Hippocampal neurons were stained with a fluorescent-bound cholera toxin that interacts with the gangloside GM1 (red) and antibodies to flotillin-1 or ceramide (green). A. A hippocampal dendrite showing immunostaining for flotillin-1, GM1 and the overlay of the two images showing a near perfect colocalization of flotillin-1 with GM1. B. An example of a hippocampal dendrite showing immunostaining for ceramide, GM1 and the overlay of the two images showing high degree of association between ceramide and GM1. Scale bar = 10 μm.
Supplementary Figure 2. TNFα did not alter cholesterol or phospholipids content of hippocampal neurons. Hippocampal neurons were treated with vehicle or TNFα (50 ng/ml) and lysed at the indicated times. Lipids were extracted and analytes were detected and quantified by ESI/MS/MS. TNFα did not alter A, the cholesterol, B, phosphatidylinositol, C, phosphatidylethanolamine, D, phosphatidylserine, or C, phosphatidylcholine content during the 5 min test period.
Supplementary Figure 3. Specificity of TNFα to promote the clustering of NR1S896 into lipid rafts. A. TNFα did not alter the total number of NR1 phosphorylated on serine 890 (NR1S890) that colocalized with GM1 at any time during the 5 min stimulation period. Numbers of GM1, NR1S890 and dual positive regions are shown. B, C. Quantitative summaries of the number of GM1+, NR1S896+ and dual positive regions present on dendritic branches following a 0 − 5 min treatment with vehicle, IL-1β or IL-6. IL-1β or IL-6 did not alter the number of GM1, NR1S896 or colocalized NR1S896 and GM1 during the 5 min test period.