

Abstract
Objective
Mice subjected to traumatic brain injury (TBI) at postnatal day (pnd) 21 show emerging cognitive deficits that coincide with hippocampal neuronal loss. Here we consider glutathione peroxidase (GPx) activity as a determinant of recovery in the injured immature brain.
Methods
Wildtype (WT) and transgenic (GPxTg) mice overexpressing GPx were subjected to TBI or sham surgery at pnd 21. Animals were euthanized acutely (3 or 24 hours postinjury) to assess oxidative stress and cell injury in the hippocampus or 4 months postinjury following behavioral assessments.
Results
In the acutely injured brains, a reduction in oxidative stress markers including nitrotyrosine was seen in the injured GPxTg group relative to WT controls. In contrast, cell injury, with marked vulnerability in the dentate gyrus, was apparent despite no differences between genotypes. Magnetic resonance imaging demonstrated an emerging cortical lesion during brain maturation that was also indistinguishable between injured genotypes. Stereologic analyses of cortical volumes likewise confirmed no genotypic differences between injured groups. However, behavioral tests beginning 3 months after injury demonstrated improved spatial memory learning in the GPxTg group. Moreover, Stereologic analysis within hippocampal subregions revealed a significantly greater number of neurons within the dentate of the GPx group.
Interpretation
Our results implicate GPx in recovery of spatial memory after TBI. This recovery may be in part attributed to a reduction in early oxidative stress and selective, long-term sparing of neurons in the dentate.
Keywords: Traumatic brain injury, controlled cortical impact, glutathione peroxidase, oxidative stress, neuronal cell loss, cognitive function, immature brain
Introduction
Traumatic brain injury (TBI) is the leading cause of death and disability among children1–3. Children less than 4 years of age have poorer motor and cognitive outcomes after TBI than do older children4–6.
To investigate the vulnerability of the immature brain, we developed a murine model of TBI at pnd 217,8, an age that approximates toddler-aged (2–4 years-old) children in terms of gliogenesis, synaptogenesis, and myelination9. This injury results in a cortical lesion and hippocampal neuronal loss, both of which continue to increase between 2 weeks and 3 months postinjury8. Importantly, cognitive dysfunction is delayed in onset and coincides with this prolonged hippocampal neuronal loss7.
Oxidative stress is a significant component of the injury cascade following TBI. The brain’s antioxidant mechanisms include superoxide dismutase (SOD), which converts free radicals to hydrogen peroxide, and catalase and glutathione peroxidase (GPx), which further metabolize hydrogen peroxide to water and oxygen10. The young brain is particularly vulnerable to oxidative stress due to its high fatty acid content and proportionately large share of total body oxygen consumption11. During development, antioxidants are inadequately expressed and do not respond to oxidative stress as robustly as they do in the adult brain, making the immature brain even more susceptible to oxidative stress-induced injury. In the CD1 mouse, SOD1 activity decreases from embryonic (E)18 to pnd 1, and then remains unchanged to pnd 21. GPx activity, on the other hand, rises steeply from E18 to pnd 1, declines sharply by pnd P4, and again from pnd 7 to pnd 14, where it is the same as at pnd 21. Protein levels for both enzymes, however, show a steady increase from El 8 to pnd 2112. We have shown that in the C57B16 mouse brain, adult levels of GPx are reached bypnd 2113. These age-dependent differences may impact the response of the immature brain to injury. Whereas SOD overexpression is protective against ischemic injury in the adult brain14, 15, SOD overexpression exacerbates hypoxic-ischemic injury in the neonatal brain16. The latter is thoughtto be due to a failure of downstream antioxidant mechanisms in the immature brain to compensate for increased hydrogen peroxide production17. While GPx is upregulated in the adult brain in response to TBI, no such upregulation occurs in the injured immature brain13, suggesting that GPx may be a key factor in the immature brain’s vulnerability to TBI. Such a possibility is reinforced by in vitro and in vivo studies. Overexpression of GPx in immature neurons in vitro is protective against hydrogen peroxide exposure18 and neonatal mice overexpressing GPx are less susceptible to hypoxic-ischemic injury than wildtype littermates19.
We hypothesized that overexpression of GPx would be protective against the long-term sequelae of traumatic injury to the immature brain. We demonstrate protection against long-term hippocampal loss associated with an acute reduction in oxidative stress and alterations in long-term behavioral functions, including improved spatial memory.
Materials and Methods
All procedures were approved by the University of California, San Francisco Institutional Animal Care and Use Committee. Analyses were conducted blinded to genotype and experimental condition.
GPx transgenic mice and surgical procedures
Glutathione peroxidase transgenic mice (GPxTg), expressing 200 copies of the human GPxl gene (gift of Dr. O. Mirochnitchenko, University of Medicine and Dentistry of New Jersey), were studied on a B6CBAF1/J background. Non-transgenic and heterozygous transgenic animals were confirmed by PCR.
Male mice at pnd 21 were anesthetized with 1.25% 2,2,2 tribromoethanol and subjected to TBI as previously described8. After a midline skin incision, a circular craniotomy was made between bregma and lambda with the medial edge of the craniotomy 0.5 mm lateral to the midline. The animal was then subjected to CCI injury using a convex impactor tip. Sham-operated controls underwent the same surgical procedures with the exception of the traumatic injury.
Western blots
Samples from the ipsilateral hippocampus were prepared for western immunoblots at 3 and 24 hours post injury (n=5/genotype and time point). To assess nitrotyrosine, GPx-1, copper-zinc SOD (CuZnSOD), manganese SOD (MnSOD), caspase-3, and MAP2, protein samples (30–40µg) were separated by 12% SDS-PAGE and transferred to PVDF membranes (Immobilon-FL, Millipore, Billerica, MA). Membranes were incubated with primary and secondary antibodies, specified in supplemental Table 1. Protein carbonyls were detected by the OxyBlot Protein Oxidation Detection Kit (Chemicon International, Temecula, CA, refer to supplemental methods).
Membranes were scanned and analyzed using the Odyssey infrared imaging system (LI-COR Biosciences). Signal intensity of each band was normalized to that of actin.
Anatomical studies
Animals were euthanized at 24 hours postinjury or at the completion of the behavioral studies. Anesthetized animals were transcardially perfused with 4% paraformaldehyde. Brains were then removed, cryoprotected, frozen, and cut into coronal sections using a cryostat.
Fluoro-Jade C labeling and semi-quantification
Fluoro-Jade C (Histo-Chem Inc. Jefferson, AR) was used to detect hippocampal injury (n=5 per genotype). Sections, 20µm in thickness, were stained with Fluoro-Jade C as previously described20 and subjected to semiquantitative analysis on a scale from 0 to 3 (refer to supplemental methods).
TUNEL labeling and quantification
Irreversible cell damage was assessed by deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) using an in situ cell death detection kit (Roche Applied Science, Indianapolis, IN, n=5 per genotype)21,22. MetaMorph (Molecular Devices, Downingtown, PA) was used to quantify TUNEL-positive cells in the hippocampus23(refer to supplemental methods).
Stereologic analyses of the hippocampus
Sections, 40µm in thickness, were incubated with a mouse monoclonal antibody to NeuN (1:1000, Chemicon) to visualize neurons. The secondary antibody was visualized using avidin-biotin-horseradish peroxidase complex.
An optical fractionator stereological design24 was used to make unbiased estimates of total neuronal numbers within hippocampal subregions25 using Stereo Investigator software (MicroBrightField, Inc., Williston, VT, n= 4/genotype).
Cortical mantle and hippocampal volumes were estimated using the NeuroLucida imaging program (MicroBrightField, refer to supplemental methods).
Magnetic resonance imaging (MRI)
MRI was conducted at 24 hours, and 7, 14, and 28 days after injury using a Bruker Omega CSI 2T system (n=6–8 per genotype). Mice were anesthetized with isoflurane mixed in 100% O2 (5% to induce, 1.5% to maintain), placed supine on a plexiglass cradle, wrapped in a water warming pad, and positioned in a birdcage radiofrequency coil (3.8×5.4cm). Multislice T1-, T2-, and diffusion-weighted images (T1WI, T2WI, and DWI, respectively) were acquired with field of view of 32×32mm, matrix 128×128 points, thickness 1.5mm, and 4 averages (refer to supplemental methods).
Behavioral Evaluation
Injured animals (n=10 WT and n=12 GPxTg) and sham controls (n=11 WT and n=11 GPxTg) were subjected to behavioral evaluations beginning 3 months after injury. Mice were tested in the open field and rotorod tasks during the first week of behavioral testing and subsequently tested in the Morris water maze the following week26. Details of the behavioral tests, including number of trials, are described in supplemental methods.
Statistical analyses
T-tests were used for 2-group comparisons. Analysis of variance (ANOVA) and, where appropriate, Tukey-Kramer posthoc tests were used to evaluate the interactions between genotype and time point or treatment (injury versus sham). Repeated-measures ANOVA using contrasts was used to assess water maze and rotorod learning. All data are expressed as means ± SEM. Significance is defined at p<0.05.
Results
CuZnSOD protein expression is reduced in the GPxTg group
As expected, protein levels of GPx were higher in GPxTg relative to WT animals (Fig. 1 and Table 2, supplemental data). MnSOD was not affected by genotype whereas there was a significant genotype effect on CuZnSOD protein expression with less CuZnSOD protein in GPxTg animals (Fig. 1 and Table 2, supplemental data).
Overexpression of GPx reduces early oxidative stress but does not alter early hippocampal injury
A 150kD protein carbonyl band was detected in the hippocampus of injured brains with a trend toward differences in genotypes (Figure 2A, Table 2, supplemental data). The nitrotyrosine signal remained consistently elevated within the first 24 hours postinjury in WT animals whereas it decreased by 24 hours in the GPxTg group(Fig. 2A, B, Table 2, supplemental data).
To determine if early reduction of oxidative stress is neuroprotective in the hippocampus, caspase-3 and MAP2 levels were evaluated at 24 hours postinjury. Pro-caspase-3 was detected at 32kD while cleaved-caspase-3 was detected at 17 and 19kD. Although pro-caspase-3 tended to decrease after injury in both genotypes, these values were not significantly different from controls (Table 2, supplemental data). Similarly, cleaved-caspase-3, the activated form of caspase-3, and three forms of MAP2 were indistinguishable from sham controls (Table 2, supplemental data).
We next evaluated cell injury, based upon Fluoro-Jade C and TUNEL staining (Fig. 3 and Fig. 4). A large number of cells and their processes in the granule cell layer of the dentate gyrus were Fluoro-Jade C-positive (Fig. 3 A–C). In contrast, there were qualitatively fewer labeled cells and their processes in CA2/CA3 (Fig. 3 D–F) and few to no cells in CA1 (data not shown). There were no differences between genotypes after injury (p= 0.42 for CA2/CA3 and p= 0.24 for the dentate, unpaired T-test).TUNEL-labeled cells were prominent inthe granule cell layer, particularly within the posterior hippocampus (supplemental Fig. 1 A–E). There was, however, no difference between genotypes.
Figure 3. Fluoro-Jade C staining in the hippocampus at 24 hours postinjury.
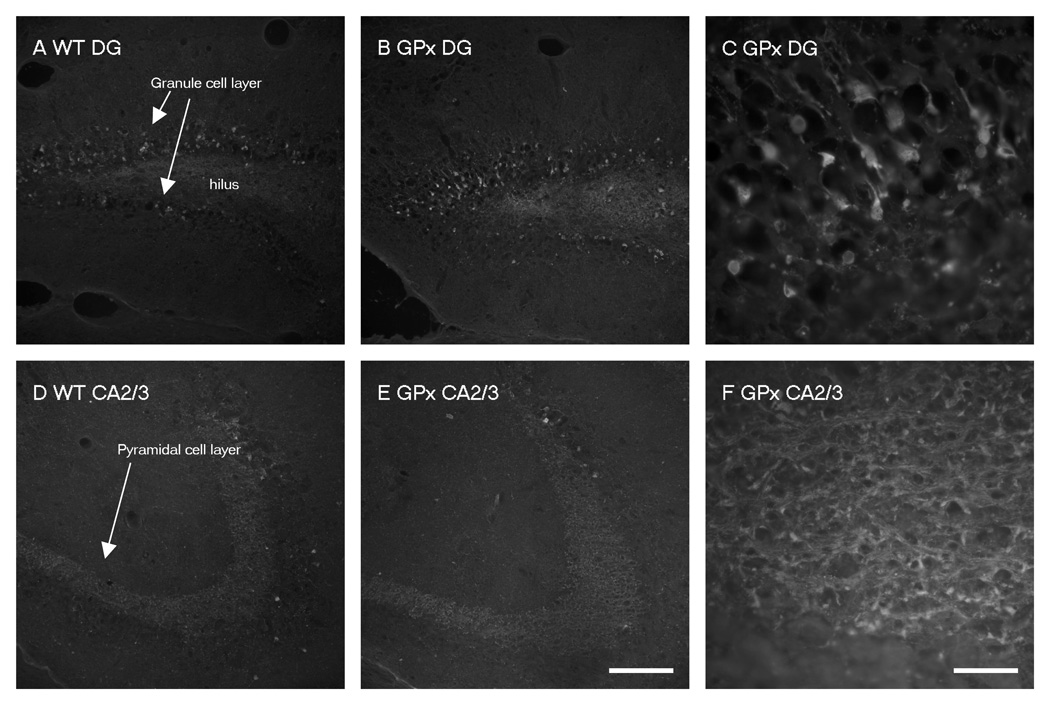
A–F). Representative photomicrographs of Fluoro-Jade C staining, an indicator of cellular degeneration. Labeled structures are apparent in the dentate gyrus (A, B, C) and the CA3 region (D, E, F) of the ipsilateral hippocampus from WT (A, D) and GPxTg (B, C, E, F) animals. (Panels C and F are images in higher magnification of panel B and E respectively). Prominently labeled cells and their processes, presumably neurons, are apparent in the granule cell layer of the dentate gyrus (A, B, C). This contrasts the CA3 region, where only a few labeled cells are evident (D, E, F).
Scale bars, A, B, D, E = 100µm, C, F = 500µm.
Figure 4. Spatial learning and memory after TBI or sham surgery.

A) Time to locate the visible (sessions 1–4)and hidden (sessions 5–10) platform locations (latency) during water maze training are shown. All groups improved with training in both the visible (F=121.219, p<0.001) and hidden (F=35.59, p<0.001) sessions. In the visible sessions, there is an overall group difference (F=7.091, p=0.001) and the GPxTg injured animals show higher latencies to locate the platform than the WT sham and GPxTg sham (p<0.05, Tukey Kramer). However, in the hidden sessions, there is also an overall group difference (F=3.953, p=0.015) where the WT injured group is significantly different from the WT sham and GPxTg sham (p<0.05, Tukey Kramer).
B) Cumulative distances to locate the visible and hidden platform locations during water maze training are shown. Similarly to the latency, all groups improve with training both in visible sessions (F=101.989, p<0.001) and hidden sessions (F=53.140, p<0.001) and there are overall group differences in visible (F=6.187, p=0.001) and hidden sessions (F=4.924, p=0.005). The GPxTg injured group shows significantly longer distance to locate the visible platform compared to the WT Sham and GPxTg Sham (p<0.05, Tukey Kramer) whereas cumulative distance of the WT injured group is significantly longer than the WT Sham and GPxTg Sham (p<0.05, Tukey Kramer).
C) WT injured animals show no spatial bias in the probe trial when the platform is removed, while all the other groups show memory retention and spend more time in the target than in any other quadrant. (* p<0.05).
GPx and evolution of the lesion
There were notable individual variations in apparent size of the injury by MRI (WT, 6.1±3.1% and GPxTg, 5.1±2.2% of ipsilateral brain pixels at 1 day), but no significant differences between genotypes. At 1 day the injured brain was manifest as a heterogenous hyperintense region on both T2W and DW images(supplemental Fig. 2A). T2 hyperintense regions exhibited 45±9% greater signal intensity than contralateral brain, consistent with substantial vasogenic edema. ADC values of the lesion were 4.56±0.21×10−4 and 4.63±0.27×10−4 mm2/second for WT and GPxTg mice, respectively, consistent with cytotoxic edema, while the contralateral brain ADC was 7.36±0.18×10−4 mm2/second. GdDTPA-BMA caused an increase in signal by 4±2% in the contralateral brain that did not increase thereafter. The injured brain, however, exhibited a steady increase in signal intensity over 30 minutes after contrast administration (supplemental Fig. 2B) with slopes of 1.65±0.67 and 1.0±0.36 hours−1 for WT and GPxTg animals, respectively, consistent with substantial barrier leakage of the contrast agent.
At 7 days postinjury, the lesion was more complex, with a hypointense blood clot adjacent to hyperintense injury on T2WI. The lesion area was hypointense on DWI, with ADC values enlarged to 19.2±6.4×10−4 mm2/second consistent with partial liquifaction. Contrast uptake was reduced to 0.92±0.63 hours−1. At 14 and 28 days postinjury, the lesion exhibited more homogeneous hyperintensity on T2WI and hypointensity on DWI compared to 7 days postinjury. ADC values increased to 23.4±2.2×10−4 mm2/second, consistent with more mature liquifaction, typically extending from the lateral ventricle to the cortical surface and surrounding CSF space. The liquefied region showed little contrast uptake (0.38±0.18 hours−1).
GPx is a determinant of behavioral outcome after injury
GPxTg animals spent more time in the center of the open field (supplemental Fig. 3A), suggesting that they are less anxious compared to WT littermates27. Total distance traveled in the open field did not differ among groups, suggesting no differences in exploratory activity (supplemental Fig. 3B). Within the GPxTg group, injured mice entered the center less (supplemental Fig. 3C) and traveled shorter distances while in the center (supplemental Fig. 3D), suggesting that TBI has a larger effect on these measures of anxiety in GPxTg group.
All groups exhibited improvement with training on the rotorod (supplemental Fig 4A). There was no effect of genotype or injury. However, the difference between performance in the first and final trials revealed that injured GPxTg mice showed less improvement (supplemental Fig. 4B) than sham-GPxTg mice, suggesting that GPx overexpression actually impairs sensorimotor learning after injury.
Spatial and non-spatial memory was assessed using the Morris water maze test. There were no differences between groups in swim speeds during the visible session (data not shown). In the visible sessions, there was anoverall group difference with the GPxTg injured animals showing higher latencies to locate the platform than the WT and GPxTg shams. However, in the hidden sessions, there was also an overall group difference with the WT injured group showing higher latencies to locate the platform than the WT and GPxTg shams (Fig. 4A). Cumulative distances to locate the platform showed a similar pattern (Fig. 4B). Following hidden platform training, WT injured animals showed no spatial bias in the first probe trial (platform removed), while all 3 other groups showed spatial memory retention in the first probe trial and searched significantly longer in the quadrant that previously contained the platform (target quadrant) than in any other quadrant (Fig. 4C). Collectively, these data show that TBI results in long-term non-spatial and spatial memory deficits. Overexpressionof GPx worsens the deficit in non-spatial memory. However, the deficit in spatial memory is rescued by GPx overexpression.
GPx overexpression modulates long-term neuronal loss within the dentate
Cortical volume was reduced to 52.2% and 54.3% in WT and GPxTg injured mice, respectively (supplemental Fig. 5A, Fig. 5B). Hippocampal volumes were reduced to 85.9% and 89.7% in WT and GPxTg injured mice, respectively.
No genotypic differences were noted in the number of neurons in CA1 or CA3 after injury (Fig. 5 and Table 3, supplemental data). In contrast, there were more neurons in the dentate gyrus of the GPxTg group relative to the WT group (Fig. 5).
Figure 5. Overexpression of GPx modulates hippocampal pathogenesis at 4 months postinjury.

A, B) Cresyl violet staining after TBI reveals prominent neuronal loss in the hippocampus of both genotypes.
Scale bar, l00µm.
C, D, E). Although mean values appear reduced in the WT relative to the GPxTg group in CA1 and CA3, these differences are not significant (unpaired T-test, p=0.12 and p=0.16, respectively). In contrast, there are more neurons in the dentate gyrus (DG) of the GPxTg group relative to the WT group (unpaired T-test, *p=0.03).
5. Discussion
This is the first study to examine the role of GPx in multiple short- and long-term outcomes after traumatic injury to the immature brain. Transgenic overexpression of GPx results in discrete, time-dependent and region-specific improvement of structural and functional integrity. GPx overexpression modulates the regulation of other antioxidants by decreasing levels of CuZnSOD in the acute postinjury phase and reducing oxidative injury. While hippocampal neuronal injury was similarly elevated in both genotypes in the acute postinjury phase, GPx overexpression resulted in higher numbers of neurons within the dentate gyrus of the hippocampus andimproved long-term spatial memory. Interestingly, anxiety and sensorimotor learning capacity were negatively affected by GPx overexpression after injury.
Antioxidants and the injured immature brain
In this study GPx protein levels in the hippocampus of GPxTg animals were higher than in WT animals and not affected by injury, consistent with our previous observation of increased GPx enzymatic activity in GPxTg mice28. We further found that GPx overexpression resulted in a compensatory reduction in CuZnSOD expression. Although CuZnSOD levels were lower in GPxTg mice than in WTs, injury did not further alter CuZnSOD levels. Interestingly, GPx activity has been shown to be upregulated in CuZnSOD-overexpressing mice19. In that study, however, ischemic injury downregulated GPx activity in both WT and CuZnSOD overexpressors. In our model of GPx overexpression, perhaps a relatively low level of SOD activity and high level of GPx activity provides the optimal balance between those two enzymes.
Role of GPx after brain injury
We focused on the hippocampus, a structure known to be vulnerable after traumatic injury to the immature brain8.In many injury models, the neonatal hippocampus sustains more severe injury than the thalamusand cortex29–31. The hippocampus has higher basal levels of Fas expression than either the thalamus or cortex and, the hippocampus has minimal baseline expression of [Fas-associated death-domain-like IL-1 beta-converting enzyme]-inhibitory protein (FLIP) relative to cortical and thalamic samples. After hypoxic ischemic injury, there is some up-regulation of Fas and FLIP in all regions30. The importance of reactive oxygen species in Fas death receptor signaling has been verified in studies showing enhanced antioxidant status protects against Fas death receptor mediated cell death32.
Protein nitration remained elevated at 24 hours post injury in the WT group. This finding contrasts that of the traumatized, adult brain, where protein nitration returns to baseline by 24 hours33. Although this difference may be related to the age of the animals at the time of injury, it may also be due to region-specific variations in protein nitration in the injured brain. Whereas Deng et al. measured protein nitration in the injured cortex, the site of maximal mechanical damage, our measurements were from the ipsilateral hippocampus. Although each of these regions shows evidence of damage, the underlying mechanisms and time course for cell injury may be quite different and as such may translate to differences in the temporal course of the nitrotyrosine signal. A second consideration relates to the instability of modified proteins due to their accelerated degradation by the proteosome, as shown in vitro by Souza et al. How this instability might translate to in vivo studies is not clear. Although protein nitration in adult brain is reduced at 24 hours after injury there is a significant rise thereafter at 48 hours33. Thus, although modified proteins may be unstable after injury, there are likely other determinants in vivo that govern temporal pattern of their instability.
Overexpression of GPx resulted in a reduction in protein nitration at 24 hours postinjury. Despite this, there were no improvements in acute molecular and histologic measures of cell damage. While we have considered the possibility that western blots may not be robust enough to detect genotypic differences34, subsequent anatomical studies, which offer greater sensitivity, likewise revealed no genotypic differences. One plausible explanation for these early negative findings may be related to the timing of reduction of oxidative stress. A reduction in oxidative stress in the GPxTg group was not apparent until 1 day postinjury. Thus, any benefit afforded by the reduction in oxidative stress may have been masked by a population of dying neurons that evolved within hours after injury.
In the injured adult brain, oxidative stress is not limited to the acutely injured brain but rather extends into the subacute period33,35. It is conceivable that the immature brain likewise shows an extended period of oxidative stress with concomitant cell loss. Although we did not evaluate subacute cell loss, we do show long-term higher numbers of neurons in the dentate of the hippocampus of brain-injured GPxTg relative to WT animals.
MRI has been employed to longitudinally analyze brain injury in many studies36–38, but few have analyzed damage beyond 2 weeks postinjury. This is the first study to use MRI to assess brain edema and to evaluate progression of the lesion over time in this model of TBI.
In general, the evolution of the cortical lesion, defined by MRI, was similar between genotypes. Stereologic analyses revealed a significant loss in cortical volume after injury but no genotypic differences, further validated the MRI findings. Collectively, these data suggest that GPx is not a determinant of the evolution of the cortical lesion. Loss of cortical volume after injury may reflect both aberrant developmental processes as well as ongoing secondary pathogenesis. That GPx failed to rescue this phenotype may reflect insensitivity of either or both processes to increased GPx activity.
GPx and behavior after TBI
Here we show that GPxTg animals show a complex response to injury that includes increased anxiety as well an improvement in spatial memory. Anxiety disorders have been reported after pediatric TBI39. Multiple studies have implicated the prefrontal cortex and several subcortical regions, including the amygdala and retrosplenial cortex in anxiety disorders40,41. Antioxidant enzymes have been linked to anxiety regulation through genetic analysis42. One such antioxidant enzyme, glutathione reductase, is responsible for replenishing the stores of glutathione for use by GPx. The associations of oxidative stress, antioxidant enzymes, and GPx in particular with anxiety, are clearly complicated and warrant further study.
We have previously reported that traumatic injury to the immature brain results in delayed deficits in hippocampal-dependent spatial memory7. Here we show that overexpression of GPx partially rescues this phenotype. The mechanism underlying this finding remains unclear. Hippocampal-dependent spatial memory is mediated in part through interactions between the dentate gyrus and CA343. Cognitive impairment may thus arise as a consequence of progressive loss of neurons in the hippocampus during maturation7. There are several scenarios that might explain the higher numbers of neurons in the dentate of brain-injured GPx animals relative to WTs. Overexpression of GPx may confer protection to this population, shown to be vulnerable, and as such improve cognitive outcome. It is also conceivable that overexpression of GPx influences ongoing neurogenesis which serves to replenish the neuronal population in the dentate. Further studies are needed to explore each of these possibilities.
Conclusion
Overall, we demonstrate that transgenic overexpression of GPx results in a complex pattern of behaviors that include both anxiety and sensorimotor impairments as well as time-dependent and region-specific improvement in the structural and functional integrity of the hippocampus, including improved spatial memory. These findings identify glutathione peroxidase as a possible therapeutic target for the prevention of TBI-induced cognitive decline with the caveat that long-term upregulation of this antioxidant may lead to untoward behaviors.
Supplementary Material
Figure 1. Characterization of antioxidant enzymes in the hippocampus by western immunoblots.

A) Representative immunoblots of GPx, CuZnSOD and MnSOD after sham surgery and TBI. There are qualitative changes in protein levels of GPx and CuZnSOD in the GPxTg group relative to shams. In contrast, MnSOD appears similar between groups (Refer to Table 2 for statistical findings.)
B) GPx protein is higher in the GPxTg group as compared to the WT group regardless of injury (2-way ANOVA, effect of genotype, # p<0.05).
C) There is a compensatory reduction in CuZnSOD in the GPxTg group relative to the WT group (2- way ANOVA, effect of genotype, ## p<0.01).
Note that the intensity of each sample was quantified and corrected as a relative intensity to loading control, actin.
Figure 2. Oxidative stress in the hippocampus after TBI.
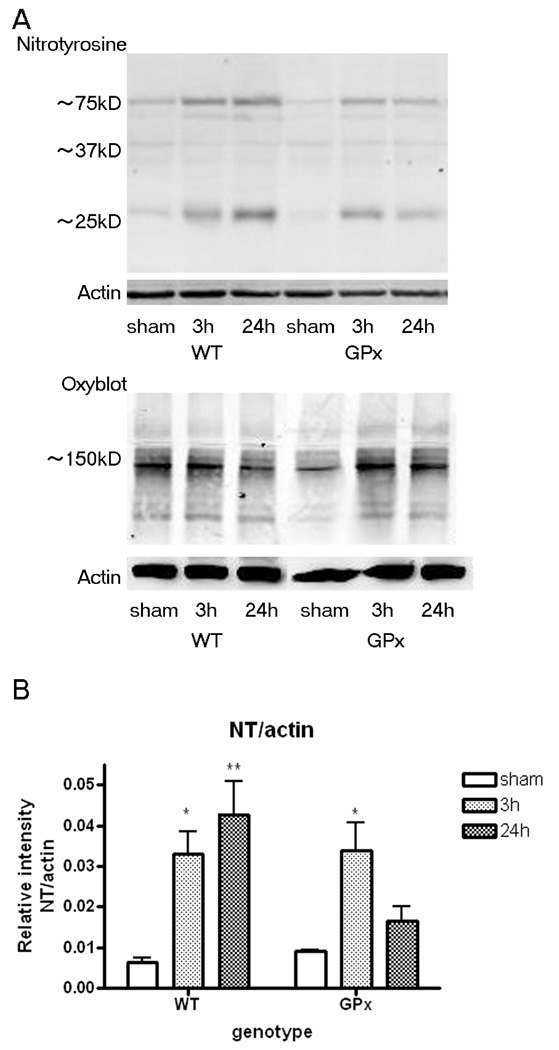
A) Representative immunoblots of oxidative stress markers, protein carbonyls (Oxyblot) and nitrotyrosine, within 24 hours postinjury. A 150kD protein carbonyl band showed a trend toward differences in genotypes (two-way ANOVA; interaction p=0.99; genotype p=0.062). Nitrated protein bands in injured brains of both genotypesare observed at 75, 37 and 25kD.
B) The 25kD nitrated protein band remains elevated 24 hours postinjury in the WT group. A similar temporal change is not seen in the GPxTg group. (ANOVA genotype × time point interaction p=0.0572; Bonferroni post-tests, * p<0.05, ** p<0.01 relative to sham)
Note that the intensity of each sample was quantified and corrected as a relative intensity to loading control, actin.
Acknowledgements
We thank Dr. Oleg Mirochnitchenko for providing us with GPxTg breeding pairs and Dr. Charles McCulloch for his invaluable assistance with our statistical analyses. This research was supported by the National Institutes of Health grant NS050159.
References
- 1.Kraus JF, Rock A, Hemyari P. Brain injuries among infants, children, adolescents, and young adults. Am J Dis Child. 1990;144:684–691. doi: 10.1001/archpedi.1990.02150300082022. [DOI] [PubMed] [Google Scholar]
- 2.Langlois JA, Rutland-Brown W, Thomas KE. The incidence of traumatic brain injury among children in the United States: differences by race. J Head Trauma Rehabil. 2005;20:229–238. doi: 10.1097/00001199-200505000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Levin HS, Eisenberg HM, Wigg NR, Kobayashi K. Memory and intellectual ability after head injury in children and adolescents. Neurosurgery. 1982;11:668–673. doi: 10.1227/00006123-198211000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Ewing-Cobbs L, Miner ME, Fletcher JM, Levin HS. Intellectual, motor, and language sequelae following closed head injury in infants and preschoolers. J Pediatr Psychol. 1989;14:531–547. doi: 10.1093/jpepsy/14.4.531. [DOI] [PubMed] [Google Scholar]
- 5.Koskiniemi M, Kyykka T, Nybo T, Jarho L. Long-term outcome after severe brain injury in preschoolers is worse than expected. Arch Pediatr Adolesc Med. 1995;149:249–254. doi: 10.1001/archpedi.1995.02170150029004. [DOI] [PubMed] [Google Scholar]
- 6.Luerssen TG, Klauber MR, Marshall LF. Outcome from head injury related to patient's age. A longitudinal prospective study of adult and pediatric head injury. J Neurosurg. 1988;68:409–416. doi: 10.3171/jns.1988.68.3.0409. [DOI] [PubMed] [Google Scholar]
- 7.Pullela R, Raber J, Pfankuch T, et al. Traumatic injury to the immature brain results in progressive neuronal loss, hyperactivity and delayed cognitive impairments. Dev Neurosci. 2006;28:396–409. doi: 10.1159/000094166. [DOI] [PubMed] [Google Scholar]
- 8.Tong W, Igarashi T, Ferriero DM, Noble LJ. Traumatic brain injury in the immature mouse brain: characterization of regional vulnerability. Exp Neurol. 2002;176:105–116. doi: 10.1006/exnr.2002.7941. [DOI] [PubMed] [Google Scholar]
- 9.Yager JY, Thornhill JA. The effect of age on susceptibility to hypoxic-ischemic brain damage. Neuroscience and biobehavioral reviews. 1997;21:167–174. doi: 10.1016/s0149-7634(96)00006-1. [DOI] [PubMed] [Google Scholar]
- 10.Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. Journal of neurotrauma. 2000;17:871–890. doi: 10.1089/neu.2000.17.871. [DOI] [PubMed] [Google Scholar]
- 11.Dringen R. Metabolism and functions of glutathione in brain. Progress in neurobiology. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 12.Khan JY, Black SM. Developmental changes in murine brain antioxidant enzymes. Pediatr Res. 2003;54:77–82. doi: 10.1203/01.PDR.0000065736.69214.20. [DOI] [PubMed] [Google Scholar]
- 13.Fan P, Yamauchi T, Noble LJ, Ferriero DM. Age-dependent differences in glutathione peroxidase activity after traumatic brain injury. Journal of neurotrauma. 2003;20:437–445. doi: 10.1089/089771503765355513. [DOI] [PubMed] [Google Scholar]
- 14.Chan PH, Kawase M, Murakami K, et al. Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci. 1998;18:8292–8299. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mikawa S, Kinouchi H, Kamii H, et al. Attenuation of acute and chronic damage following traumatic brain injury in copper, zinc-superoxide dismutase transgenic mice. J Neurosurg. 1996;85:885–891. doi: 10.3171/jns.1996.85.5.0885. [DOI] [PubMed] [Google Scholar]
- 16.Ditelberg JS, Sheldon RA, Epstein CJ, Ferriero DM. Brain injury after perinatal hypoxia-ischemia is exacerbated in copper/zinc superoxide dismutase transgenic mice. Pediatr Res. 1996;39:204–208. doi: 10.1203/00006450-199602000-00003. [DOI] [PubMed] [Google Scholar]
- 17.Fullerton HJ, Ditelberg JS, Chen SF, et al. Copper/zinc superoxide dismutase transgenic brain accumulates hydrogen peroxide after perinatal hypoxia ischemia. Ann Neurol. 1998;44:357–364. doi: 10.1002/ana.410440311. [DOI] [PubMed] [Google Scholar]
- 18.McLean CW, Mirochnitchenko O, Claus CP, et al. Overexpression of glutathione peroxidase protects immature murine neurons from oxidative stress. Dev Neurosci. 2005;27:169–175. doi: 10.1159/000085989. [DOI] [PubMed] [Google Scholar]
- 19.Sheldon RA, Jiang X, Francisco C, et al. Manipulation of antioxidant pathways in neonatal murine brain. Pediatr Res. 2004;56:656–662. doi: 10.1203/01.PDR.0000139413.27864.50. [DOI] [PubMed] [Google Scholar]
- 20.Schmued LC, Stowers CC, Scallet AC, Xu L. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 2005;1035:24–31. doi: 10.1016/j.brainres.2004.11.054. [DOI] [PubMed] [Google Scholar]
- 21.Bonthius DJ, McKim R, Koele L, et al. Use of frozen sections to determine neuronal number in the murine hippocampus and neocortex using the optical disector and optical fractionator. Brain research. 2004;14:45–57. doi: 10.1016/j.brainresprot.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 22.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. The Journal of cell biology. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lebesgue D, LeBold DG, Surles NO, et al. Effects of estradiol on cognition and hippocampal pathology after lateral fluid percussion brain injury in female rats. Journal of neurotrauma. 2006;23:1814–1827. doi: 10.1089/neu.2006.23.1814. [DOI] [PubMed] [Google Scholar]
- 24.West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 25.Witgen BM, Lifshitz J, Grady MS. Inbred mouse strains as a tool to analyze hippocampal neuronal loss after brain injury: a stereological study. Journal of neurotrauma. 2006;23:1320–1329. doi: 10.1089/neu.2006.23.1320. [DOI] [PubMed] [Google Scholar]
- 26.Benice TSRA, Kohama S, et al. Sex-differences in age-related cognitive decline in C57BL/6J mice associated with increased brain microtubule-associated protein 2 and synaptophysin immunoreactivity. Neuroscience. 2006;137:413–423. doi: 10.1016/j.neuroscience.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 27.Lister RG. Ethologically-based animal models of anxiety disorders. Pharmacol Ther. 1990;46:321–340. doi: 10.1016/0163-7258(90)90021-s. [DOI] [PubMed] [Google Scholar]
- 28.Igarashi T, Potts MB, Noble-Haeusslein LJ. Injury severity determines Purkinje cell loss and microglial activation in the cerebellum after cortical contusion injury. Exp Neurol. 2006. [DOI] [PubMed]
- 29.Ferriero DM, Holtzman DM, Black SM, Sheldon RA. Neonatal mice lacking neuronal nitric oxide synthase are less vulnerable to hypoxic-ischemic injury. Neurobiol Dis. 1996;3:64–71. doi: 10.1006/nbdi.1996.0006. [DOI] [PubMed] [Google Scholar]
- 30.Graham EM, Sheldon RA, Flock DL, et al. Neonatal mice lacking functional Fas death receptors are resistant to hypoxic-ischemic brain injury. Neurobiol Dis. 2004;17:89–98. doi: 10.1016/j.nbd.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 31.Sheldon RA, Hall JJ, Noble LJ, Ferriero DM. Delayed cell death in neonatal mouse hippocampus from hypoxia-ischemia is neither apoptotic nor necrotic. Neuroscience letters. 2001;304:165–168. doi: 10.1016/s0304-3940(01)01788-8. [DOI] [PubMed] [Google Scholar]
- 32.Payton KS, Sheldon RA, Mack DW, et al. Antioxidant status alters levels of Fas-associated death domain-like IL-IB-converting enzyme inhibitory protein following neonatal hypoxia-ischemia. Dev Neurosci. 2007;29:403–411. doi: 10.1159/000105481. [DOI] [PubMed] [Google Scholar]
- 33.Deng Y, Thompson BM, Gao X, Hall ED. Temporal relationship of peroxynitrite-induced oxidative damage, calpain-mediated cytoskeletal degradation and neurodegeneration after traumatic brain injury. Exp Neurol. 2007;205:154–165. doi: 10.1016/j.expneurol.2007.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tweedie D, Milman A, Holloway HW, et al. Apoptotic and behavioral sequelae of mild brain trauma in mice. Journal of neuroscience research. 2007;85:805–815. doi: 10.1002/jnr.21160. [DOI] [PubMed] [Google Scholar]
- 35.Hall ED, Detloff MR, Johnson K, Kupina NC. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. Journal of neurotrauma. 2004;21:9–20. doi: 10.1089/089771504772695904. [DOI] [PubMed] [Google Scholar]
- 36.Assaf Y, Holokovsky A, Berman E, et al. Diffusion and perfusion magnetic resonance imaging following closed head injury in rats. Journal of neurotrauma. 1999;16:1165–1176. doi: 10.1089/neu.1999.16.1165. [DOI] [PubMed] [Google Scholar]
- 37.Bao F, Liu D. Peroxynitrite generated in the rat spinal cord induces apoptotic cell death and activates caspase-3. Neuroscience. 2003;116:59–70. doi: 10.1016/s0306-4522(02)00571-7. [DOI] [PubMed] [Google Scholar]
- 38.Onyszchuk G, Al-Hafez B, He YY, et al. A mouse model of sensorimotor controlled cortical impact: characterization using longitudinal magnetic resonance imaging, behavioral assessments and histology. J Neurosci Methods. 2007;160:187–196. doi: 10.1016/j.jneumeth.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vasa RA, Grados M, Slomine B, et al. Neuroimaging correlates of anxiety after pediatric traumatic brain injury. Biol Psychiatry. 2004;55:208–216. doi: 10.1016/s0006-3223(03)00708-x. [DOI] [PubMed] [Google Scholar]
- 40.Camargo EE. Brain SPECT in neurology and psychiatry. J Nucl Med. 2001;42:611–623. [PubMed] [Google Scholar]
- 41.Panksepp J, Siviy S, Normansell L. The psychobiology of play: theoretical and methodological perspectives. Neuroscience and biobehavioral reviews. 1984;8:465–492. doi: 10.1016/0149-7634(84)90005-8. [DOI] [PubMed] [Google Scholar]
- 42.Hovatta I, Tennant RS, Helton R, et al. Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature. 2005;438:662–666. doi: 10.1038/nature04250. [DOI] [PubMed] [Google Scholar]
- 43.Kesner R. Behavioral functions of the CA3 subregion of the hippocampus. Learn Mem. 2007;14:127–136. doi: 10.1101/lm.688207. [DOI] [PubMed] [Google Scholar]
- 44.Sterio DC. The unbiased estimation of number and sizes of arbitrary particles using the disector. J Microsc. 1984;134:127–136. doi: 10.1111/j.1365-2818.1984.tb02501.x. [DOI] [PubMed] [Google Scholar]
- 45.Gundersen HJ, Jensen EB, Kieu K, Nielsen J. The efficiency of systematic sampling in stereology--reconsidered. J Microsc. 1999;193:199–211. doi: 10.1046/j.1365-2818.1999.00457.x. [DOI] [PubMed] [Google Scholar]
- 46.Gundersen HJ, Jensen EB. The efficiency of systematic sampling in stereology and its prediction. J Microsc. 1987;147:229–263. doi: 10.1111/j.1365-2818.1987.tb02837.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.