

Abstract
Animal models of premature aging are often defective for DNA repair. Ku80-mutant mice are disabled for nonhomologous end joining; a pathway that repairs both spontaneous DNA double-strand breaks (DSBs) and induced DNA DSBs generated by the action of a complex composed of Rag-1 and Rag-2 (Rag). Rag is essential for inducing DSBs important for assembling V(D)J segments of antigen receptor genes that are required for lymphocyte development. Thus, deletion of either Rag-1 or Ku80 causes severe combined immunodeficiency (scid) leading to chronic inflammation. In addition, Rag-1 induces breaks at non-B DNA structures. Previously we reported Ku80-mutant mice undergo premature aging, yet we do not know the root cause of this phenotype. Early aging may be caused by either defective repair of spontaneous DNA damage, defective repair of Rag-1-induced breaks or chronic inflammation caused by scid. To address this issue, we analyzed aging in control and Ku80-mutant mice deleted for Rag-1 such that both cohorts are scid and suffer from chronic inflammation. We make two observations: 1) chronic inflammation does not cause premature aging in these mice and 2) Ku80-mutant mice exhibit early aging independent of Rag-1. Therefore, this study supports defective repair of spontaneous DNA damage as the root cause of early aging in Ku80-mutant mice.
Keywords: nonhomologous end joining, Rag-1, DNA double-strand breaks, V(D)J recombination, opportunistic infection
1. Introduction
DNA DSBs are cytotoxic lesions generated by both exogenous and endogenous agents that may be repaired by one of two major pathways: homologous recombination and nonhomologous end joining (NHEJ). Homologous recombination repairs the DSB by annealing the broken strand to a homologous template usually provided by the sister chromatid during DNA replication (West, 2003) while NHEJ repairs the DSB by simply joining the ends together without the use of a homologous template (Lieber et al., 2004). Both pathways are frequently used in mammalian cells (Jasin, 2000) and disruption of either leads to gross chromosomal rearrangements that may cause cancer (Hoeijmakers, 2001) and perhaps aging (Hasty et al., 2003; Lombard et al., 2005; Vijg and Dolle, 2002).
NHEJ repairs spontaneous DSBs that are generated by a variety of sources including by-products of metabolism (reactive oxygen species, ROS), stalled replication forks and exogenous clastogenic agents. Ku80-deletion disables the repair of these spontaneous DSBs causing hypersensitivity to agents that generate DSBs (Lim et al., 2000).
NHEJ also repairs DSBs induced by the Rag heterodimer. Rag-1 and Rag-2 expression is largely restricted to developing T and B lymphocytes (Yamamoto et al., 1992). Rag binds to recombination signal sequences (RSS) to induce a DSB that initiates V(D)J [Variable(Diverse)Joining] recombination for assembling antigen receptor genes (Lieber et al., 2004). Both Rag-1 and Rag-2 are essential for generating this break and deletion of either completely ablates this activity impairing lymphocyte development and causing scid (Mombaerts et al., 1992; Shinkai et al., 1992). Similarly, Ku80-mutant mice suffer from scid as a result of failed V(D)J recombination (Nussenzweig et al., 1996; Zhu et al., 1996). Thus, deletion of Rag-1 stops the generation of DSBs that initiate V(D)J recombination while deletion of Ku80 disrupts the repair of Rag-induced DSBs.
Rag-1 is also expressed in the murine central nervous system causing speculation that Rag induces DSBs in nonlymphoid tissue (Chun et al., 1991) though this speculation is controversial (Schatz and Chun, 1992). Reports showing Ku80-deletion causes neuronal apoptosis in the brain (Gu et al., 2000) and retina (Karanjawala et al., 2003) present the possibility that Rag-1 induced breaks stimulate apoptosis for these tissues in the absence of Ku-mediated repair. However, these potential DSBs would likely be at locations other than RSS. Rag is capable of generating multiple nicks at non-B DNA structures at the major breakpoint region in the Bcl-2 gene (Raghavan et al., 2005b; Raghavan et al., 2004). Rag-induced breaks in this region lead to NHEJ-dependent translocations that lead to cancer (Raghavan et al., 2005a). In addition, current data supports the possibility that the neuronal cell genome is dynamic due to Rag mediated retrotransposition in collaboration with NHEJ by a manner reminiscent to V(D)J recombination (Roth and Craig, 1998). Thus, it is possible that Rag-1-induced breaks at diverse sequences contribute to the Ku80-mutant phenotype in nonlymphoid tissues.
In addition to scid and neuronal apoptosis, Ku80-mutant mice exhibit an early onset of aging characteristics that include kyphosis, poor skin and fur coat condition and shortened life span (Vogel et al., 1999). Microscopically, Ku80-mutant mice exhibit a variety of age-related changes that include an early onset of osteopenia, epiphysis closure and skin atrophy (Vogel et al., 1999). Importantly, Ku80-mutant mice do not show these gross and microscopic phenotypes early in life demonstrating their age-related nature. Furthermore, control mice show the very same gross and microscopic characteristics at a later age than Ku80-mutant mice suggesting these mice prematurely undergo normal aging (Hasty and Vijg, 2004).
At this time the root cause of early aging is not certain for Ku80-mutant mice. It could be due to either defective repair of spontaneous DNA damage or due to debilitative chronic inflammation caused by opportunistic infections that result from scid or possibly Rag-1-induced breaks in nonlymphoid tissue. Defects in DNA damage repair are proposed to cause early aging. DNA may be damaged by numerous sources including ROS, replication errors including stalled replication forks and exposure to exogenous genotoxins. To support this proposition, numerous early aging models in humans and mice are defective for a variety of DNA repair pathways (Hasty et al., 2003; Lombard et al., 2005; Martin, 1978). Furthermore, longevity models show increased resistance to DNA damaging agents (Holzenberger et al., 2003; Migliaccio et al., 1999) and decreased levels of age-related genomic damage (Matheu et al., 2007). Based on these observations, the primary cause for early aging in Ku80-mutant mice has been widely assumed to be defective repair of spontaneous DNA damage. However, some or all of the early aging phenotypes could result from opportunistic infection that causes a chronic stress response and malnutrition (Lombard et al., 2005). In addition, Rag-1-induced DSBs in non-B DNA structures may also contribute to age-related phenotypes, in particular to neuronal tissues. This potential activity in neuronal cells could impact other tissues since aging is influenced by cell-nonautonomous systems in some species; in particular defects in neuronal tissue may impact the entire animal (Kenyon, 2005). Thus, failure to efficiently repair Rag-1-induced breaks in neurons could influence general aging.
Here we compare Ku80 Rag-1-double mutant mice (ku80−/− rag-1−/−) mice to Rag-1-mutant mice (Ku80+/+(+/−) rag-1−/−) for aging. All these mice are scid since all are defective for Rag-1; thus, we are able to determine the importance chronic inflammation and Rag-1-induced breaks in non-B DNA structures have on aging. We observe early aging in only the double-mutant cohort, but not the Rag-1-mutant cohort. Thus, we make two conclusions. First we show that chronic inflammation does not cause premature aging in these mice since both cohorts have clear signs of chronic stress but the Rag-1-mutant cohort does not exhibit premature aging. Second, we show that Rag-1-induced breaks fail to influence aging since the Ku80 Rag-1 double mutant cohort exhibits a premature aging phenotype that is nearly identical to our previous reports for Ku80-mutant mice (Hasty and Vijg, 2004; Vogel et al., 1999). These observations suggest the root cause of premature aging in Ku80-mutant mice is defective repair of spontaneous DNA damage.
2. Materials and Methods
2.1 Mouse husbandry
We crossed Ku80+/− rag-1−/− mice to establish our double mutant (ku80−/− rag-1−/−) and Rag-1–mutant control (Ku80+/+(+/−) rag-1−/−) cohorts. Rag-1+/+ and Rag-1+/− mice were not studied since previous papers describing deletion of either Rag-1 or Rag-2 did not report a phenotype in the heterozygous condition and since the Rag heterozygote crosses result in a Mendelian pattern of inheritance (Mombaerts et al., 1992; Shinkai et al., 1992). In addition, we did not observe a phenotype for Rag-1+/− mice up to nine months.
Mice were housed in microisolator cages in a specific pathogen-free environment. Serum samples from sentinel mice are tested twice a year for EDIM, MHV, MVM, M. pulmonis, MPV, Parvo NS-1, Polyoma, PVM, REO3, Sendai and TMEV. Pinworms are detected by the anal tape test and cecum examination. The fur is microscopically examined for mites. The rodent diet is irradiated, the water is acidified to 2.5 to 3.0, the bedding and the whole cage set up is autoclaved including wire top, isolator cage, card holders, water and water bottles.
Mice were observed 5–6 times a week for the entire course of their life spans. Moribund mice (loosing weight and responsiveness) were observed multiple times a day and all mice were euthanatized when they were immobile and could no longer reach the water source. Morbidities were scored by Kaplan-Meier analysis and measured for statistical significance by the log-rank test. Euthanatized mice were observed by necropsy and organs removed and fixed for histology. All mouse procedures were done in accordance with the principles of laboratory animal care (NIH publication No. 86-23, revised 1985) and approved by the institutional IACUC and.
2.2 Genotyping by PCR
Total DNA was isolated from mouse-tails using Tail Lysis Buffer (100mM Tris-Cl pH8.5, 5mM EDTA, 0.2%SDS, 200mM NaCl), followed by ethanol precipitation. The DNA was resuspended in water. PCR reactions were carried out using Taq polymerase (PGC Scientific, Frederick, Maryland) on an Eppendorf Mastercycler Gradient (Eppendorf, Westbury, New York). Sense primer 5′GAGAGTCTACGACAACTGTGC 3′ and antisense primer 5′-AGAGGGACTGCAGCCATATTA-3′ detects the Ku80 wild-type allele while sense primer 5′-GGTTGCCAGTCATGCTACGGT-3′ and antisense primer 5′-CCAAAGGCCTACCCGCTTCCATT-3′ detects the Ku80-mutant allele; conditions: 29 cycles of 94°C for 30 seconds, 59°C for 1 minute, 72°C for 30 seconds. Sense primer 5′-CCGGACAAGTTTTTCATCGT-3′ detects both Rag-1 wild-type and mutant alleles, antisense primer 5′-GAGGTTCCGCTACGACTCTG-3′ detects only Rag-1 wild-type allele while antisense primer 5′-TGGATGTGGAATGTGTGCGAG-3′ detects only Rag-1-mutant allele; conditions: 35 cycles of 94°C for 30 seconds, 58°C for 45 seconds, 72°C for 45 seconds.
2.3 Length measurement
Mice were anesthetized with isoflurane by inhalation and then positioned on their side and measured from eye to the base of tail.
2.4 Histology Quantification
Mouse skin sections (dorsal region over cranial to mid-thorax) were observed at 100X magnification. Pictures were captured and printed; 4-inch sections of collagen, adipose and skeletal muscle layers were cut from the printout and weighed in grams for double mutant and control mice. Weights for each skin layer were averaged per genotype and percent change in tissue layer surface area calculated by dividing the average double-mutant mouse weight by the average control weight times 100%.
Mouse femurs were photographed at 40X magnification and printed. In order to evaluate the changes in cortical wall surface area between double-mutant and control mice, 2-inch length-wise sections of cortical wall (from the growth plate towards the metaphysis) were cut from the bone pictures and weighed in grams. Averages were taken of like genotypes and percent change calculated by dividing the average cortical wall weight of double-mutant mice by the control average times 100%. In order to evaluate the changes in trabeculae surface area between double-mutant and control mice, all trabeculae of from the growth plate to the articular surface was cut from the bone printout and weighed in grams. Percent change in trabeculae surface area for the double-mutant mice was calculated by dividing the average trabeculae weight of double-mutant mice by the control average times 100%.
The growth plate of the femur was analyzed at 200X magnification in order to visualize the chondrocytes. Each individual chondrocyte was counted along the entire length of the growth plate for both double mutant and control mice.
2.5 Histology
Tissues were fixed in 10% neutral buffered formalin for 24 hours and then in 70% ethanol until embedded in paraffin, cut into 4 mM sections and stained with haematoxylin and eosin by standard procedures.
2.6 Cell proliferation
To generate mouse skin fibroblasts (MSFs), one ear was removed from double mutant and Rag-1–mutant control mice and then cut into small mm-sized pieces and seeded on to a 3.5 cm plate in M10 with fresh antibiotics at 20% O2 [Minimum Essential Medium with 10% fetal bovine serum, 2 mM glutamine, 30 mg penicillin/ml, 50 mg streptomycin/ml]. This is passage zero. For the proliferation curve, 2000 MSFs (passage 2) were seeded per 1.5 cm well on a 24-well plate in two ml of M10. Cells were counted using a hemacytometer on days 4, 6, 8, 10 and 12. For the 3T3 analysis, 3.7 × 104 cells (passage 2) were seeded per 3.5 cm well of a six-well plate in M10 media for a total of two wells. Cells were counted every 3.5 days using a hemacytometer and then re-seeded at the original concentration onto two wells. As cell number declined below 7.4 × 104, then 3.7 × 104 cells were seeded onto a single well. Passaging was discontinued when cell number declined below 3.7 × 104 cells. For the picture, passage 4 cells were seeded at 2.5 × 104 per 3.5 cm well and then stained with 0.2% methylene blue 5 days later.
3. Results
To determine if the Ku80-mutant aging phenotype is caused by defective repair of spontaneous DNA damage, chronic inflammation caused by scid or defective repair of Rag-1-induced DSBs, we crossed Ku80+/− (Zhu et al., 1996) and Rag-1+/− (Mombaerts et al., 1992) mice to generate a ku80−/− rag-1−/− cohort (simply called double mutants) and a control cohort (Ku80+/+ rag-1−/− and Ku80+/− rag-1−/−, simply called Rag-1–mutant controls) These mice are in a mixed 129 x C57Bl/6J background and were housed in a specific pathogen-free environment. All cohorts were evaluated for life span and age-related phenotypes. The Ku80+/+ and Ku80+/− mice deleted for Rag-1 exhibit the same phenotype and are therefore combined into one cohort. These cohorts were evaluated for possible gender-specific differences, but none were observed; therefore, male and females are combined for simplicity.
3.1 Early aging
Previously we showed that Ku80-mutant mice are small and exhibit an early aging phenotype (Hasty and Vijg, 2004; Vogel et al., 1999; Zhu et al., 1996); therefore, double mutant mice and their Rag-1–mutant control littermates were observed for size and age-related characteristics. Here we observe a similar size phenotype (Fig. 1). Double mutant mice are about half the weight of Rag-1–mutant control mice at 4 and 10 weeks (Fig. 1C, p<0.001 F-test). In addition, double mutant mice are ~ 25% shorter than Rag-1–mutant control mice as measured from eye to base of tail (Fig. 1D, p<0.001 F-test). When viewed grossly, we also observe a similar aging phenotype as to our previously described Ku80-mutant mice (Hasty and Vijg, 2004; Vogel et al., 1999). Typically seen is kyphosis, an abnormal convexity in the curvature of the thoracic spine from a lateral perspective. None of these mice exhibit gross aging signs for their first 30 weeks (not shown). However, double mutant mice, but not their Rag-1–mutant control littermates, start to exhibit kyphosis by ~30 weeks and all double mutant mice exhibit kyphosis by 55 weeks (9 out of 9 mice, Fig. 1A, B). Rag-1–mutant control mice exhibit kyphosis at a later age (4 out of 4 mice at weeks 79, 87, 105 and 116, not shown). These observations of size and kyphosis are consistent with our previous reports describing Ku80-mutant mice (Hasty and Vijg, 2004; Vogel et al., 1999). Thus, size and kyphosis occur in Ku80-mutant mice independent of Rag-1 status.
Fig. 1.
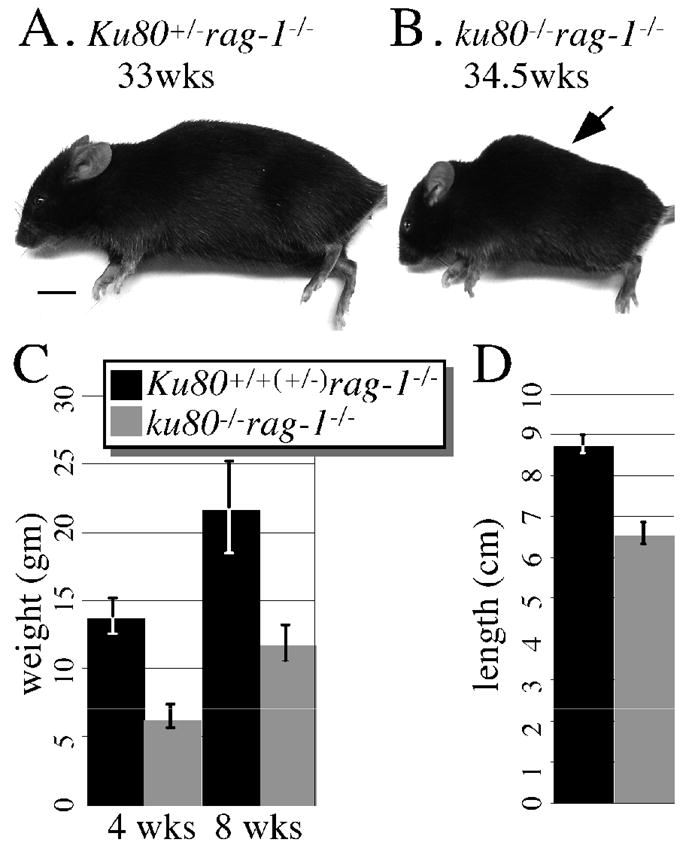
Gross analysis. Lateral view of a (A) 33 week-old Ku80+/− rag-1−/− and a (B) 34.5 week-old ku80−/− rag-1−/− mouse. Only the ku80−/− rag-1−/− mouse exhibits kyphosis (arrow). Scale: 1 cm. (C) Weight of Ku80+/+(+/−) rag-1−/− (black, 21 mice) and ku80−/− rag-1−/− (gray, 12 mice) mice at 4 and 10 weeks. (D) Total length from eye to base of tail for Ku80+/+(+/−) rag-1−/− (black; 5 mice at 32.8, 36.2, 39.1, 39.1, 40.1 weeks) and ku80−/− rag-1−/− (gray; 5 mice at 19.4, 32.8, 36.2, 40, 40.1 weeks).
We investigated three age-related characteristics by histology that were commonly observed in Ku80-mutant mice: osteopenia (the cause of kyphosis), epiphysis closure and skin atrophy (Hasty and Vijg, 2004; Vogel et al., 1999). Previously, we showed no variance between these tissues between Ku80-mutant mice and their control littermates for the first 15 weeks of life (Hasty and Vijg, 2004; Vogel et al., 1999); therefore, we concentrate our studies from weeks 43 to 55, a time when only the Ku80-mutant mice exhibit these aging characteristics. We observe tissues from three double mutant mice (45–48 weeks) and four Rag-1–mutant control littermates (43–55 weeks). To evaluate cortical wall thickness, the distal part of the femur was observed at the point proximal to the epiphysis. All 3 double mutant femurs exhibit about 50% reduced cortical wall thickness compared to 4 Rag-1–mutant control femurs (Fig. 2A, B, G, p=0.003 F-test). The surface area of trabeculae was evaluated in the distal femur medullary cavity between the epiphysis and the joint. All 3 double mutant femurs exhibit about 65% reduced trabecular surface area compared to 4 Rag-1–mutant control femurs (Fig. 2A, B, G, p=0.008 F-test). For epiphysis closure, the same distal part of the femur was observed. All 3 double mutant epiphyses exhibit poor columnar organization of chondrocytes with about a 50% reduction in chondrocyte number compared to 4 Rag-1–mutant control epiphyses (Fig. 2C, D, G, p=0.04 F-test). For skin atrophy, a skin section from the dorsal region over the cranial to mid thorax was examined. All 3 double mutant skin sections are reduced for all subcutaneous elements (superficial collagen, subcutaneous adipose and skeletal muscle) compared to 4 Rag-1–mutant control skin sections (Fig. 2E, F, G). The collagen, adipose and skeletal muscle layers were reduced by about 50% (p=0.02 F-test), 85% (p=0.09 F-test), and 70% (p=0.008 F-test), respectively. Therefore, by gross and histological analysis the double mutant mice, but not their control littermates, show aging signs from 45–48 weeks. These age-related phenotypes are identical to our previous reports (Hasty and Vijg, 2004; Vogel et al., 1999) and; therefore, occur in Ku80-mutant mice independent of Rag-1-status.
Fig. 2.
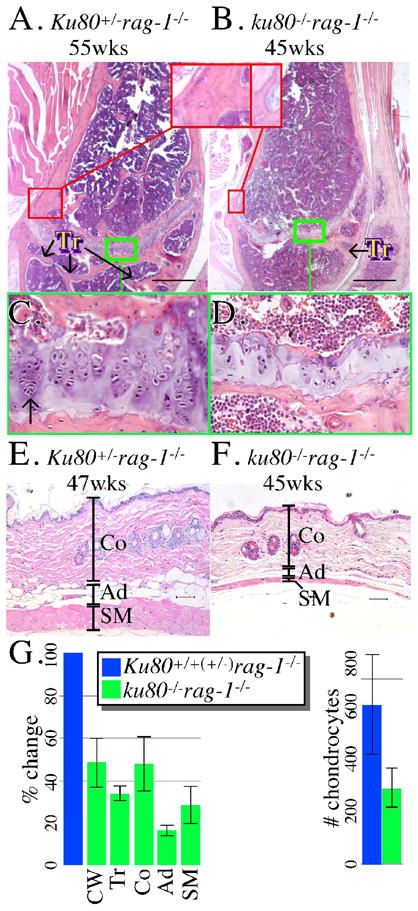
Microscopic analysis. (A, B) Femur from a (A) 55 week-old Ku80+/− rag-1−/− and a (B) 45 week-old ku80−/− rag-1−/− mouse. Compare thickness of the bone wall (red boxes) and number of trabeculae (Tr). (C, D) Compare the columnar organization and number of chondrocytes from (C) Ku80+/− rag-1−/− and (D) ku80−/− rag-1−/− mouse. Green box from A, B. Arrow indicates chondrocytes. Scale: 500 mm. (E, F) Skin taken from the dorsal–cervico-thoracic region from a (E) 47 week-old Ku80+/− rag-1−/− and a (F) 45 week-old ku80−/− rag-1−/− mouse. Compare thickness of the three skin layers. (Co, Collagen; Ad, Adipose; SM, Skeletal Muscle) Scale: 50 mm. (G) Quantitation of results for Ku80+/+(+/−) rag-1−/− (blue) and ku80−/− rag-1−/− (green) mice. Three control and 3 ku80−/− rag-1−/− mice observed for trabeculae. Four control and 3 ku80−/− rag-1−/− mice observed for cortical wall thickness, skin layers and chondrocytes. Left: % change compared to the mean of the control. Right: the mean number of chondrocytes in the epiphysis. Refer to Experimental procedures for quantification methods.
3.2 Proliferation and senescence in fibroblasts
Previously we showed Ku80-mutant fibroblasts undergo premature replicative senescence (Lim et al., 2000); a term that refers to the limited proliferative potential and eventual arrest exhibited by normal mammalian cells in tissue culture (Hayflick, 1965). Here we determine if chronic inflammation or Rag-1 activity influences this phenotype by observing mouse skin fibroblasts (MSFs) derived from the ears of six double mutant mice and seven Rag-1–mutant control littermates between the ages of 32.4–39.5 weeks. All experiments were performed in atmospheric oxygen. We find that the double mutant MSFs, proliferate less than Rag-1–mutant control MSFs as measured by a proliferation curve (Fig. 3A, p=0.02). After 12 days, all double mutant MSFs proliferate less than Rag-1–mutant control MSFs. We also find that double mutant MSFs, but not Rag-1–mutant control MSFs, are resistant to transformation as determined by a modified 3T3 assay that measures cell number after serial passages (Fig. 3B, p=0.003). In addition, double mutant MSFs (Fig. 3C), but not Rag-1–mutant control MSFs (Fig. 3D), exhibit morphological characteristics typical for replicative senescence, they appeared post-mitotic with increased surface area, spreading and extension of the plasma membrane. Thus, resistance to spontaneous transformation and senescence are not influenced by Rag-1 status since the double mutant fibroblasts exhibit the same phenotype as described for Ku80-mutant fibroblasts (Lim et al., 2000).
Fig. 3.
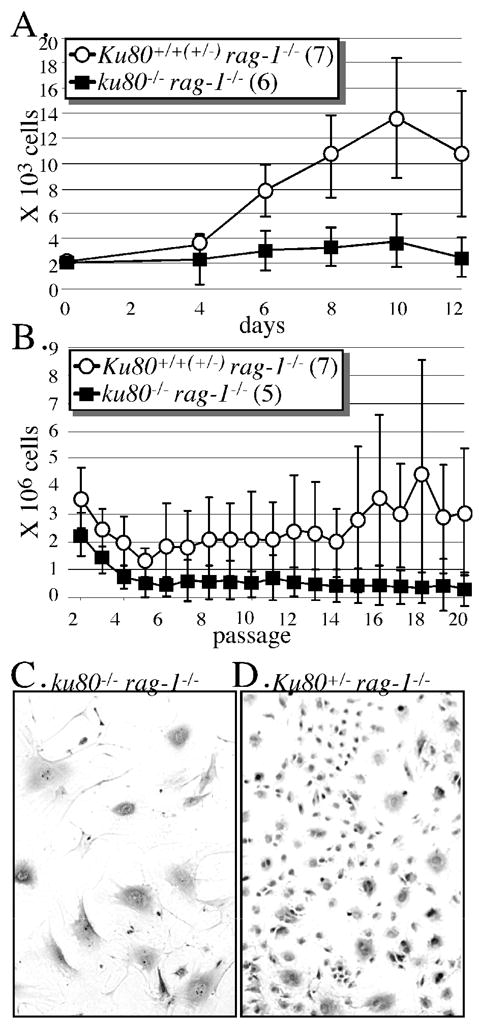
Mouse skin fibroblast analysis. All cells derived from the ears of mice between 32.4–39.5 weeks of age and incubated in atmospheric O2. (A) MSF proliferation curve. Cells (passage 2) derived from seven Rag-1-mutant controls and six double mutant mice. (B) Modified 3T3 analysis to measure lifetime proliferation capacity. MSF (passage 2) are seeded onto two 3.5 cm plates (3.7 × 104 cells/3.5 cm plate). Every 3.5 days cells were trypsinized, combined, counted and seeded at their original concentration onto another 2 plates. As total cell number declined below 7.4 × 104, then cells were seeded onto a single plate at the original concentration. Passaging was discontinued when less than 3.7 × 104 cells remained. (C) Picture of cells (passage 4) derived from the ears of a double mutant mouse. Note the flattened appearance for the double mutant MSF. (D) Picture of cells (passage 4) derived from the ears of a Rag-1-mutant control mouse. Note the spindle-shaped appearance for the Rag-1-mutant control MSF.
3.3 Severe inflammation
So far we have shown that double mutant mice and cells, but not Rag-1-mutant mice and cells, exhibit early aging and premature replicative senescence. Thus, Rag-1 status is not important for these phenotypes. Now we investigate double mutant and Rag-1-mutant mice for chronic inflammation to determine if a chronic stress response could contribute to premature aging as previously proposed (Lombard et al., 2005).
Double mutant mice and their Rag-1–mutant control littermates were observed for illness and potential cause of death since deletion of Rag-1 causes scid that could lead to chronic inflammation. We typically observe malaise beginning as early as 10 weeks but more commonly around 30 weeks for both cohorts. Frequently these mice exhibit gross characteristics typical for infection that include labored breathing, skin ulcers, abscesses and suppurative conjunctivitis (Fig. 4A, B). Moribund mice were euthanatized and evaluated by necropsy and histology. Most moribund mice showed signs of severe purulent inflammation [Ku80+/+ rag-1−/− (8/9 mice between the ages of 38 to 116 weeks), Ku80+/− rag-1−/− (14/18 mice between the ages of 27 to 79 weeks), ku80−/− rag-1−/− (7/8 mice between the ages of 45 to 67 weeks)]. Otitis media was commonly observed (Fig. 4C, D). In addition, meningitis, encephalitis, ventriculitis, osteomyelitis, periostitis, esophagitis, pharyngitis, pleuritis and pneumonia was observed. Intralesional bacterial colonies were visualized by histology. Additionally enterococcus, alpha hemolytic streptococcus, coagulase negative staphylococcus and gram-positive cocci were cultured from either the middle ear or retroorbital tissues from the Rag-1-mutant controls. Signs of dehydration and starvation were commonly observed that include hemorrhage and ulceration in the lumen of the stomach and small intestine that reflects reduced intake of food and water (likely due to sickness caused by infection). In addition to the moribund mice, we euthanatized 10 Rag-1-mutant control and 6 double mutant mice without gross signs of inflammation at 15 weeks of age; many of these mice also exhibited histological signs of inflammation that include otitis media and pneumonia [Ku80+/+ rag-1−/− (3/6), Ku80+/− rag-1−/− (1/4), ku80−/− rag-1−/− (3/6)]. These data illustrate the early onset and severity of inflammation that leads to chronic stress and malnutrition.
Fig. 4.
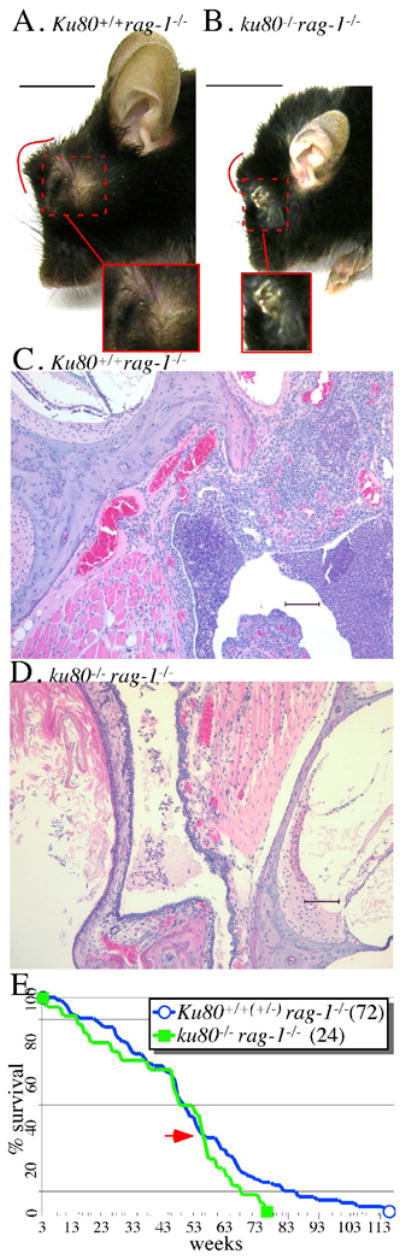
Opportunistic infections and life span. Suppurative conjunctivitis from a (A) 51-week old Rag-1-mutant control mouse and a (B) 38-week old double mutant mouse. The red line outlines the abnormal shape of the face in profile due to the protruding periocular abscess. The red dashed box shows the abscess adjacent to the eye that has broken through the skin. Scale bar: 1 cm. Otitis media from a (C) 21-week old Rag-1-mutant control mouse and a (D) 16-week old double mutant mouse. Scale bar: 100 mm. (E) Life span of Rag-1-mutant control (blue) and double mutant (green) mice. There is no statistically significant difference between these life spans (log-rank test, Kaplan-Meier survival analysis: p=0.213); however, there is a statistically significant difference when considering the last point their curves meet (red arrow) to the end (p=0.017).
The life span was measured for double mutant mice and their Rag-1–mutant control littermates (Fig. 4E). Both cohorts exhibit a similar median life span (49 wks; p=0.213), albeit the Rag-1-mutant cohort has a longer maximum life span (116 vs. 76 wks; p=0.017). Previously we showed 50% of the Ku80-mutant mice died 66 weeks earlier than their Ku80+/+ and Ku80+/− control littermates that are functional for Rag-1 (Vogel et al., 1999). However, for this study, the double mutant mice live longer than our previous report for Ku80-mutant mice (median life span of 36 vs. 48 weeks). We commonly observe differences in Ku80-mutant life span from one experiment to another (a range of median life span from 36–70 weeks) that are likely due to changes in genetic background; however, the Ku80-mutant mice always have a much shorter life span than Rag-1-functional controls (VH, PH personal observations). More strikingly Rag-1–mutant control mice die much earlier than Rag-1-functional control mice from our previous study (median life span of 50 vs. 102 weeks) (Vogel et al., 1999). Early death is likely due to opportunistic infections since all mice are scid in this study and since these mice experience severe inflammation likely due to opportunistic infection. Thus, the Rag-1-mutant mice present with signs of severe inflammation and malnutrition, but do not show early sings of aging; therefore, a scid-related phenotype does not cause early aging.
4. Discussion
We investigate the impact severe chronic inflammation and Rag-1-induced DSBs have on life span and aging for double mutant and Rag-1–mutant control mice. These mice were observed throughout the course of their life span. We show that both cohorts experience severe chronic inflammation, but only the double mutant cohort experiences early aging. We make two conclusions.
First, we conclude that a scid-related phenotype does not cause early aging in Ku80-mutant mice since the Rag-1–mutant control mice exhibit severe chronic inflammation but not premature aging. Previously, Rag-1 deletion was shown to cause nonleaky scid due to an arrest in B and T cell differentiation caused by defective V(D)J recombination (Mombaerts et al., 1992). Surprisingly, this is the first life span study reported for Rag-1-mutant mice and we clearly show these mice are predisposed to opportunistic infection that causes severe inflammation and malnutrition. These maladies have been proposed as a potential cause for early aging (Lombard et al., 2005). Even though these mice are subject to a severe chronic stress response and malnutrition, there are no signs of premature aging in these mice. Thus, chronic infection and malnutrition do not cause early aging.
Second, we conclude Rag-1-induced DSBs do not influence early aging for Ku80-mutant mice since the double mutant cohort exhibits early aging identical to Ku80 single mutant mice (Hasty and Vijg, 2004; Vogel et al., 1999). With or without Rag-1, Ku80-mutant mice present with an early onset of gross and microscopic changes that include kyphosis, osteopenia, epiphysis closure and skin atrophy. These changes are commonly observed in both studies by week 40. Therefore, Rag-1 induced DSBs have no impact on these aging phenotypes. Even though Rag-1 is also expressed in neuronal tissue (Chun et al., 1991) and Ku80-mutant mice exhibit neuronal apoptosis in the developing brain (Gu et al., 2000) and retina (Karanjawala et al., 2003), we find Rag-1 does not impact the Ku80-mutant phenotype in nonlymphoid tissue. Therefore, Rag-1’s influence on the Ku80-mutant phenotype is only detected in developing lymphocytes where Rag-1-induced DSBs are essential for pro-B cell lymphomas (Holcomb et al., 2006; Lim et al., 2000).
These observations show that chronic inflammation and Rag-1-induced DSBs are not responsible for early aging in Ku80-mutant mice and therefore, suggest the primary cause for early aging is improper repair of spontaneous genomic damage. This means the cause of early aging in Ku80-mutant mice is similar to other early aging models (Hasty et al., 2003; Lombard et al., 2005).
Mouse aging models are typically defective for DNA repair or some aspect of chromosomal maintenance. These models show an early onset of similar aging characteristics even though the specific defective pathway may be very different from one model to another. Included in these aging models are defects in homologous recombination (Cao et al., 2003), nucleotide excision repair and transcription (de Boer et al., 2002), mitochondrial DNA maintenance (Kujoth et al., 2005; Trifunovic et al., 2004), mitotic chromosomal segregation (Baker et al., 2004) and helicase activity in association with telomere maintenance (Chang et al., 2004; Du et al., 2004; Rudolph et al., 1999). While the root cause may be defective chromosomal maintenance, the aging phenotype may actually be caused by cellular responses that are common to all these defects (Maier et al., 2004; Tyner et al., 2002). To this end we show that chronic stress caused by severe inflammation and Rag-1-induced DSBs do not cause early aging. Furthermore, premature replicative senescence observed in Ku80-mutant skin fibroblasts is independent of a scid-related phenotype and Rag-1-induced DSBs (note these fibroblasts were derived from mice experiencing chronic inflammation). Therefore, the initial defect that causes Ku80-mutant mice to undergo early aging is likely due to defective repair of spontaneous DNA damage that leads to persistent cellular responses and mutations.
Acknowledgments
We thank Dr. Wayne Kornegay for interpreting pathology, Mr. Gary Chisholm for statistical analysis and Ms. Charnae Williams for technical assistance. This work was supported by grants NIH UO1 ES11044, P01 AG17242 and R01 CA76317-05A1 to PH and DOD W81XWH-04-1-0325 to VBH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, van Deursen JM. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–749. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003;17:201–213. doi: 10.1101/gad.1050003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, Lombard D, Pathak S, Guarente L, DePinho RA. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet. 2004;36:877–882. doi: 10.1038/ng1389. [DOI] [PubMed] [Google Scholar]
- Chun JJ, Schatz DG, Oettinger MA, Jaenisch R, Baltimore D. The recombination activating gene-1 (RAG-1) transcript is present in the murine central nervous system. Cell. 1991;64:189–200. doi: 10.1016/0092-8674(91)90220-s. [DOI] [PubMed] [Google Scholar]
- de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H, Weeda G, van der Horst GT, van Leeuwen W, Themmen AP, et al. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296:1276–1279. doi: 10.1126/science.1070174. [DOI] [PubMed] [Google Scholar]
- Du X, Shen J, Kugan N, Furth EE, Lombard DB, Cheung C, Pak S, Luo G, Pignolo RJ, DePinho RA, et al. Telomere shortening exposes functions for the mouse werner and bloom syndrome genes. Mol Cell Biol. 2004;24:8437–8446. doi: 10.1128/MCB.24.19.8437-8446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Sekiguchi J, Gao Y, Dikkes P, Frank K, Ferguson D, Hasty P, Chun J, Alt FW. Defective embryonic neurogenesis in Ku-deficient but not DNA-dependent protein kinase catalytic subunit-deficient mice. Proc Natl Acad Sci U S A. 2000;97:2668–2673. doi: 10.1073/pnas.97.6.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J. Aging and genome maintenance: lessons from the mouse? Science. 2003;299:1355–1359. doi: 10.1126/science.1079161. [DOI] [PubMed] [Google Scholar]
- Hasty P, Vijg J. Accelerating aging by mouse reverse genetics: a rational approach to understanding longevity. Aging Cell. 2004;3:55–65. doi: 10.1111/j.1474-9728.2004.00082.x. [DOI] [PubMed] [Google Scholar]
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Experimental Cell Research. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- Holcomb VB, Vogel H, Marple T, Kornegay RW, Hasty P. Ku80 and p53 suppress medulloblastoma that arise independent of Rag-1-induced DSBs. Oncogene. 2006;25:7159–7165. doi: 10.1038/sj.onc.1209704. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Jasin M. Chromosome breaks and genomic instability. Cancer Invest. 2000;18:78–86. doi: 10.3109/07357900009023065. [DOI] [PubMed] [Google Scholar]
- Karanjawala ZE, Hinton DR, Oh E, Hsieh CL, Lieber MR. Developmental retinal apoptosis in Ku86−/− mice. DNA Repair (Amst) 2003;2:1429–1434. doi: 10.1016/j.dnarep.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Lieber MR, Ma Y, Pannicke U, Schwarz K. The mechanism of vertebrate nonhomologous DNA end joining and its role in V(D)J recombination. DNA Repair (Amst) 2004;3:817–826. doi: 10.1016/j.dnarep.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Lim DS, Vogel H, Willerford DM, Sands AT, Platt KA, Hasty P. Analysis of ku80-mutant mice and cells with deficient levels of p53. Mol Cell Biol. 2000;20:3772–3780. doi: 10.1128/mcb.20.11.3772-3780.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005;120:497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, Sutherland A, Thorner M, Scrable H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004;18:306–319. doi: 10.1101/gad.1162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GM. Genetic syndromes in man with potential relevance to the pathobiology of aging. Birth defects: Original article series. 1978:5–39. [PubMed] [Google Scholar]
- Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, Flores JM, Vina J, Blasco MA, Serrano M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448:375–379. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals [see comments] Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Nussenzweig A, Chen C, da Costa Soares V, Sanchez M, Sokol K, Nussenzweig MC, Li GC. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature. 1996;382:551–555. doi: 10.1038/382551a0. [DOI] [PubMed] [Google Scholar]
- Raghavan SC, Hsieh CL, Lieber MR. Both V(D)J coding ends but neither signal end can recombine at the bcl-2 major breakpoint region, and the rejoining is ligase IV dependent. Mol Cell Biol. 2005a;25:6475–6484. doi: 10.1128/MCB.25.15.6475-6484.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan SC, Swanson PC, Ma Y, Lieber MR. Double-strand break formation by the RAG complex at the bcl-2 major breakpoint region and at other non-B DNA structures in vitro. Mol Cell Biol. 2005b;25:5904–5919. doi: 10.1128/MCB.25.14.5904-5919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan SC, Swanson PC, Wu X, Hsieh CL, Lieber MR. A non-B-DNA structure at the Bcl-2 major breakpoint region is cleaved by the RAG complex. Nature. 2004;428:88–93. doi: 10.1038/nature02355. [DOI] [PubMed] [Google Scholar]
- Roth DB, Craig NL. VDJ recombination: a transposase goes to work. Cell. 1998;94:411–414. doi: 10.1016/s0092-8674(00)81580-9. [DOI] [PubMed] [Google Scholar]
- Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96:701–712. doi: 10.1016/s0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- Schatz DG, Chun JJ. V(D)J recombination and the transgenic brain blues. New Biol. 1992;4:188–196. [PubMed] [Google Scholar]
- Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall AM, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- Vijg J, Dolle ME. Large genome rearrangements as a primary cause of aging. Mech Ageing Dev. 2002;123:907–915. doi: 10.1016/s0047-6374(02)00028-3. [DOI] [PubMed] [Google Scholar]
- Vogel H, Lim DS, Karsenty G, Finegold M, Hasty P. Deletion of Ku86 causes early onset of senescence in mice. Proc Natl Acad Sci U S A. 1999;96:10770–10775. doi: 10.1073/pnas.96.19.10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Atsuta M, Hamatani K. Restricted expression of recombination activating gene (RAG-1) in mouse lymphoid tissues. Cell Biochem Funct. 1992;10:71–77. doi: 10.1002/cbf.290100202. [DOI] [PubMed] [Google Scholar]
- Zhu C, Bogue MA, Lim DS, Hasty P, Roth DB. Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell. 1996;86:379–389. doi: 10.1016/s0092-8674(00)80111-7. [DOI] [PubMed] [Google Scholar]