

Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs) have been shown to reduce the risk of colorectal cancer (CRC). They are also known to induce the regression of colorectal adenomas, which are precursors to CRC. Despite these evidences, the exact mechanism by which NSAIDs exerts its anti-oncogenic effect is not completely understood. Using a focus formation assay, here we show that sulindac sulfide, a NSAID, specifically inhibits cell transformation mediated by oncogenic Ha-Ras, but not by other established oncogene products such as v-Src, Gα 12, and Gα13. Our results suggest that the ability of sulindac sulfide to suppress transformation is confined to specific oncogenic pathways. Further studies of the sulindac-resistant oncogenic pathways may lead to identification of novel therapeutic agents that are effective in the prevention or treatment of CRC.
Keywords: Chemoprevention, Non-steroidal anti-inflammatory drugs, Oncogene, Ha-Ras, v-Src, Gα12, Gα13, Focus formation assay, Transformation
1. Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are routinely prescribed to reduce swelling and pain in patients suffering from ailments such as arthritis and headache. These compounds are thought to exert their effects by interfering with the cyclooxygenase pathway, thus inhibiting the synthesis of pro-inflammatory prostaglandins [1]. In addition to their prescribed role, NSAIDs have been shown to reduce the risk of developing colorectal cancer (CRC) and adenoma [2–4]. Of particular interest, sulindac, a specific NSAID, has been shown to have a significant chemopreventive effect in reducing the size and number of adenomatous polyps in patients afflicted with familial adenomatous polyposis (FAP), an autosomal dominant genetic disorder characterized by the development of hundreds of colorectal adenomas during the second to third decade of life [5,6]. Sulindac is a pro-drug when administered orally and is metabolized to sulindac sulfide and sulindac sulfone by colonic bacteria [7]. Sulindac sulfide is considered the active metabolite and exerts its function by non-selectively inhibiting cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) [7,8]. COX-2 is overexpressed in human colorectal cancers [9], and its inactivation is associated with decreased carcinogenesis [10]. Although much less active in inhibiting COX activities, sulindac sulfone has also been shown to induce apoptosis and can inhibit mammary carcinogenesis [11,12].
While its molecular mechanisms remain largely unknown, substantial evidence exists that the antiproliferative and antineoplastic effect of sulindac is not solely dependent on eicosanoid metabolism [13]. Colon cancer cells lacking COX activity are inhibited to the same degree as colon cancer cells with COX activity [14]. Furthermore, the addition of prostaglandins does not rescue COX-producing colon cancer cells from sulindac-associated growth arrest [15]. Other signal transduction pathways have been identified as possible targets of sulindac. Experiments have shown that sulindac sulfide can directly affect Ras-mediated signal transduction by inhibiting Ras/Raf dependent transactivation [16]. In addition, sulindac can inhibit the activation of the nuclear factor-κB pathway (NF-κB) [17]. Finally, studies have identified the peroxisome proliferator-activated receptor δ as a target for sulindac sulfide [18].
The present study examines the effect of sulindac sulfide on cellular transformation caused by several established oncogene products in an attempt to specific signaling pathways targeted by it. The results indicate that sulindac sulfide exclusively inhibits Ha-Ras-mediated transformation of four oncogenes tested, despite the shared targets that exist among the other signal transduction pathways. These observations illustrate the specific nature by which sulindac sulfide exerts its antineoplastic effect and open the possibility of developing novel therapeutic agents that target the sulindac sulfide-resistant pathways of oncogenesis.
2. Materials and methods
2.1. Drugs and plasmid constructs
Sulindac sulfide was purchased from Biomol (Plymouth Meeting, PA). The drug was dissolved in dimethyl sulfoxide (DMSO) in stock solutions of 250 μM. Expression plasmids containing Ha-Ras, v-Src, Gα12, and Gα13 were kindly provided by Dr Raul Urrutia (Mayo Clinic, Rochester, MN) [19].
2.2. Cell proliferation/cytotoxicity assay
Assays were performed as described by the CellTiter 96® Aqueous One Solution Cell Proliferation Assay Protocol (Promega, Madison, WI). NIH3T3 fibroblasts (obtained from American Type Culture Collection) were cultured in 96-well plates in 200 μl Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS), 100 IU penicillin/ml, and 1 mg/ml streptomycin. One thousand cells were seeded into each well and maintained for 3 days in culture with media changed every 24 h. At the end of cultivation, 20 μl of CellTiter 96® Aqueous One Solution (MTS tetrazolium compound, [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium), inner salt]) was added to each well. After 3 h of incubation, the plates were analyzed for absorption at 490 nm using a microplate reader. Media with vehicle (DMSO) was used as reference. Each sample was present in quadruplicate on each plate.
2.3. Transformation assay
Focus formation assays were used to analyze the transformation of NIH3T3 fibroblasts [20]. Cells (106) plated in a 100-mm tissue culture dish were transfected using 20 μl of LipofectAMINE™ reagent (GIBCO-BRL). Cells were transfected with 5 μg of Ha-Ras, v-Src, Gα12, or Gα13. After 24 h, sulindac sulfide was added to a final concentration of 50 μM. Media and drug were replaced every 48 h. The control dishes contained either no drug or vehicle (DMSO) alone. After 14 days, cells were stained with 0.1% methylene blue, and foci were counted as described [20]. Each experiment was performed in triplicate in at least four experiments. After subtracting appropriate background from spontaneous foci formation, the mean numbers of foci and standard deviations were calculated. Two-tailed Student’s t-test was used to determine statistical significance.
3. Results
We first determined the optimal concentration of sulindac sulfide with which to conduct the transformation experiments. This was accomplished by incubating NIH3T3 fibroblasts in the presence of increasing concentrations of sulindac sulfide and measuring their proliferative potential by a cell proliferation assay (Fig. 1). As shown, sulindac sulfide did not begin to inhibit cell growth until it reached a concentration of 200 μM. However, in 100-mm tissue culture dishes, treatments with over 100 μM sulindac sulfide for the 14-day period of the transformation assay caused extensive peeling of cells, suggesting a generalized cytotoxic effect. We therefore chose a concentration of 50 μM for all subsequent experiments, which did not cause any cell death over the course of the experiments. This concentration is consistent with that used in similar previous studies [16].
Fig. 1.

Proliferation of NIH3T3 fibroblasts in the presence of sulindac sulfide. NIH3T3 cells were seeded in 96-well plates and maintained in media containing increasing concentrations of sulindac sulfide for 72 h at which time MTS proliferation assays were performed as described in Section 2. Absorbance at 490 nm linearly correlated to cell number. Shown are the means of four independent experiments performed in quadruplicate. Bars indicate standard deviations.
The ability of sulindac sulfide to inhibit Ha-ras mediated transformation [16] warrants examination whether sulindac sulfide can inhibit transformation by other oncogenic products that are dependent on Ras or share targets with it. For example, Gα12 and Gα13 have been shown to induce transformation in fibroblasts [21]. Effectors of Gα12 and Gα13 can activate Ras and other small monomeric G proteins [22,23], providing a link from heterotrimeric to monomeric G proteins. In addition, stimulation of Gα12 and Gα13 induce expression of COX-2 [24]. v-Src is another potent transforming oncogene that activates the Ras-MAP kinase (MAPK) pathway. v-Src activates Raf, and induces phosphorylation of MAPK and tyrosine residues of the RasGAP-associated proteins p62 and p190 [25–27]. Moreover, activation by Gα12 and Gα13 has been shown to in part depend on Src [28]. To investigate whether sulindac sulfide similarly inhibits the transforming activities of these other established oncogenes, we conducted focus formation assays in the presence or absence of sulindac sulfide using expression plasmids containing each of the three stated oncogenes. In each experiment, foci formation was counted without drug, with vehicle (0.5% DMSO), and with 50 μM sulindac sulfide dissolved in DMSO. As shown in Fig. 2, while sulindac sulfide inhibited foci formation caused by Ha-Ras (P < 0.04 compared to control), it had little or no effect on transformation caused by the other three oncogenes.
Fig. 2.

Focus formation assays of NIH3T3 fibroblasts. Cells (106) cultured in 100-mm culture dishes were transfected in triplicate with 5 μg of plasmid DNA containing the respective oncogenes. Cells were maintained in the absence of any drugs, or in the presence of DMSO only or 50 μM sulindac sulfide in DMSO. Foci were counted 14 days after transfection. *P < 0.04 compared to no treatment or DMSO only. N = 4.
4. Discussion
In this study, we investigated whether sulindac sulfide could protect against neoplastic transformation elicited by Ha-Ras, Gα12, Gα13, and v-Src. Strong evidence exists that sulindac sulfide directly inhibits p21Ras activation of Raf and Ha-Ras-induced foci formation [16]. Therefore, we chose to examine those oncoproteins that are linked to eicosanoid metabolism or have been shown to modulate the activity of Ras or its targets. Effectors of Gα12 and Gα13 activate Ras and other monomeric G proteins such as Rho and Rac [22,23,30]. Previous experiments have also demonstrated a synergistic effect on foci formation when constitutively active mutants of Gα12 are cotransfected with c-Raf-1 [29]. In addition, studies have shown that Gα12 stimulates fibroblast cell proliferation through the COX-2 signaling pathway and increases arachidonic acid secretion [24]. Similarly, v-Src stimulates activation of MAPK and certain RasGAP proteins [26,27]. Despite the overlapping or shared nature of these transforming proteins with Ras, the results of our current study demonstrate that sulindac sulfide cannot inhibit the transformation induced by v-Src, Gα12 and Gα13. Part of the reasons that transformation mediated by these three oncoproteins are resistant to sulindac sulfide may be due to additional cellular pathways that bypass the inhibitory effect of sulindac sulfide (Fig. 3).
Fig. 3.
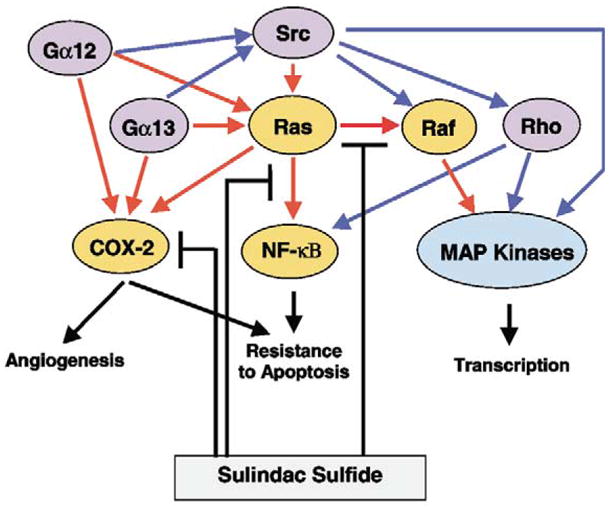
Proposed model to illustrate the selective inhibition of Ras-mediated transformation by sulindac sulfide. The relationships among the four oncogenes examined in the present study are shown. The sulindac sulfide-sensitive proteins and pathways are shown in tan and red, respectively. The sulindac sulfide-insensitive proteins and pathways are shown purple and blue, respectively.
The selectivity of sulindac sulfide in inhibiting Ha-Ras-, but not Gα12- and Gα13-, mediated transformation does not contradict the role of sulindac sulfide as a COX-2 and p21Ras inhibitor. In addition to Ras, Gα12 and Gα13 can activate other monomeric G protein signaling cascades such as Rho that are not inhibited by sulindac sulfide [16,23]. Such activation is thought to be dependent upon Src, and has been implicated in JNK activation [28]. Additional targets important for Ha-Ras-mediated transformation include the activation of the NF-κB pathway by p21Ras. Inhibition of the NF-κB pathway reduces the cell proliferation of Ha-Ras-transformed fibroblasts and renders non-transformed fibroblasts resistant to Ha-Ras-mediated transformation [31]. Sulindac sulfide has been shown to inhibit the activation of NF-κB in colon cancer cells and other cell lines [17]. Considering that Ras mutations are present in approximately 50% of colon cancers [32], inhibition of the NF-kB pathway by sulindac sulfide may be linked to the direct inhibitory effect on p21Ras. In contrast, Rho has been shown to activate the NF-κB pathway [33] in a mutually independent fashion from Ras [34]. Thus, it is possible that while the selective inhibition of Ha-Ras-mediated transformation by sulindac sulfate may be linked to the Ras-dependent activation of NF-κB, the Rho-dependent activation of NF-κB may account for the resistance to sulindac sulfide in v-Src-, Gα12- and Gα13-mediated transformation (Fig. 3). Additional experiments are necessary to define this relationship.
The concentration at which sulindac sulfide inhibits Ha-Ras-mediated transformation in the present study does not affect cell proliferation (Fig. 1). In contrast, at a higher concentration, sulindac sulfide acts mainly as a cytotoxic agent, perhaps by inducing apoptosis [11,13]. The ability of sulindac sulfide in inhibiting Ha-Ras-mediated transformation is therefore primarily exerted at a biochemical level. This may explain the observation that despite a relatively low plasma concentration of 10–15 μM, sulindac sulfide was effective in regressing polyps in patients with familial adenomatous polyposis [35]. The differential effect of sulindac sulfide on cell transformation and proliferation at different concentrations may also be important in further understanding the mechanisms of action of sulindac sulfide in cancer chemoprevention and in identifying newer compounds that are more potent and specific than sulindac sulfide.
Acknowledgments
We would like to thank Dr Raul Urrutia for providing the plasmids used in this study. This work is supported by National Institutes of Health grants DK52230 and CA84197.
Abbreviations
- COX
cyclooxygenase
- CRC
colorectal cancer
- DMEM
Dulbecco’s modified Eagle medium
- DMSO
dimethyl sulfoxide
- FBS
fetal bovine serum
- MTS
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium), inner salt]
- NSAID
non-steroidal anti-inflammatory drug
References
- 1.Damas J, Volon G. Effect of oxamethacin and sulindac on prostaglandin synthesis. CR Seances Soc Biol Fil. 1979;173:849–853. [PubMed] [Google Scholar]
- 2.Martinez ME, McPherson RS, Levin BFJ. Aspirin and other nonsteroidal anti-inflammatory drugs and risk of colorectal adenomatous polyps among endoscoped individuals. Cancer Epidemiol Biomarkers Prev. 1995;4:703–707. [PubMed] [Google Scholar]
- 3.Thun MJ. NSAID use and decreased risk of gastrointestinal cancers. Gasteroenterol Clin North Am. 1996;25:333–348. doi: 10.1016/s0889-8553(05)70250-8. [DOI] [PubMed] [Google Scholar]
- 4.DuBois RN, Giardiello FM, Smalley WE. Nonsteroidal anti-inflammatory drugs, eicosanoids, and colorectal cancer prevention. Gastroenterol Clin North Am. 1996;25:773–791. doi: 10.1016/s0889-8553(05)70274-0. [DOI] [PubMed] [Google Scholar]
- 5.Giardiello FM, Hamilton SR, Krush AJ, Piantadosi S, Hylind L, Celano P, Booker SV, Robinson CR, Offerhaus GJ. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med. 1993;328:1313–1316. doi: 10.1056/NEJM199305063281805. [DOI] [PubMed] [Google Scholar]
- 6.Giardiello FM, Offerhaus JA, Tersmette AC, Hylind LM, Krush AJ, Brensinger JD, Booker SV, Hamilton SR. Sulindac induced regression of colorectal adenomas in familial adenomatous polyposis: evaluation of predictive factors. Gut. 1996;38:578–581. doi: 10.1136/gut.38.4.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duggan DE, Hooke KF, Risley E, Shen TY, Arman CG. Identification of the biologically active form of sulindac. J Pharmacol Exp Ther. 1977;201:8–13. [PubMed] [Google Scholar]
- 8.Vane JR, Botting RM. Mechanism of action of nonsteroidal anti-inflammatory drugs. Am J Med. 1998;104:2S–8S. doi: 10.1016/s0002-9343(97)00203-9. [DOI] [PubMed] [Google Scholar]
- 9.Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–1188. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 10.Sheng H, Shao J, Kirkland SC, Isakson P, Coffey RJ, Morrow J, Beauchamp RD, DuBois RN. Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J Clin Invest. 1997;99:2254–2259. doi: 10.1172/JCI119400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piazza GA, Rahm AL, Krutzsch M, Sperl G, Paranka NS, Gross PH, Brendel K, Burt RW, Alberts DS, Pamukcu R. Antineoplastic drugs sulindac sulfide and sulfone inhibit cell growth by inducing apoptosis. Cancer Res. 1995;55:3110–3116. [PubMed] [Google Scholar]
- 12.Thompson HJ, Jiang C, Lu J, Mehta RG, Piazza GA, Paranka NS, Pamukcu R, Ahnen DJ. Sulfone metabolite of sulindac inhibits mammary carcinogenesis. Cancer Res. 1997;57:267–271. [PubMed] [Google Scholar]
- 13.Shiff SJ, Qiao L, Tsai LL, Rigas B. Sulindac sulfide, an aspirin-like compound, inhibits proliferation, causes cell cycle quiescence, and induces apoptosis in HT-29 colon adenocarcinoma cells. J Clin Invest. 1995;96:491–503. doi: 10.1172/JCI118060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanif R, Pittas A, Feng Y, Koutsos MI, Qiao L, Staiano-Coico L, Shiff SI, Rigas B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem Pharmacol. 1996;52:237–245. doi: 10.1016/0006-2952(96)00181-5. [DOI] [PubMed] [Google Scholar]
- 15.Chan TA, Morin PJ, Vogelstein B, Kinzler KW. Mechanisms underlying nonsteroidal antiinflammatory drug-mediated apoptosis. Proc Natl Acad Sci USA. 1998;95:681–686. doi: 10.1073/pnas.95.2.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herrmann C, Block C, Geisen C, Haas K, Weber C, Winde G, Moroy T, Muller O. Sulindac inhibits Ras signaling. Oncogene. 1998;17:1769–1776. doi: 10.1038/sj.onc.1202085. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto Y, Yin MJ, Lin KM, Gaynor RB. Sulindac inhibits activation of the NF-kappaB pathway. J Biol Chem. 1999;274:27307–27314. doi: 10.1074/jbc.274.38.27307. [DOI] [PubMed] [Google Scholar]
- 18.He TC, Chan TA, Vogelstein B, Kinzler KW. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99:335–345. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gebelein B, Fernadez-Zapico M, Imoto M, Urrutia R. KRAB-independent suppression of neoplastic cell growth by the novel zinc finger transcription factor KS1. J Clin Invest. 1998;102:1911–1919. doi: 10.1172/JCI1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cox A, Der CJ. Biological assays for cellular transformation. Methods Enzymol. 1994;238:277–294. doi: 10.1016/0076-6879(94)38026-0. [DOI] [PubMed] [Google Scholar]
- 21.Xu N, Bradley L, Ambdukar I, Gutkind JS. A mutant alpha subunit of G12 potentiates the eicosanoid pathway and is highly oncogenic in NIH 3T3 cells. Proc Natl Acad Sci USA. 1993;90:6741–6745. doi: 10.1073/pnas.90.14.6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins LR, Minden A, Karin M, Brown JH. Galpha12 stimulates c-Jun NH2-terminal kinase through the small G proteins Ras and Rac. J Biol Chem. 1996;29:17349–17353. doi: 10.1074/jbc.271.29.17349. [DOI] [PubMed] [Google Scholar]
- 23.Buhl AM, Johnson NL, Dhanasekaran N, Johnson GL. G alpha 12 and G alpha 13 stimulate Rho-dependent stress fiber formation and focal adhesion assembly. J Biol Chem. 1995;270:24631–24634. doi: 10.1074/jbc.270.42.24631. [DOI] [PubMed] [Google Scholar]
- 24.Dermott JM, Reddy MR, Onesime D, Reddy EP, Dhanasekaran N. Oncogenic mutant of Galpha12 stimulates cell proliferation through cycloxygenase-2 signaling pathway. Oncogene. 1999;18:7185–7189. doi: 10.1038/sj.onc.1203345. [DOI] [PubMed] [Google Scholar]
- 25.Morrison DK. Mechanisms regulating Raf-1 activity in signal transduction pathways. Mol Reprod Dev. 1995;42:507–514. doi: 10.1002/mrd.1080420420. [DOI] [PubMed] [Google Scholar]
- 26.Gardner AM, Vaillancourt RR, Johnson GL. Activation of mitogen-activated protein kinase/extracellular signal-regulated kinase by G protein and tyrosine kinase oncoproteins. J Biol Chem. 1993;268:17896–17901. [PubMed] [Google Scholar]
- 27.Ellis C, Moran M, McCormick F, Pawson T. Phosphorylation of GAP and GAP-associated proteins by transforming and mitogenic tyrosine kinases. Nature. 1990;343:377–381. doi: 10.1038/343377a0. [DOI] [PubMed] [Google Scholar]
- 28.Nagao M, Kaziro Y, Itoh H. The Src family tyrosine kinase is involved in Rho-dependent activation of c-Jun N-terminal kinase by Galpha12. Oncogene. 1999;18:4425–4434. doi: 10.1038/sj.onc.1202832. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Saez R, Leal MA, Chan AM. Synergism between two growth regulatory pathways: cooperative transformation of NIH3T3 cells by G alpha 12 and c-raf-1. Oncogene. 1996;12:2377–2383. [PubMed] [Google Scholar]
- 30.Fromm C, Coso O, Montaner S, Xu N, Gutkind JS. The small GTP-binding protein Rho links G protein-coupled receptors and Galpha12 to the serum response element and to cellular transformation. Proc Natl Acad Sci USA. 1997;94:10098–10103. doi: 10.1073/pnas.94.19.10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jo H, Zhang R, Zhang H, McKinsey TA, Shao J, Beauchamp RD, Ballard DW, Liang P. NF-kappa B is required for H-ras oncogene induced abnormal cell proliferation and tumorigenesis. Oncogene. 2000;19:841–849. doi: 10.1038/sj.onc.1203392. [DOI] [PubMed] [Google Scholar]
- 32.Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 33.Perona R, Montaner S, Saniger L, Sanchez-Perez I, Bravo R, Lacal JC. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997;11:463–475. doi: 10.1101/gad.11.4.463. [DOI] [PubMed] [Google Scholar]
- 34.Montaner S, Perona R, Saniger L, Lacal JC. Multiple signalling pathways lead to the activation of the nuclear factor kappaB by the Rho family of GTPases. J Biol Chem. 1998;273:12779–12785. doi: 10.1074/jbc.273.21.12779. [DOI] [PubMed] [Google Scholar]
- 35.Swanson BN, Boppana VK, Vlasses PH, Holmes GI, Monsell K, Ferguson RK. Sulindac disposition when given once and twice daily. Clin Pharmacol Ther. 1982;32:397–403. doi: 10.1038/clpt.1982.178. [DOI] [PubMed] [Google Scholar]