

Abstract
The retroviral cyclin protein (rv-cyclin) of walleye dermal sarcoma virus contains two known functional domains, a cyclin box motif and a carboxy terminal transcription activation domain (AD). The AD contacts TATA-binding protein-associated factor 9 (TAF9), and this action is necessary for both positive and negative regulation of transcription from host and viral promoters. Negative regulation occurs via interference with TAF9 binding by transcriptional activators. Transcription factors that share a functional TAF9-binding motif include NF-κB. Rv-cyclin down regulates NF-κB-dependent transcription, whether induced by TNFαor by direct phosphorylation of IκB by expressed MEKK1. In rv-cyclin-expressing cells NF-κB p65 is phosphorylated and translocates to the nucleus, where it forms heterodimers with p50 and binds NF-κB response elements. Furthermore, interference with NF-kB is dependent upon an intact TAF9-binding motif in rv-cyclin. The outcome of this NF-κB down regulation is likely to be important in the control of virus replication and tumorigenesis.
INTRODUCTION
Walleye dermal sarcoma virus (WDSV) is a complex retrovirus etiologically associated with dermal sarcomas in walleye fish (Sander vitreus) (Martineau et al., 1992; Martineau et al., 1991; Walker, 1969; Yamamoto, Kelly, and Nielsen, 1985; Yamamoto et al., 1976). The seasonal nature of dermal sarcoma is characterized by low levels of spliced accessory gene transcripts, A1 and B, during tumor development and high levels of full-length and spliced transcripts during tumor regression (Bowser et al., 1988; Bowser and Wooster, 1991; Bowser et al., 1996; Martineau et al., 1991; Quackenbush et al., 1997; Rovnak, Casey, and Quackenbush, 2001). Infectious virus is only found in regressing tumors.
The A1 transcript encodes a retroviral cyclin (rv-cyclin, Orf A protein), which contains a cyclin box motif and a transcription activation domain (AD) (LaPierre, Casey, and Holzschu, 1998; Rovnak et al., 2005). Rv-cyclin localizes in the nucleus where it is associated with active transcription complexes and with cofactors of transcription including components of the Mediator complex (Rovnak, Casey, and Quackenbush, 2001; Rovnak et al., 2005; Rovnak and Quackenbush, 2002; Rovnak and Quackenbush, 2006). Rv-cyclin inhibits transcription from the WDSV promoter in luciferase reporter systems, and mutations within the AD diminish this activity (Rovnak et al., 2005; Rovnak and Quackenbush, 2002; Zhang and Martineau, 1999). The rv-cyclin AD directly interacts with TATA-binding protein-associated factor 9 (TAF9) (Rovnak and Quackenbush, 2006). The herpes simplex virus transcription factor, VP16, also binds TAF9, and rv-cyclin blocks VP16/TAF9 interaction both physically and functionally (Choi, Asada, and Uesugi, 2000; Rovnak and Quackenbush, 2006; Uesugi et al., 1997). In addition to VP16 and rv-cyclin, seven other transcription factors are known to contain the conserved TAF9 binding motif, FXXØØ, one of which is the NF-κB subunit, p65 (Choi, Asada, and Uesugi, 2000; Uesugi and Verdine, 1999). NF-κB regulates a wide array of genes involved in inflammatory, anti-apoptotic, and immune responses. The NF-κB family is comprised of five members, p50, p52, p65 (Rel A), Rel B, and c-Rel (recently reviewed in (Hayden and Ghosh, 2008). Each member contains a Rel homology domain (RHD) near the N-terminus of the protein that is responsible for the formation of homo- and heterodimers, nuclear localization, and DNA binding. p65, Rel B, and c-Rel have transcription activation domains located in their C-terminus. Inactive NF-κB is sequestered in the cytoplasm in a complex with IκB. Upon exposure of cells to a diverse array of stimuli, IκB is phosphorylated and targeted for degradation resulting in the release of NF-κB from the complex. NF-κB is post-translationally modified and translocates to the nucleus where it binds to promoter sequences of target genes to activate their transcription.
Most viruses have evolved strategies to influence the NF-κB signaling pathway (recently reviewed in Hiscott et al., 2006). Transcription of some viruses, such as human immunodeficiency virus (HIV) and cytomegalovirus (CMV), is dependent on NF-κB activation and binding to NF-κB consensus sites in the viral promoters. Persistent activation of the NF-κB pathway in Epstein Barr virus (EBV) infection and transduction of c-rel by an avian retrovirus are associated with tumor formation. Many viruses encode proteins that inhibit NF-κB and interfere with the innate immune response. Inhibition of NF-κB signaling by viral proteins may occur at numerous steps in the transduction pathway. For example, adenovirus type 12 E1A inhibits phosphorylation of NF-κB, and African swine fever virus protein, A238L, prevents acetylation of p65 (Granja, Perkins, and Revilla, 2008; Guan, Jiao, and Ricciardi, 2008).
We reasoned that rv-cyclin could interfere with NF-κB-dependent transcription via its TAF9 binding motif as observed previously with VP16 transcriptional activation (Rovnak et al., 2005; Rovnak and Quackenbush, 2006). In this study, we show that the rv-cyclin AD functions to inhibit NF-κB p65-dependent transcription.
RESULTS
rv-cyclin inhibits NF-κB-dependent transcription in both HeLa and walleye cell lines
Previous studies showed that rv-cyclin affects transcription from various promoters and that this regulation is due, in part, to the interaction of the rv-cyclin AD with TAF9 (Rovnak et al., 2005; Rovnak and Quackenbush, 2002; Rovnak and Quackenbush, 2006). The AD of NF-κB p65 has also been reported to bind TAF9 (Buss et al., 2004; Lu and Levine, 1995; Uesugi and Verdine, 1999). Therefore, we evaluated the effects of rv-cyclin on NF-κB-dependent transcription.
HeLa cells and the walleye cell line, W12, were co-transfected with an NF-κB luciferase reporter vector (pNF-κB-luc) and a plasmid encoding constitutively active MEKK1 (pFC-MEKK). MEKK1 phosphorylates IκBα, targeting it for degradation, which results in the release of NF-κB (Lee et al., 1997). Over-expression of MEKK1 resulted in 182- and 5-fold activation of transcription from the NF-κB reporter construct in HeLa and W12 cells, respectively. No increased activation was observed in cells transfected with control vector, pFC (Fig. 1A and B). However, in cells co-transfected with a plasmid encoding rv-cyclin (pKH3-rv-cyclin) and MEKK1, significant inhibition of NF-κB-dependent transcription in HeLa cells (11.8 fold) and W12 cells (5.8 fold) was evident (P ≤ 0.001) (Fig. 1A and B).
Figure 1. WDSV rv-cyclin inhibits NF-κ B-dependent transcription in HeLa and walleye cells.
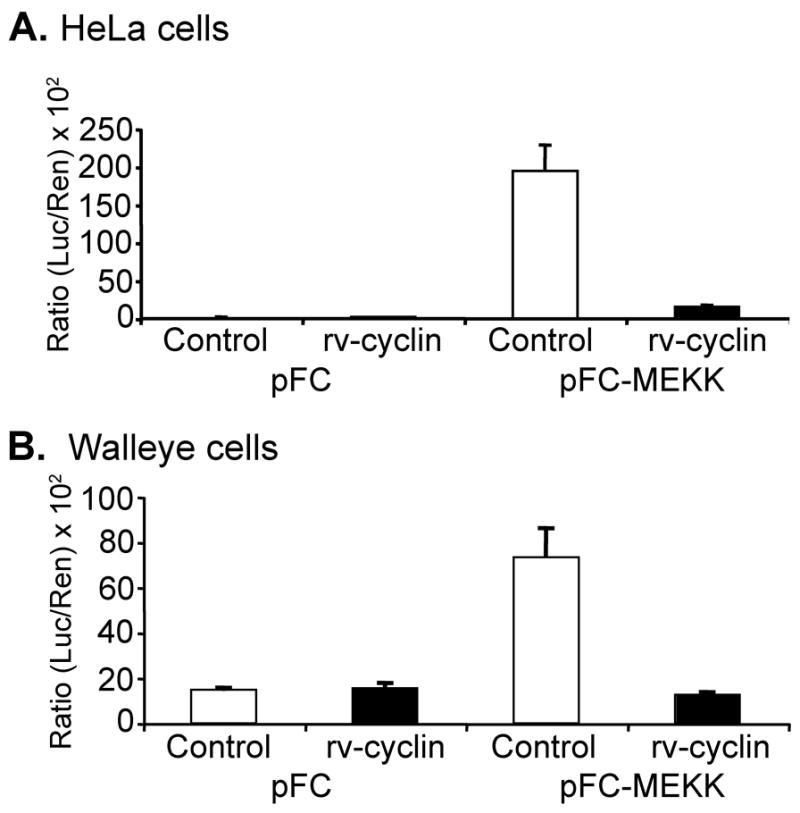
HeLa cells (A) and walleye W12 cells (B) were co-transfected with NF-κB-luciferase reporter vector (pNF-κB-luc), control or MEKK expression vectors (pFC or pFC-MEKK), and control or rv-cyclin expression vectors (pKH3 (white bars) or pKH3-rv-cyclin (black bars)). A Renilla expression vector with a thymidine kinase promoter (pRL-Tk) was also included. Results are reported as mean ± standard deviation of the ratio of luciferase activity versus Renilla activity from triplicate wells. The data presented in this figure are representative of four independent experiments.
To further evaluate the effect of rv-cyclin on NF-κB-dependent transcription, cells were treated with tumor necrosis factor alpha (TNFα), a known inducer of NF-κB signaling cascade. Treatment of HeLa cells with TNFα resulted in activation of luciferase expression from the NF-κB reporter, and again expression of the rv-cyclin significantly reduced NF-kB-dependent transcription 2.2 fold (P ≤ 0.001) (Fig. 2).
Figure 2. WDSV rv-cyclin inhibits NF-κB transcription after TNFα treatment.
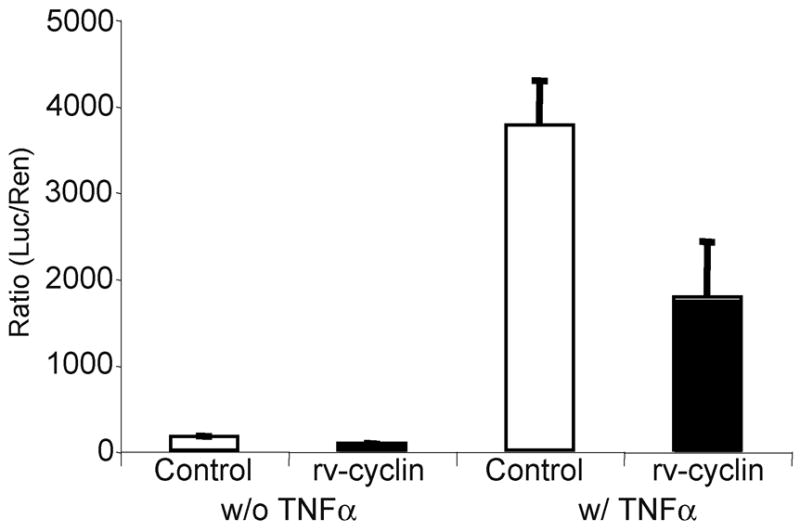
HeLa cells were co-transfected with a NF-κB-luciferase reporter vector (pNF-κB-luc), a control expression vector, pKH3 (white bars), or rv-cyclin expression vector, pKH3-rv-cyclin (black bars), and Tk promoter-Renilla vector (pRL-Tk). Cells were treated with TNFα at 24 hrs post-transfection and harvested 5 hrs later. Results are reported as mean ± standard deviation of the ratio of luciferase activity versus Renilla activity from quadruplicate wells.
WDSV rv-cyclin does not affect phosphorylation or nuclear localization of p65
Treatment of cells with TNFα activates the IKK complex resulting in phosphorylation and degradation of IκBα NF-κB p65 is then phosphorylated and translocated to the nucleus. Studies by Buss et al (Buss et al., 2004) showed that binding of NF-κB p65 to TAF9 was favored by phosphorylation of serine 536 (p65-Ser536).
We established an rv-cyclin-inducible cell line (HeLa Tet-Off/myc-rv-cyclin). Uninduced and induced cells were either left untreated or were treated with 15 ng/ml of TNFα. Whole cell lysates were assayed by western blot with a phospho-specific NF-κB p65 antibody, which showed that p65 was phosphorylated on residue Ser536 after TNFα treatment regardless of whether rv-cyclin was present (Fig. 3A). Likewise, analysis of nuclear extracts from TNFα-treated cells demonstrated successful nuclear translocation of p65 in the presence or absence of rv-cyclin (Fig. 3B). Together, these data exclude rv-cyclin interference with Ser536 phosphorylation and nuclear translocation as mechanisms of transcription inhibition.
Fig. 3. WDSV rv-cyclin does not affect phosphorylation or nuclear translocation of NF-κB p65.
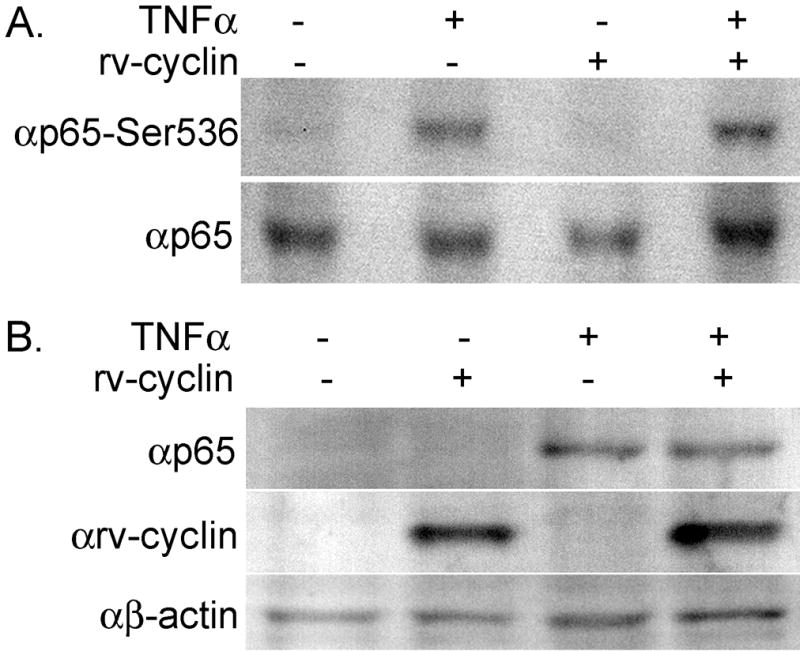
(A) Whole cell lysates from cells with or without induced rv-cyclin expression and untreated or treated with 15 ng/ml TNFα were examined by western blot. Blots were probed with the indicated antibodies: anti-p65 and anti-p65-Ser536 (phosphoserine 536). (B) Nuclear extracts from untreated or TNFα-treated cells with or without induced rv-cyclin expression were probed for p65, β-actin and rv-cyclin.
WDSV rv-cyclin does not affect binding of NF-κB to DNA
Electrophoretic mobility shift assays (EMSA) were employed to evaluate whether rv-cyclin inhibition of NF-κB-dependent transcription was due to an affect on NF-κB-DNA binding. Nuclear extracts from cells with or without induced rv-cyclin expression and TNFα treatment (Fig. 3B) were incubated with a κB specific probe, and the resulting complexes were separated on a non-denaturing polyacrylamide gel. Very little binding to the κB-specific probe was observed with nuclear extracts from cells without TNFα treatment (Fig. 4A, lanes 2 and 3). In contrast, nuclear extracts from cells treated with TNFα with or without induced rv-cyclin expression formed a prominent protein-DNA complex (Fig. 4A, lanes 4 and 5). The specificity of protein binding to the NF-κB probe was demonstrated by competition with a 100-fold molar excess of unlabeled NF-κB probe (Fig. 4A, lanes 6 and 8). Competition of nuclear extract binding to the NF-κB probe was not observed with an unlabeled Oct1 competitor probe (Fig. 4A, lanes 7 and 9). To assess the composition of the NF-κB binding complex, formed after TNFα stimulation, nuclear extracts were incubated with normal rabbit serum or with polyclonal antiserum against p65, p50, or rv-cyclin. When antibody to NF-κB subunit, p50 or p65, was added to nuclear extracts, the formation of a supershifted protein-DNA complex was observed (Fig. 4B, lanes 1, 2, 5, 6). Addition of anti-rv-cyclin or normal rabbit serum did not cause a supershift of the NF-κB-DNA complex (Fig. 4B, lanes 3, 4, 7, 8). Anti-rv-cyclin sera is effective in binding the native rv-cyclin protein (Rovnak, Casey, and Quackenbush, 2001; Rovnak et al., 2007; Rovnak and Quackenbush, 2002; Rovnak and Quackenbush, 2006). These results indicate that the complex formed on the κB probe contains a p50/p65 heterodimer and further demonstrate that rv-cyclin is not associated with the NF-κB that is bound to DNA and is not interfering with NF-κB binding to DNA.
Fig. 4. WDSV rv-cyclin does not affect binding of NF-κB to DNA.
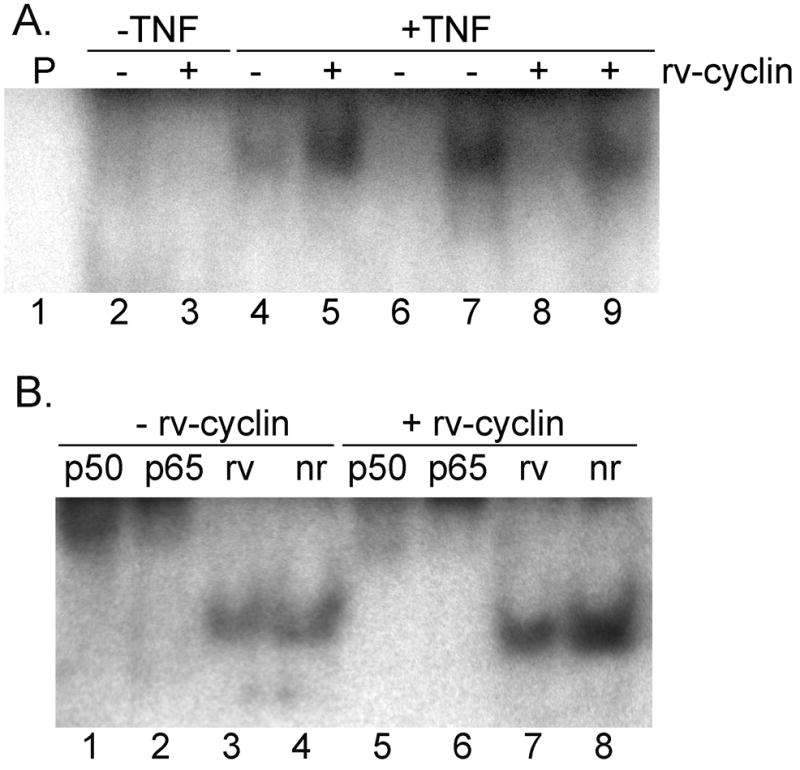
(A) Nuclear extracts from untreated or from cells treated with TNFα and with or without induced rv-cyclin expression were incubated with a 32P-labeled oligonucleotide containing a κB binding site (lanes 2–9), and the reactions separated on a 6% nondenaturing polyacrylamide gel for autoradiography. Lane 1 contains oligonucleotide probe (P) alone. Unlabeled NF-κB probe (competitor probe) was added to the reactions in lanes 6 and 8. Unlabeled Oct1 probe (nonspecific competitor probe) was added to the reactions in lanes 7 and 9. (B) Nuclear extracts from cells treated with TNFα and with or without induced rv-cyclin were incubated with the κB binding site probe and anti-NF-κB p50 (p50) (lanes 1 & 5), anti-NF-κB p65 (p65) (lanes 2 & 6), anti-rv-cyclin (rv) (lanes 3 & 7) or normal rabbit serum (nr) (lanes 4 & 8). Reaction products were separated on a 6% gel for autoradiography.
Mutations within the AD of rv-cyclin alter the inhibitory effects of NF-κB-dependent transcription
Previous studies demonstrated that mutations within the AD of rv-cyclin affected the function of rv-cyclin and its protein-protein interactions (Rovnak et al., 2005; Rovnak and Quackenbush, 2006). To assess whether these AD mutations altered the effects of rv-cyclin on NF-κB-dependent transcription, HeLa cells were co-transfected with the NF-κB-luc reporter vector and pFC-MEKK1 and with either wild type rv-cyclin (WT) or rv-cyclin containing mutations within the TAF9-binding motif at valine 260, V260F or V260S. V260F is a conservative mutation that maintains TAF9 binding and rv-cyclin function, and V260S is a loss-of-function mutation (Rovnak and Quackenbush, 2006). The wild type rv-cyclin significantly inhibited NF-κB-dependent luciferase activity to 12% of the activity found in cells that do not express rv-cyclin (control value set at 100% activity) (P ≤ 0.01) (Fig. 5). Rv-cyclin with the conservative V260F mutation significantly reduced luciferase activity to 33% of control values (P ≤ 0.01) (Fig. 5). In contrast, rv-cyclin with the V260S mutation had no significant effect on NF-κB-dependent transcription. These results suggest that the rv-cyclin TAF9-binding motif is primarily responsible for the inhibitory effects on NF-κB-dependent transcription.
Fig. 5. Mutation of rv-cyclin AD at Val260 affects the inhibition of NF-κB-dependent transcription.

HeLa cells were co-transfected with a NF-κB-luciferase reporter vector (pNF-κB-luc), pFC or pFC-MEKK constructs, and control vector, pKH3 (white bars), or pKH3-rv-cyclin wild type (WT) or rv-cyclin AD mutations (V260F or V260S) (black bars). A Renilla expression vector with a thymidine kinase promoter (pRL-Tk) was also included. Results are reported as mean ± standard deviation of the ratio of luciferase activity versus Renilla activity from quadruplicate wells. The data presented in this figure are representative of two independent experiments.
Expression of rv-cyclin AD interferes with the activation domain of NF-κB p65
When fused to the GAL4 DNA binding domain (DBD), the p65 activation domain (p65AD416–551) activates transcription from a GAL4 luciferase reporter vector (pFR-luc) (Fig. 6A & B, white bars). The isolated rv-cyclin AD was localized to the nucleus by fusion to a nuclear localization sequence (nls) in an expression construct, pKH3nls-rv-cyclin240–270 (Rovnak and Quackenbush, 2006). Co-expression of the rv-cyclin AD in cells with the GAL4DBD-p65AD416–551 construct resulted in a significant reduction in luciferase activity compared to the activity in cells without rv-cyclin expression and titration of pKH3nls-rv-cyclin240–270 demonstrated specific inhibition of the activity of GAL4DBDp65AD416–551 (P ≤ 0.001, 0.0 μg rv-cyclin240–270 vs 0.2 μg rv-cyclin240–270; P ≤ 0.01, 0.0 μg rv-cyclin240–270 vs 0.1 μg rv-cyclin240–270) (Fig. 6A).
Fig. 6. The rv-cyclin AD interferes with the activation of transcription by GAL4-p65AD.
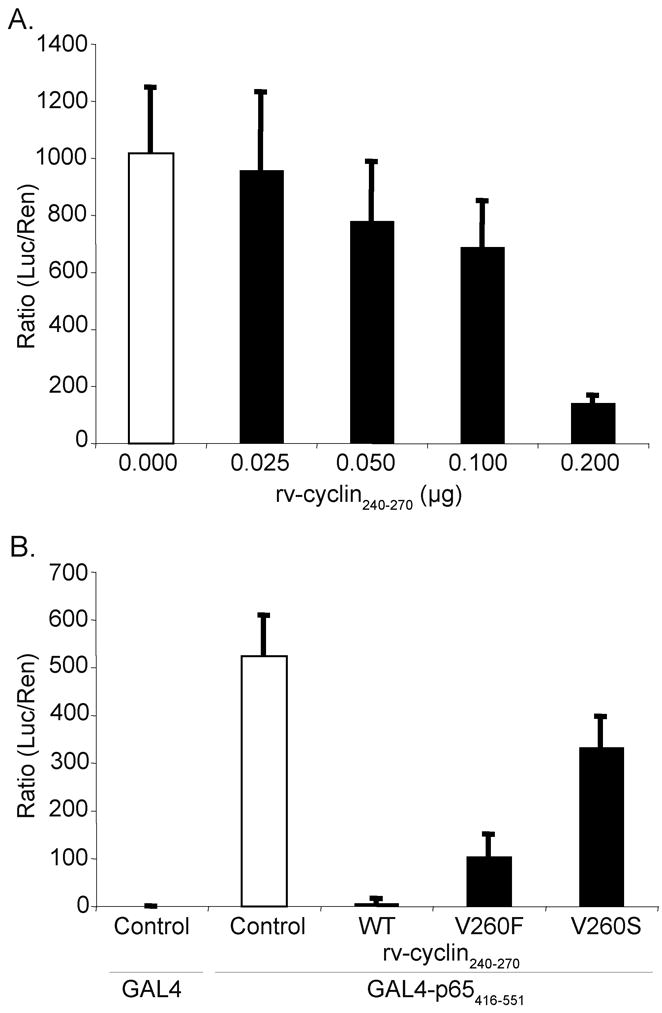
(A) HeLa cells were transfected with a GAL4-luciferase reporter construct (pFR-luc), a vector expressing a GAL4-DNA binding domain fusion with NF-kB p65 AD amino acids 416–551 (pGAL4-p65416–551), and a control expression vector, pKH3 (white bars), or the indicated quantities of expression construct pKH3-rv-cyclin240–270 (black bars) and pRL-Tk. Results are reported as ratios of luciferase versus Renilla activity. (B) HeLa cells transfected with GAL4 reporter vector (pFR-luc), pGAL4-p65416–551 fusion construct, and control expression vector, pKH3 (white bars), pKH3-rv-cyclin240–270 wild type (WT), or pKH3-rv-cyclin240–270 mutations (V260F or V260S) (black bars) and pRL-Tk. Results are reported as mean ± standard deviation of the ratio of luciferase activity versus Renilla activity from quadruplicate wells. The data presented in this figure are representative of two independent experiments.
Further evaluation of the rv-cyclin AD’s inhibitory effect on p65 activation of transcription was tested with mutations V260F and V260S. AD-V260F inhibited GAL4DBD-p65AD activity to 20% of control values (Fig. 6B). AD-V260S reduced the ability of the wild type rv-cyclin AD to inhibit expression from the GAL4-luciferase reporter construct to 64% of control. These results demonstrate that the rv-cyclin AD is responsible for the inhibition of function of the p65 AD and support a mechanism for inhibition that is dependent upon a critical residue within the TAF9-binding motif of the rv-cyclin AD.
Over-expression of TAF9 partially restores NF-κB dependent transcription in the presence of rv-cyclin
The data suggests that the TAF9-binding motif of the rv-cyclin is functional in the inhibition of NF-κB-dependent transcription, and we hypothesize that such inhibition is based on interference with necessary NF-κB interaction with TAF9 via a common TAF9-binding motif. Over-expression of TAF9 should reverse rv-cyclin inhibition of NF-κB dependent transcription by occupying the rv-cyclin TAF9-binding motif. However, excess TAF9 would be expected to bind both NF-κB and rv-cyclin at and away from transcription sites, thereby leading to overall inhibition of NF-κB-induced transcription with increasing TAF9 concentrations. This effect was observed in control preparations co-transfected with a TAF9 expression vector, pKH3TAF9. There was however an apparent relief of the rv-cyclin-mediated inhibition of NF-κB dependent transcription (1.9-fold increase, P ≤ 0.08, 0.0 μg TAF9 vs 0.013 μg TAF9; 2.5-fold increase, P ≤ 0.01, 0.0 μg TAF9 vs 0.05 μg TAF9) (Fig. 7). These results further support the role of the rv-cyclin TAF9-binding motif in the inhibition of transcription promotion by other factors, which share this motif.
Fig. 7. Over-expression of TAF9 partially restores NF-κB dependent transcription in the presence of rv-cyclin.

HeLa cells were co-transfected with a NF-κB-luciferase reporter vector (pNF-κB-luc), pFC or pFC-MEKK constructs, and control vector, pKH3 (white bars), or pKH3-rv-cyclin wild type (WT) and the indicated quantities of expression construct pKH3-hTAF9. A Renilla expression vector with a thymidine kinase promoter (pRL-Tk) was also included. Results are reported as mean ± standard deviation of the ratio of luciferase activity versus Renilla activity from quadruplicate wells. The data presented in this figure are representative of two independent experiments.
DISCUSSION
These studies indicate that WDSV has developed a strategy to manipulate the NF-κB signaling pathway by interfering with the function of the p65 activation domain. Rv-cyclin inhibited an NF-κB reporter gene when cells were treated with TNFα or when MEKK1 was over-expressed. Binding of TNFα to its receptor leads to IKK activation, and activated MEKK1 results in activation of the NF-κB pathway by phosphorylation of IκBα (Lee et al., 1997; Zhao and Lee, 1999). Phosphorylation of IκBα results in its degradation and the release of NF-κB. NF-κB is then phosphorylated and translocates to the nucleus where it binds to target gene promoters. Viruses encode proteins that affect NF-κB signaling at multiple positions in the pathway such as blocking IKK activation or inhibiting IκB degradation or NF-κB phosphorylation or DNA binding (recently reviewed in (Hiscott et al., 2006). Increased levels of phosphorylated NF-κB p65 were found in lysates from cells, with or without rv-cyclin, after treatment with TNFα, and EMSA demonstrated that rv-cyclin did not affect the formation of NF-κB p50/p65 heterodimers on a κB-specific probe.
Posttranslational modifications of NF-κB p65 regulate its binding to IκB and its binding to κB sites in promoters and interactions with coactivators and repressors (Hayden and Ghosh, 2008). Phosphorylation at serine residues 276 or 536 enhances the interaction of p65 with the histone acetyltransferases, p300 and CBP, and results in acetylation of lysine 310, which increases its transcriptional activity (Chen et al., 2005). We focused our attention on residue 536, because NF-kB p65 phosphorylated at serine 536 participates in binding to TAF9 (Buss et al., 2004), and the WDSV rv-cyclin protein binds TAF9 (Rovnak and Quackenbush, 2006). Buss et al. (Buss et al., 2004) suggest that the hydrogen bond formed between Ser536 and Asp533 regulates binding to either TAF9 or to the protein, amino terminal enhancer of split (AES); Upon phosphorylation of Ser536 the hydrogen bond is disrupted, and TAF9 binding is favored. In the absence of Ser536 phosphorylation, p65 preferentially binds AES, and AES represses NF-κB transcriptional activity (Tetsuka et al., 2000). Exposure of cells to TNFα with or without rv-cyclin, resulted in phosphorylation of p65 Ser536, indicating that p65 is in the correct context for binding to TAF9. A similar mechanism, of phospho-regulation of binding to TAF9 or to a co-repressor, has been shown with p53 and the co-repressor MDM2 (Jabbur et al., 2002; Uesugi and Verdine, 1999). MDM2 binding to the FXXΦΦ motif in p53 prevents binding to TAF9. Phosphorylation of p53 at residue Thr18 disrupts a hydrogen bond between Thr18 and Asp21 and attenuates MDM2 interaction to facilitate recruitment of TAF9 (Jabbur et al., 2002; Kussie et al., 1996; Uesugi and Verdine, 1999).
The phosphorylation of p65 at Ser536 and its subsequent interaction with TAF9 has been shown to be required for transcription of interleukin 8 (Buss et al., 2004), but recently, Sasaki et al. (Sasaki et al., 2007) showed that phosphorylation of p65 Ser536 was not required for expression of Traf1. Their data suggested that the distance of the κB response element from the transcription start site influences which NF-κB-responsive genes are induced by phosphorylated and non-phosphorylated p65. It is anticipated that this higher degree of NF-κB gene regulation will influence the number and type of NF-κB-responsive cellular genes that can be affected by rv-cyclin.
The wild type rv-cyclin AD and the conservative V260F AD mutation inhibited the transcriptional activity of a GAL4-p65 AD fusion protein, while the V260S AD mutation greatly reduced these inhibitory effects. These results are similar to those previously observed with herpesvirus VP16 and with the WDSV promoter (Rovnak et al., 2005; Rovnak and Quackenbush, 2002; Rovnak and Quackenbush, 2006). Interaction of rv-cyclin AD V260S with purified TAF9 was reduced in pull down assays, suggesting that the inhibitory effect of rv-cyclin on NF-κB-dependent transcription is due to competition for binding to TAF9. However, the reductions of inhibition in the case of the conservative V260F mutation (Figs. 5 and 6B) were unexpected based on previous results, which demonstrated enhanced TAF9 binding with this mutation (Rovnak and Quackenbush, 2006) and enhanced inhibition of the WDSV promoter in walleye fibroblasts (Rovnak et al., 2005). This variation indicates that factors in addition to TAF9 interaction may be influencing the rv-cyclin inhibition of NF-κB-induced transcription. We previously showed that a GST-rv-cyclin AD fusion protein pulls down p300 and CBP from nuclear extracts and could compete with VP16C for binding to p300 (Rovnak et al., 2005; Rovnak and Quackenbush, 2006). It is possible that rv-cyclin binds directly to p300/CBP and in turn blocks or alters interaction with p65 and affects its activity. An example of such a mechanism is the African swine fever virus protein, A238L, which binds p300/CBP and inhibits NF-κB acetylation and transcriptional activity (Granja, Perkins, and Revilla, 2008). Pursuit of such interactions will be the subject of future investigations.
These studies focused on the role of the rv-cyclin AD in NF-κB-dependent transcription. However, we cannot rule out the contribution of the cyclin box motif and its affect on transcription. Rv-cyclin interacts with cyclin-dependent kinase 8 (cdk8), a component of the Mediator complex, (Rovnak et al., 2005; Rovnak and Quackenbush, 2002)(Rovnak unpublished). The rv-cyclin/cdk8 interaction may affect the presence or absence of the cdk8 module of Mediator at a specific promoter and affect the eventual outcome, i.e. inhibition or activation of transcription. The HIV promoter serves as an example of cdk8 association with gene regulation; Studies by Kim et al. (Kim et al., 2006) demonstrate that in cells harboring HIV, which has an NF-κB-regulated promoter, the cdk8 module of Mediator is lost upon activation of transcription. The combination of cdk8 function in the Mediator complex with TAF9 interference may further influence rv-cyclin’s effects on individual promoters. The work presented here extends our previous observations and suggests that the rv-cyclin AD, through binding to TAF9, inhibits the binding of select transcription factors with TAF9-dependent activation domains.
Activation of the NF-κB pathway is essential for the initiation of an immune response, and is often a target for viral interference (Hiscott et al., 2006). Type I interferon is the major antiviral defense mechanism of the innate immune response in mammals and fish. Viral infection of host cells leads to the activation of transcription factors, NF-κB and interferon regulatory factor (IRF), which are required for optimal activation of IFN promoters. Type I IFN genes have been identified in several species of fish and function in ways similar to mammalian type I IFNs (Robertsen, 2006; Whyte, 2007). We have tested a zebrafish type I IFN promoter reporter (zIFN-luc) in walleye cells and found that rv-cyclin inhibited luciferase expression after induction with poly I:C treatment (data not shown). These results support the hypothesis that the WDSV rv-cyclin functions to inhibit the innate immune response in infected cells. We have shown previously that the rv-cyclin also inhibits transcription from the WDSV promoter in walleye fibroblasts (Rovnak and Quackenbush, 2002). This combination of immune interference and suppression of viral antigen expression during the period of tumor growth suggests a concerted effort to evade an effective immune response to tumor development.
MATERIALS AND METHODS
Cells and generation of rv-cyclin inducible cell line
HeLa cells were maintained in Dulbecco’s modified Eagle medium at 37°C in 5% CO2. W12 cells were maintained in Liebowitz 15 media at 20°C in closed flasks. All media was supplemented with 10% fetal bovine calf sera (Gibco), 4 mM glutamine, 100 units penicillin mL−1, and 100 μg streptomycin mL−1.
The inducible rv-cyclin cell line was generated using the Tet-Off Gene Expression System (BD Biosciences). WDSV rv-cyclin was sub-cloned from the pKH3-OrfA vector into the pTRE-Myc vector at HindIII and XbaI sites after modification of termini of the open reading frame by PCR. The HeLa Tet-Off cell line (BD Biosciences) was co-transfected with pTRE-Myc-rv-cyclin and pTK-Hyg plasmids and selected with G418 and hygromycin. Individual colonies were expanded and screened for inducible expression of myc-tagged rv-cyclin when cultured in the absence of doxycycline.
Plasmid constructs
All rv-cyclin clones have been described previously (Rovnak 2001, 2005 and 2006). The p65 AD (a.a. 416–551) was amplified by RT-PCR from HeLa cell RNA with forward primer, CGGGATCCGGCCCTCCTCAGGCTGTGGC, and reverse primer, GGAATTCTTAGGAGCTGATCTGACTCAG, incorporating BamH1 and EcoR1 restriction sites, respectively. After restriction digestion, amplicons were cloned into the GAL4 DNA binding domain fusion vector, pFA-CMV (Stratagene). Full-length human TAF9 was amplified by RT-PCR from HeLa cell RNA and cloned into the expression construct pKH3 (pKH3-hTAF9). All constructs were confirmed by sequence analysis.
Reporter-gene constructs and luciferase assays
Cells seeded in 24-well plates were transfected with 0.3 μg of the pNF-κB-Luc vector (Stratagene), 0.01 μg (W12) or 0.0001 μg (HeLa) of pRL-TK plasmid (Promega), 0.05 μg pFC or pFC-MEKK (Stratagene), and 0.05 μg pKH3 or pKH3-rv-cyclin wild type or pKH3-rv-cyclin V260F or V260S using FuGENE6 (Roche) according to the manufacturers’ suggestions. NF-κB-Luc contains five tandem repeats of the κB consensus site upstream of the luciferase gene. pFC-MEKK expresses constitutively activated MEKK1. Experiments were performed using the dual-luciferase reporter assay system (Promega). Cell lysates were harvested at 48 hr (HeLa) or 72 hr (W12) post-transfection with passive lysis buffer and centrifuged at 20,000 × g for 5 min at 4°C. For TNFα stimulation, 15 ng/ml of TNFα was added 24 hrs after transfection with pNF-κB-luc, pRL-TK, and pKH3 or pKH3-rv-cyclin, and luciferase activity was measured 5 hrs later. Luciferase activities were obtained with a Beckman Coulter LD400 luminometer. Luciferase activity from the reporter vector was normalized for transfection efficiency with values from the co-transfected pRL-TK vector. All transfections were performed in triplicate or quadruplicate. The data is presented as the mean ± standard deviation of the ratio of luciferase activity versus Renilla activity. Student’s t test and 95% confidence intervals based on a t distribution were used for statistical analyses. A P value of less than 0.01 was considered significant.
For assays of GAL4-DBD fusion constructs, cells were transfected with 0.3 μg of pFR-Luc, 0.025 μg or pFA-CMV or pFA-p65AD416–551, and 0.2 μg pKH3nls, pKH3nls-rv-cyclin240–270, pKH3nls-rv-cyclin240–270V260S, or pKH3nls-rv-cyclin240–270V260F using FuGENE6 (Roche) according to the manufacturers’ suggestions. The pFR-Luc reporter vector (Stratagene) contains five tandem repeats of the yeast GAL4 DNA binding site upstream of the luciferase gene. Lysates were harvested 48 hrs after transfection and luciferase activity was measured as above.
Nuclear extracts and gel mobility shift assays
HeLa Tet-Off myc-tagged rv-cyclin cells were cultured in the presence (uninduced expression) or absence (induced expression) of 2 μg/ml doxycycline and either left untreated or treated with 20 ng/ml TNFα for 10 min. Nuclear extracts were prepared by hypotonic lysis and KCl extraction of nuclei as previously described (Dignam, Lebovitz, and Roeder, 1983). A double-stranded NF-kB specific oligonucleotide probe (5′ AGTTGAGGGGACTTTCCCA GGC 3′) (Promega) was end-labeled with γ-32P ATP and T4 polynucleotide kinase (New England Biolabs). 3.5 μg of nuclear extract and 1 μg poly (dI:dC) (GE Healthcare) were added to EMSA reaction buffer (10 mM HEPES pH 7.9, 50 mM KCl, 2.5 mM DTT, 0.1 mM EDTA, 0.05% NP-40, and 10% (v/v) glycerol) in a total volume of 20 μl and incubated for 10 min at room temperature. 20,000 cpm of labeled oligonucleotide probe was then added, and incubated for another 20 min. Where indicated, 2 μg of antibody was added after the incubation with probe, and the reaction was further incubated at 4°C for 1 hr. Samples were then separated on a 6% DNA Retardation Gel (Invitrogen) in 0.5X TBE (45 mM Tris-base pH 8.3, 45 mM Boric acid, 1 mM EDTA). The gels were run at 150 V for 45 min, dried and placed on x-ray film at −80°C. A double-stranded Oct 1 oligonucleotide (5′ TGTCGAATGCAAATCACTAGAA 3′) (Promega) was used as a nonspecific competitor for EMSA.
Antibodies
Anti-myc (clone 9E10, sc-40), anti-p65 (H-286X), and anti-p50 (sc7178X) were supplied by Santa Cruz Biotechnology, Inc.. Anti-NF-kB p65 (#3034), anti-phospho-NF-kB p65 Ser 536 (#3036S) and κ-Actin (#4967) were from Cell Signaling Technology. Anti-Orf A antisera for mobility shift assays and preimmune sera were previously described (Rovnak, Casey, and Quackenbush, 2001).
Acknowledgments
This work was supported by a National Institute of Health grant CA095056 from the National Cancer Institute. We thank Carol H. Kim (University of Maine) for the zebrafish interferon promoter reporter construct.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bowser PR, Wolfe MJ, Forney JL, Wooster GA. Seasonal prevalence of skin tumors from walleye (Stizostedion vitreum) from Oneida Lake, New York. Journal of Wildlife Diseases. 1988;24:292–298. doi: 10.7589/0090-3558-24.2.292. [DOI] [PubMed] [Google Scholar]
- Bowser PR, Wooster GA. Regression of dermal sarcoma in adult walleyes (Stizostedion vitreum) Journal of Aquatic Animal Health. 1991;3:147–150. [Google Scholar]
- Bowser PR, Wooster GA, Quackenbush SL, Casey RN, Casey JW. Comparison of fall and spring tumors as inocula for experimental transmission of walleye dermal sarcoma. Journal of Aquatic Animal Health. 1996;8:78–81. [Google Scholar]
- Buss H, Dorre A, Schmitz ML, Hoffmann E, Resch K, Kracht M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-kB at serine 536 is mediated by multiple protein kinases including IkB (IKK)-a, IKKb, IKKe, TRAF family member-associated (TANK)=binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediate interleukin-8 transcription. The Journal of Biological Chemistry. 2004;279:55633–55643. doi: 10.1074/jbc.M409825200. [DOI] [PubMed] [Google Scholar]
- Chen LF, Williams SA, Mu Y, Nakano H, Duerr JM, Buckbinder L, Greene WC. NF-κB RelA phosphorylation regulates RelA acetylation. Molecular and Cellular Biology. 2005;25:7966–7975. doi: 10.1128/MCB.25.18.7966-7975.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Asada S, Uesugi M. Divergent hTAFII31-binding motifs hidden in activation domains. J Biol Chem. 2000;275(21):15912–6. doi: 10.1074/jbc.275.21.15912. [DOI] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Research. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granja AG, Perkins ND, Revilla Y. A238L inhibits NF-ATc2, NF-kB, and c-Jun acitvation through a novel mechanism involving protein kinase C-ø-mediated up-regulation of the amino-terminal transactivation domain of p300. The Journal of Immunology. 2008;180:2429–2442. doi: 10.4049/jimmunol.180.4.2429. [DOI] [PubMed] [Google Scholar]
- Guan H, Jiao J, Ricciardi RP. Tumorigenic adenovirus type 12 E1A inhibits phosphorylation of NF-kB by PKAc, causing loss of DNA binding and transactivation. Journal of Virology. 2008;82:40–48. doi: 10.1128/JVI.01579-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Hiscott J, Nguyen TL, Arguello M, Nakhaei P, Paz S. Manipulation of the nuclear factor-kB pathway and the innate immne response by viruses. Oncogene. 2006;25:6844–6867. doi: 10.1038/sj.onc.1209941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbur JR, Tabor AD, Cheng X, Wang H, Uesugi M, Lozano G, Zhang W. Mdm-2 binding and TAFII31 recruitment is regulated by hydrogen bond disruption between the p53 resiudes Thr18 and Asp21. Oncogene. 2002;21:7100–7113. doi: 10.1038/sj.onc.1205856. [DOI] [PubMed] [Google Scholar]
- Kim YK, Bourgeois CF, Pearson R, Tyagi M, West MJ, Wong J, Wu SY, Chiang CM, Karn J. Recruitment of TFIIH to the HIV LTR is a rate limiting step in the emergence of HIV from latency. The EMBO Journal. 2006;25:3596–3604. doi: 10.1038/sj.emboj.7601248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussie PH, Goria S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich PH. Structure of the MDM2 oncoprotein bound to the p53 tumor supressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- LaPierre LA, Casey JW, Holzschu DL. Walleye retroviruses associated with skin tumors and hyperplasias encode cyclin D homologs. Journal of Virology. 1998;72:8765–8771. doi: 10.1128/jvi.72.11.8765-8771.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FS, Hagler J, Chen ZJ, Maniatis T. Activation of the IκBα kinase complex by MEKK1, a kinase of the JNK pathway. Cell. 1997;88:213–222. doi: 10.1016/s0092-8674(00)81842-5. [DOI] [PubMed] [Google Scholar]
- Lu H, Levine AJ. Human TAFII31 protein is a transcriptional coactivator of the p53 protein. Proc Natl Acad Sci, USA. 1995;92:5154–5158. doi: 10.1073/pnas.92.11.5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau D, Bowser PR, Renshaw RR, Casey JW. Molecular characterization of a unique retrovirus associated with a fish tumor. Journal of Virology. 1992;66(1):596–599. doi: 10.1128/jvi.66.1.596-599.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau D, Renshaw R, Williams JR, Casey JW, Bowser PR. A large unintegrated retrovirus DNA species present in a dermal tumor of walleye Stizostedion vitreum. Diseases of Aquatic Organisms. 1991;10:153–158. [Google Scholar]
- Quackenbush SL, Holzschu DL, Bowser PR, Casey JW. Transcriptional analysis of walleye dermal sarcoma virus (WDSV) Virology. 1997;237:107–112. doi: 10.1006/viro.1997.8755. [DOI] [PubMed] [Google Scholar]
- Robertsen B. The interferon system of teleost fish. Fish & Shellfish Immunology. 2006;20:172–191. doi: 10.1016/j.fsi.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Rovnak J, Casey JW, Quackenbush SL. Intracellular targeting of walleye dermal sarcoma virus Orf A (rv-cyclin) Virology. 2001;280:31–40. doi: 10.1006/viro.2000.0731. [DOI] [PubMed] [Google Scholar]
- Rovnak J, Casey RN, Brewster CD, Casey JW, Quackenbush SL. Establishment of productively infected walleye dermal sarcoma explant cells. Journal of General Virology. 2007;88(9):2583–2589. doi: 10.1099/vir.0.82967-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovnak J, Hronek BW, Ryan SO, Cai S, Quackenbush SL. An activation domain within the walleye dermal sarcoma virus retroviral cyclin protein is essential for inhibition of the viral promoter. Virology. 2005;342(2):240–51. doi: 10.1016/j.virol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovnak J, Quackenbush SL. Walleye dermal sarcoma virus cyclin interacts with components of the Mediator complex and the RNA polymerase II holoenzyme. Journal of Virology. 2002;76:8031–8039. doi: 10.1128/JVI.76.16.8031-8039.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovnak J, Quackenbush SL. Walleye dermal sarcoma virus retroviral cyclin directly contacts TAF9. Journal of Virology. 2006;80(24):12041–12048. doi: 10.1128/JVI.01425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki CY, Slemenda CF, Ghosh P, Barberi TJ, Longo DL. Traf1 induction and protection from tumor necrosis factor by nuclear factor-κB p65 is independent of serine 536 phosphorylation. Cancer Research. 2007;67:11218–11225. doi: 10.1158/0008-5472.CAN-07-0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsuka T, Uranishi H, Imai H, Ono T, Sonta S-i, Takahashi N, Asamitsu K, Okamoto T. Inhibition of nuclear factor-κB-mediated transcription by association with the amino-terminal enhancer of split, a groucho-related protein lacking WD40 repeats. The Journal of Biological Chemistry. 2000;275:4383–4390. doi: 10.1074/jbc.275.6.4383. [DOI] [PubMed] [Google Scholar]
- Uesugi M, Nyanguile O, Lu H, Levine AJ, Verdine GL. Induced α helix in the VP16 activation domain upon binding to a human TAF. Science. 1997;277:1310–1313. doi: 10.1126/science.277.5330.1310. [DOI] [PubMed] [Google Scholar]
- Uesugi M, Verdine GL. The α-helical FXXΦΦ motif in p53: TAF interaction and discrimination by MDM2. Proc Natl Acad Sci, USA. 1999;96:14801–14806. doi: 10.1073/pnas.96.26.14801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker R. Virus associated with epidermal hyperplasia in fish. National Cancer Institute Monograph. 1969;31:195–207. [PubMed] [Google Scholar]
- Whyte SK. the innate immune response of finfish- A reivew of current knowledge. Fish & Shellfish Immunology. 2007;23:1127–1151. doi: 10.1016/j.fsi.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Kelly RK, Nielsen O. Morphological differentiation of virus-associated skin tumors of walleye (Stizostedion vitreum vitreum) Fish Pathology. 1985;20:361–372. [Google Scholar]
- Yamamoto T, MacDonald RD, Gillespie DC, Kelly RK. Viruses associated with lymphocystis and dermal sarcoma of walleye (Stizostedion vitreum vitreum) Journal Fish Research Board Canada. 1976;33:2408–2419. [Google Scholar]
- Zhang Z, Martineau D. Walleye dermal sarcoma virus: OrfA N-terminal end inhibits the activity of a reporter gene directed by eukaryotic promoters and has a negative effect on the growth of fish and mammalian cells. Journal of Virology. 1999;73(10):8884–8889. doi: 10.1128/jvi.73.10.8884-8889.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Lee FS. Mitogen-activated protein kinase/ERK kinase kinases 2 and 3 activate nuclear factor-κB through IκB kinase-α and IκB kinase-β. The Journal of Biological Chemistry. 1999;274:8355–8358. doi: 10.1074/jbc.274.13.8355. [DOI] [PubMed] [Google Scholar]