

Abstract
The uptake of neurotransmitter by plasma membrane transporters is a principal method for regulating extracellular transmitter levels. Neurotransmitter-mediated signals in turn are able to regulate transporter expression and function. Thus, there is a continual interplay between transporters and the transmitters they transport. Previously we showed that extracellular γ-aminobutyric acid (GABA) increases the expression of the GABA transporter 1 (GAT1) on a time scale of minutes by acting via the transporter to slow transporter internalization. This mechanism requires in part direct tyrosine phosphorylation of the transporter. In the present study we show that the presence of GABA on a longer time scale causes a net decrease in GAT surface expression. The decrease in expression represents the contributions of transporter-mediated up-regulation and a more substantial GABA-receptor-mediated down-regulation. This receptor-mediated down-regulation is the result of both changes in the rates of transporter trafficking and in the number of transporters available for trafficking. As with transporter-mediated regulation of GAT1, the receptor-mediated regulation is associated with changes in the direct phosphorylation of GAT1. These data suggest that multiple pathways, perhaps converging upon mechanisms involving protein phosphorylation, act to regulate GAT1 expression in neurons.
Keywords: GABA receptor, GAT1, recycling, trafficking, uptake
1. Introduction
GABA is the major inhibitory neurotransmitter in the mammalian central nervous system. Following its release, the activity of GABA is terminated primarily via the binding of GABA to high affinity GABA transporters and subsequent transport. There are three subtypes of GABA transporter, GAT1–3, although a fourth, the betaine transporter, also transports GABA. GAT1 is the predominant GABA transporter in neurons (Guastella et al., 1990). The physiological significance of GAT1 is evidenced by both the prolonged decay time of evoked IPSPs in the presence of GAT1 inhibitors in CA1 pyramidal cells (Engel et al., 1998; Roepstorff and Lambert, 1994) and by behavioral changes in GAT1 knockout mice resulting from the inhibition of GABA uptake (Chiu et al., 2005; Jensen et al., 2003). The finding that the transport rate of GAT1 is much slower than the time course of receptor-mediated synaptic transmission suggests that GAT1 may exert its effects by acting as a diffusion sink and sequestering neurotransmitter from receptor binding sites (Mager et al., 1993; Wadiche et al., 1995). Therefore, the number of plasma membrane transporters may contribute to neurotransmitter signaling. In the basal state, GAT1 expression is on the order of 1000 transporters/μm2, with approximately 30 – 50% of them on the plasma membrane, thus providing ample binding sites for released GABA (Chiu et al., 2002; Wang and Quick, 2005).
GAT1 is subject to multiple forms of regulation, many of which alter transporter surface expression in a trafficking-dependent manner. Since GAT1 continuously traffics to and from the plasma membrane (Deken et al., 2003), the regulation can exert its effects via changes in endocytosis and exocytosis rates, and/or the number of transporters available for recycling. Multiple trafficking modulators have been identified. For example, protein kinase C (PKC)-mediated decreases in GAT1 surface expression are due to increases in the rate of GAT1 internalization, with no changes in the number of recycling transporters. In contrast, depletion of extracellular calcium results in a greater than 50% reduction in the number of GAT1 molecules in the recycling pool (Wang and Quick, 2005). The soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein syntaxin 1A binds the cytoplasmic N-terminal tail of GAT1 and results in a decrease in GABA translocation rates (Deken et al., 2000).
Previously, we showed that extracellular GABA increases GAT1 surface expression on a time scale of minutes through a slowing of the GAT1 internalization rate (Bernstein and Quick, 1999). This substrate-mediated regulation acts via a transporter-dependent mechanism and requires direct tyrosine phosphorylation of GAT1 (Whitworth and Quick, 2001). In the present study, we identify a second mechanism by which GAT1 expression is regulated; namely, through a GABAA receptor-dependent process. This down-regulation of GAT1 expression occurs with longer GABA exposure times, is induced by reagents that change neuronal excitability, and correlates with an increase in the serine phosphorylation of GAT1.
2. MATERIALS AND METHODS
2.1. Reagents
Biotinylation reagents were obtained from Pierce Chemical (Rockford, IL). Immunoblotting reagents and [3H]GABA were obtained from Amersham Biosciences Inc (Piscataway, NJ). GAT1 antibody AB1570W and anti-phosphoserine antibodies were obtained from Chemicon International Inc (Temecula, CA). Protease inhibitor cocktail tablets were obtained from Roche Diagnostics GmbH (Mannheim, Germany). Protein A agarose was obtained from Upstate (Lake Placid, NY). Neurobasal medium and B27 supplement were purchased from Invitrogen (Carlsbad, CA). All other reagents were obtained from Sigma-Aldrich (St. Louis, MO).
2.2. Cell culture
Primary cortical neuron cultures were prepared from E18 rat embryos as described previously (Wang and Quick, 2005). Briefly, cortex from the embryos was carefully dissected and minced in ice-cold Earle’s balanced salt solution (EBSS) (CaCl2 0.29, MgCl2 0.10, KCl 0.37, NaCl 9.35, pH 7.4, in g/L) suspended with 100 units papain. Tissue was incubated for 30 min at 37 °C followed by gentle trituration, dilution, and plating onto poly-L-lysine coated plates to a concentration of 106 cells per 60-mm2 dish. Neurons were maintained in Neurobasal medium with B27 supplement and 0.5 mM L-glutamine. Medium was replaced twice weekly. Cultured neurons were assayed at 21 days.
2.3. [3H]GABA uptake assays
Cultured neurons were washed three times in Hank’s balanced salt solution (HBSS): CaCl2 0.14, KCl 0.40, KH2PO4 0.06, MgCl2 0.10, MgSO4 0.10, NaCl 8.06, NaHCO3 0.35, Na2HPO4 0.09, pH 7.4 (in g/L), and allowed to equilibrate for 5 min in the final wash. Drugs were then applied to the HBSS buffer for 15–30 min and removed before [3H]GABA was applied. For long-term treatment, cells were incubated in culture medium containing particular drugs and kept at 37 °C for 24 hr. The concentration of [3H]GABA was 30 nM. In order to minimize changes in transporter expression, the assay time was 5 min. The assay was terminated by quickly rinsing the cells three times with HBSS. Cells were lysed in 0.005% SDS and divided into half. One half was used for measuring protein concentration and the other half was used to determine the amount of [3H]GABA uptake.
2.4. Surface biotinylation
The Standard surface biotinylation experiments were performed as described previously (Whitworth and Quick, 2001). Briefly, following drug treatment, neuron cultures were incubated in HBSS containing 1 mg/ml biotin on ice for 30 min with gentle shaking. Drugs were present throughout all the steps except the 4 °C biotinylation step. The biotin reaction was then quenched with 100 mM glycine dissolved in phosphate-buffered saline/Ca2+/Mg2+ (NaCl 8.06, KCl 0.20, Na2HPO4 1.36, KH2PO4 0.2, CaCl2 0.01, MgCl2 0.20, pH 7.4, in g/L) for 15 min on ice. The cells were lysed with 500 ml radioimmunoprecipitation assay (RIPA) buffer (10 mM Tris-Cl, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, pH 7.4) supplemented with protease inhibitor cocktail on ice for 60 min. One tenth of the cell lysate was saved for the analysis of protein concentrations, and the remaining was mixed with 100 μl avidin-agarose beads at 4 °C overnight. Protein absorbed by the beads was isolated from the remaining cell lysate by three washes with ice-cold RIPA buffer and then eluted from the beads with SDS loading buffer (62.5 mM Tris-Cl, 2% SDS, 100 mM β-mercaptoethanol, pH 6.8). The protein samples were later processed using standard Western blotting procedures.
To measure GAT1 exocyotsis, intact cells were first surface labeled by biotinylation reagents at 4 °C as described above. Cells were then switched to 37 °C in the presence of biotinylation reagents to allow membrane trafficking for various time periods (Wang and Quick, 2005). The remaining steps were the same as described above. The amount of GAT1 that underwent trafficking was determined by measuring the band density at each time point (Tt) and subtracting the amount of transporter protein that initially was found on the plasma membrane (T0). The exoctyosis time constants (τ) were determined by fitting data to the exponential function Tt=T0 + A (1-e (−t/τ)), where A is the saturated amount of exocytic GAT1 that was labeled by biotinylation reagents. The exocytosis rate is the inverse of τexo (1/τexo).
To measure the endocytosis of GAT1, after cells were surface labeled by biotinylation reagents at 4 °C, the extra biotinylation reagents were removed by repeated washes with ice-cold HBSS. Then cells were then switched to 37 °C in the presence of biotinylation reagents for various time periods to allow membrane trafficking. At a given time point, cells were chilled with ice-cold HBSS. Biotin bound to plasma membrane proteins was cleaved by incubating the cells with the membrane impermeant reducing agent 2-mercaptoethane sulfonic acid (MESNA; 250 mM) in ice-cold PBS, pH 8.2 for 25 minutes. Excess MESNA was then oxidized by 5 mg/ml iodoacetic acid for 20 minutes. Cell membrane preparation and protein precipitation were as described above. The rate obtained by fitting the equation Tt=T0 + A (1-e (−t/τ)) is the sum of the exocytosis and endocytosis rates: τsum= τexo + τendo, from which the rate of endocytosis was estimated.
2.5 Immunoprecipitation
After drug treatment, cells were lysed in RIPA buffer supplemented with protease inhibitor cocktail on ice for 60 min. GAT1 in the cell lystate was immunoprecipitated by anti-GAT1 antibody and protein A-agarose at 4 °C overnight. Nonimmune serum was used as a negative control. The protein-antibody-agarose complex was then washed three times with ice-cold RIPA buffer. Protein eluted from the agrose by SDS-loading buffer was subject to Western blotting and probed by anti-phosphoserine or anti-phosphotyrosine antibodies.
To determine the phosphorylation states of surface GAT1, a surface biotinylation assay was first performed on cortical neurons pretreated with either control medium or 10 μM GABA for 24 h. Biotinylated (surface) GAT1 protein was precipitated by avidin-agarose beads and eluted with 10 mM dithiothreitol. Both biotinylated protein and total protein were then incubated with either nonimmune serum or anti-GAT1 antibody at 4 °C for 1 hr before mixing with protein A-agarose at 4 °C overnight. The immunoprecipitates eluted from agarose beads were further resolved by SDS-PAGE and immunoblotted by anti-phosphoserine or anti-phosphotyrosine antibodies.
2.6 Statistical analyses
Statistical analyses were performed using SPSS (SPSS Inc.). Student’s t-test was used to compare differences between two samples. One-way analysis of variance (ANOVA) followed by Tukey’s Honestly Significant Difference test was used to analyze differences across multiple samples.
3. RESULTS
To examine the short-term effect of extracellular GABA on GAT1 function, primary neuronal cultures from embryonic rat cortex were exposed to different concentrations of GABA ranging from 0.1 to 10 μM for 30 min. The results are shown in Fig. 1A and 1B. In the basal state where cells were maintained in normal culture medium without drug treatment, surface GAT1 accounted for 43 ± 5% of the total amount of cellular GAT1. Compared to the basal state, cells treated with exogenous GABA resulted in a dose-dependent, 45% increase in surface GAT1 expression (Fig. 1A). Using actin to quantify protein levels, the total amount of cellular GAT1 remained unchanged. This indicated that the increase in surface expression of GAT1 was due to a relative subcellular redistribution of GAT1 to the plasma membrane. The increase in surface GAT1 was comparable to the increase in [3H]GABA uptake (Fig. 1B). Approximately 90% of the [3H]GABA uptake was mediated through GAT1, as indicated by the inclusion of the GAT1-specific inhibitor SKF89976A (Larsson et al., 1988) during the assay. These results were consistent with our previous findings in cultured hippocampal neurons that endogenously express GAT1 and in cell lines heterologously expressing GAT1(Bernstein and Quick, 1999), indicating that short-term GABA treatment elevates the transport capacity of GAT1 by increasing its surface expression.
Fig. 1.

Extracellular GABA regulates the function of GABA transporter GAT1. A, short-term exposure to exogenous GABA increases surface expression of GAT1. Dissociated cortical neuron cultures were pre-treated with different concentrations of GABA for 30 min at 37 °C before surface biotinylation at 4 °C. Immunoblots show GAT1 and actin immunoreactivity in 0.2 volume of total cell lysate (upper panel) and GAT1 immunoreactivity in one volume of surface fraction (lower panel). The histogram illustrates the quantified results from eight experiments for surface GAT1 (white bar) and total GAT1 immunoreactivity (relative to actin, black bar). Surface GAT1 in the experimental conditions (white bars) that were significantly different from the control are denoted by the asterisk (p < 0.05). B, GABA uptake through GAT1 increases after short-term exposure to exogenous GABA. Cortical neuron cultures were pre-incubated in medium containing different concentrations of GABA for 30 min at 37 °C prior to uptake assay. Drug concentrations (in μM) are shown below the abscissa; the value underlined indicates that the drug was added only during the assay. Data are from five experiments, four wells/condition/experiment. GABA uptake under control conditions was 308 ± 121 fmol/min/mg of protein. GABA uptake that was significantly different from the control is denoted by the asterisk (p < 0.05). C, long-term GABA treatment decreases the surface expression of GAT1. Different concentrations of GABA were applied in culture medium for 24 hr at 37 °C before surface biotinylation. Immunoblots show GAT1 and actin immunoreactivity in 0.1 volume of total cell lysate (upper panel) and GAT1 immunoreactivity in one volume of surface fraction (lower panel). Data are from ten experiments and symbols are presented as in A. D, GABA uptake through GAT1 decreases after long-term exposure to exogenous GABA. Cortical neuron cultures were pre-incubated in medium containing different concentrations of GABA for 24 hr at 37 °C prior to uptake assay. Data are from five experiments, four wells/condition/experiment, and symbols are presented as in B. GABA uptake under control conditions was 315 ± 104 fmol/min/mg of protein. E, time-dependent changes in GAT1 surface expression due to GABA treatment. Dissociated cortical neurons were incubated in culture medium containing 10 μM GABA at 37 °C for different time periods prior to surface biotinylation at 4 °C. The time periods during which GABA was present in the medium are indicated on the abscissa. Data are from six separate experiments.
Next, we treated cultures with exogenous GABA for 24 hr and examined GAT1 subcellular distribution. We found the surface expression of GAT1 was down-regulated to approximately 70% of control in a dose-dependent manner (Fig. 1C). This decrease in GAT1 surface expression correlated well with decreased [3H]GABA uptake in these cells (Fig 1D), consistent with the hypothesis that longer-term GABA application decreases the number of surface GAT1 molecules while the transport efficiency of individual transporters remains unchanged. This decrease in GAT1 surface expression did not occur in the presence of exogenous glutamate or glycine, indicating that the down-regulation in GAT1 surface expression is due to a GABA-mediated process (data not shown). Moreover, since the total amount of cellular GAT1 remained unchanged after 24 hr GABA treatment (Fig. 1C, upper immunoblot), the reduced surface expression of GAT1 under long-term GABA treatment was not due to loss of total cellular GAT1 protein, but to shifts in the GAT1 subcellular distribution.
Taken together, two effects are observed when exogenous GABA is introduced to cultured cortical neurons: increased surface GAT1 expression with 30-minute GABA treatment (Figure 1A open column); and decreased surface GAT1 expression after 24-hour GABA treatment (Figure 1C, grey column). In both situations, the total amount of cellular GAT1 remains unchanged. If these two effects are compared, a difference of 60% – 80% in GAT1 surface expression is revealed. To determine the time course over which there is a switch from surface expression increase to surface expression decrease, we sampled GAT1 surface expression at multiple time intervals. The result is shown in Fig. 1E. Biotin-labeled (surface) GAT1 in the presence of GABA peaked at 15 to 30 min of GABA application and then the surface expression of GAT1 steadily decreased until it reached approximately 70% of baseline after 12–24 hr exposure to exogenous GABA. Further exposure failed to reveal additional decrease in the surface GAT1 expression up to 48 h (data not shown).
The biphasic effect of GABA on GAT1 surface expression suggested that two different mechanisms might be responsible. We have previously shown that the short-term increase in GAT1 surface expression is due to a transporter-mediated mechanism (Whitworth and Quick, 2001). To determine the role of a GAT1-mediated mechanism on long-term expression, we applied nipecotic acid, a GAT1 specific substrate (Solis and Nicoll, 1992), to cortical neurons for 24 hr prior to surface biotinylation. Contrary to the long-term GABA effect, long-term nipecotic acid application induced a dose-dependent increase in the surface expression of GAT1 to approximately 180% of control, similar to that seen with short-term GABA application (Fig. 2A).
Fig. 2.
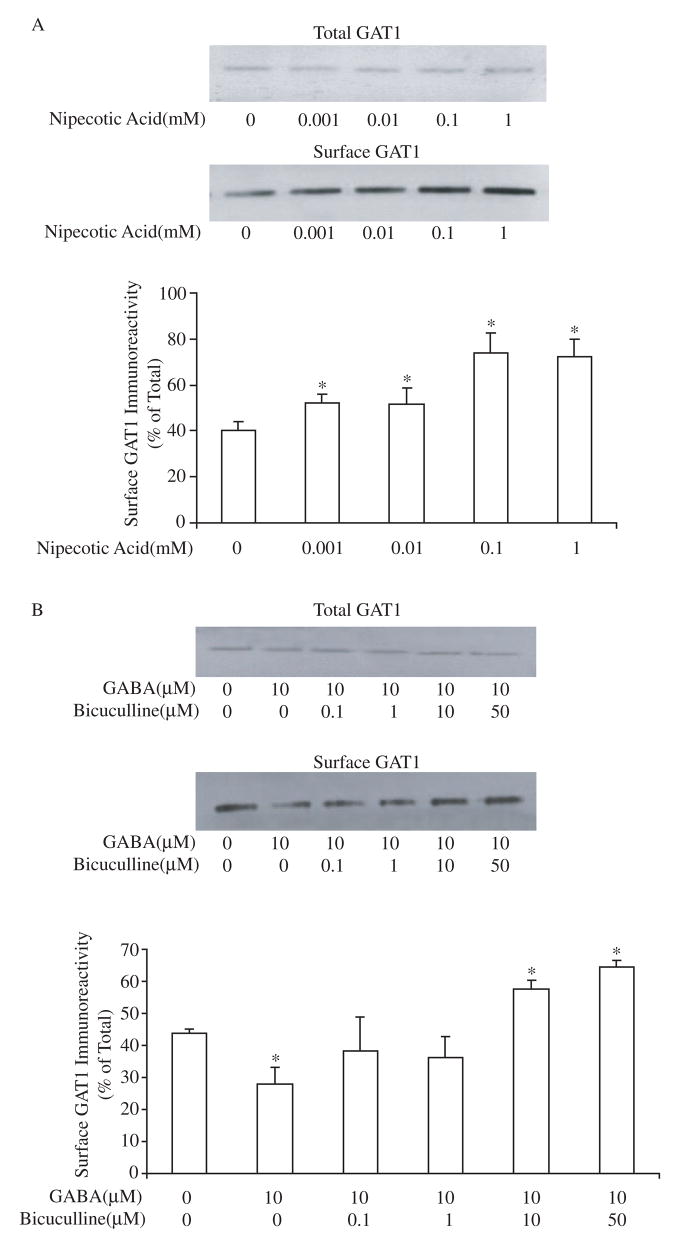
The long-term effect of GABA on GAT1 subcellular distribution is the result of two mechanisms. A, long-term application of nipecotic acid increases the surface expression of GAT1. Different concentrations of nipecotic acid were applied to the culture medium for 24 hr prior to surface biotinylation. The immunoblots of GAT1 show GAT1 immunoreactivity in 0.1 volume of total cell lysate (upper panel) and one volume of surface fraction (lower panel). Experimental conditions with a significant difference from the control are denoted with an asterisk (p<0.05). Data are from four experiments. B, bicuculline reverses long-term GABA-mediated decreases in GAT1 surface expression. Cortical neurons were incubated in 10 μM GABA with or without bicuculline for 24 hr prior to surface biotinylation. The immunoblots of GAT1 show GAT1 immunoreactivity in 0.1 volume of total cell lysate (upper panel) and one volume of surface fraction (lower panel). Data are from five experiments. Asterisks indicate statistical significance (p<0.05) when comparing the drug treatment groups to the control group (no drug treatment).
The nipecotic acid data suggested that the long-term GABA-induced down-regulation effect is likely not due to action of substrates on GAT1 itself. The fact that extracellular GABA but not specific GAT1 substrates decreased GAT1 surface expression at 24 hr suggested that GABA receptors might be involved. To test this hypothesis, neurons were maintained in medium containing 10 μM GABA alone or in combination with different concentrations of bicuculline (bicuculline methiodide), a GABAA receptor inhibitor, prior to measurement of GAT1 surface expression. The results are shown in Fig. 2B. The long-term GABA-induced decrease of GAT1 surface expression was blocked by bicuculline in a dose-dependent manner. Moreover, with the blockade of GABAA receptors by bicuculline, long-term GABA treatment was able to increase the surface fraction of GAT1 to greater than 150%. Bicuculline itself had no effect on GAT1 surface expression (data not shown). These data suggest that two mechanisms work together to determine long-term GABA-mediated expression of GAT1: a transporter-mediated effect that increases surface expression and a receptor-mediated effect that decreases surface expression.
The finding that GABA receptor activation reduced the surface expression of GAT1 without significantly changing total cellular GAT1 (Fig. 1C) suggested that GAT1 trafficking, rather than GAT1 protein expression, was being modulated by GABA. To examine this, we measured the exocytosis and endocytosis rates of GAT1 and estimated the size of the GAT1 recycling pool in both the basal state and after 24 h GABA treatment. These results are shown in Figures 3 and 4. In the basal state, GAT1 underwent robust internalization and reinsertion to the plasma membrane. In examining the exocytosis process, we found that GAT1 continuously trafficked to the cell surface, reaching a plateau such that 35 ± 10% of GAT1 was on the plasma membrane. A single exponential function was used to fit the time course of GAT1 exocytosis and the time constant (τ) of exocytosis was estimated to be 1.43 ± 0.13 min (rate of exocytosis: 0.70 ± 0.06 min−1, Fig. 3A, filled circles). At the same time, GAT1 was rapidly internalized, with an endocytosis time constant of 1.09 ± 0.04 min (rate of endocytosis: 0.92 ± 0.03 min−1, Fig. 3B, filled circles). The ratio of the endocytosis and exocytosis rates (rendo/rexo ≈ 0.92/0.70 ≈ 1.28) explains the GAT1 surface expression in the basal state of approximately 40% ([GAT1intra]/[GAT1surface] ≈ 57%/43% ≈ 1.33). These are consistent with our previous estimates (Wang and Quick, 2005).
Fig. 3.

Long-term GABA treatment alters GAT1 endocytosis and exocytosis rates. A, the exocytosis rate of GAT1. Data from six experiments show the time course of surface GAT1 expression in the basal state (filled circles) and after treatment with GABA for 24 h (open squares). Transporters on the plasma membrane, after incubation for different time periods at 37 °C, were compared to the calibrated standards of total surface GAT1 to quantify exocytosis. Time constant τ1 (basal state) and τ2 (24 hr) were obtained by fitting data to the exponential function Tt=T0+A(1-e (−t/τ). B, the endocytosis rate of GAT1. Transporters internalized after incubating for different time periods at 37 °C are compared to the calibrated standards of total surface GAT1. Data are collected from seven experiments. The values and symbols are as described in A.
Fig. 4.

Changes in the size of the GAT1 recycling pool. A, long-term GABA treatment decreases the size of the GAT1 recycling pool. Transporters resident on the plasma membrane were labeled with biotin at 37 °C for 30 min. Drugs were presented prior to and throughout all biotinylation steps. Immunoblots show the GAT1 immunoreactivity in 0.3 volume of total cellular lysate fraction (upper panel) and one volume of surface fraction (lower panel). Data are from eight experiments. Experimental groups that were significantly different from the control group are indicated by one asterisk; groups that were significantly different (p<0.05) from the group treated with 10 μM GABA alone are indicated by two asterisks (p < 0.05). B, TTX decreases the size of GAT1 recycling pool. Cells were maintained in medium containing TTX (1 μM) and/or K+ (20 mM) for 24 hr before measuring the size of GAT1 recycling pool. Data are from six experiments. Immunoblots show the GAT1 immunoreactivity in 0.2 volume of total cellular lysate fraction (upper panel) and one volume of surface fraction (lower panel). Experimental groups that were significantly different from the control group are indicated by one asterisk; groups that were significantly different from the group treated with 1 μM TTX alone are indicated by two asterisks (p<0.05).
When exogenous GABA was present in the medium for 24 hours, the decrease in GAT1 surface expression was indicated by a significantly reduced exocytosis rate (0.12 ± 0.02 min−1; time constant of 8.16 ± 1.00 min) and decreased maximal surface expression (15 ± 7%) (Fig. 3A, open squares). The rate of GAT1 endocytosis was also reduced significantly, with a time constant of 2.67 ± 0.43 min (rate of endocytosis: 0.38 ± 0.06 min−1, Fig. 3B, open squares). The difference between the endocytosis and exocytosis rates (rendo/rexo ≈ 0.38/0.12 ≈ 3.17) reflects the ratio of intracellular and surface GAT1 ([GAT1intra]/[GAT1surface] ≈ 75%/25% ≈ 3) when cells are treated with GABA for 24 hours.
The fact that long-term GABA treatment differentially interfered with the exocytosis and endocytosis rates indicated that such treatment might also alter the size of GAT1 recycling pool. To estimate the changes in the pool size under drug treatment, we first measured the size of the GAT1 recycling pool in the basal state using modified biotinylation assays (Wang and Quick, 2005). As shown in Fig. 4A, 31 ± 3% of cellular GAT1 molecules continuously trafficked to and from the plasma membrane in the basal state within 30 minutes of biotin labeling. When 10 μM GABA was applied for 24 h, the pool size underwent a significant decrease to 24 ± 1% of control. Bicuculline treatment did not significantly impact the size of GAT1 recycling pool (32 ± 3%). However, when it was co-applied with GABA for 24 h, it prevented the GABA-mediated decrease of the pool size (29 ± 2%).
The reversal of long-term GABA-initiated changes by bicuculline raised the possibility of an activity-mediated mechanism underlying the long-term GABA effect. The size of the GAT1 recycling pool decreased to 21 ± 2% when we applied the Na+ channel blocker TTX to the medium. K+ (20 mM) did not significantly change the pool size (27 ± 2%); however, it prevented the down-regulation effect of TTX on the GAT1 pool size (Fig. 4B).
Previously, we showed that GAT1 can be phosphorylated on either serine or tyrosine residues. Increases in GAT1 serine phosphorylation correlate with a down-regulation of GAT1 surface expression, whereas increases in GAT1 tyrosine phosphorylation correlate with an up- regulation of GAT1 surface expression. The relative abundance of these two mutually exclusive phosphorylation states determines at least in part the subcellular distribution of GAT1 (Quick et al., 2004). Moreover, we found previously that short-term treatment of cultured hippocampal neurons with GAT1 substrates altered GAT1 surface expression on a time scale of minutes, and this effect required direct tyrosine phosphorylation of GAT1 (Whitworth and Quick, 2001). These data suggested that the observed changes in trafficking resulting from long-term GABA treatment might involve alterations in GAT1 serine and tyrosine phosphorylation states as well. To test this hypothesis, co-immunoprecipitation assays were performed to examine the serine and tyrosine phosphorylation states of GAT1 following substrate treatment. As shown in Fig. 5A, long-term exposure to exogenous GABA increased GAT1 serine phosphorylation to 130 ± 14% of control. Bicuculline alone did not significantly alter the serine phosphorylation of GAT1; however, when it was applied in addition to exogenous GABA, bicuculline was able to prevent the GABA-mediated increase in GAT1 serine phosphorylation levels. GAT1 tyrosine phosphorylation remained unchanged after either GABA or bicuculline treatment (data not shown). In order to determine whether changes in GAT1 serine phosphorylation were also associated with substrate-initiated GAT1 internalization, transporter biotinylation assays were performed, followed by serine phosphorylation quantification (Fig. 5B). The immunoprecipitation from the biotinylated fraction represents the amount of serine phosphorylation of surface GAT1, whereas immunoprecipitation from the whole cell lysates represents the amount of serine phosphorylation of total cellular GAT1. In the basal state, approximately 79 ± 6% of GAT1 serine phosphorylation was on surface-resident GAT1. When treated with exogenous GABA for 24 hr, the amount of serine phosphorylation of the total cellular GAT1 increased approximately 1.3 fold such that nearly 100% of surface GAT1 was serine phosphorylated. These data suggest that the GABA-receptor mediated decrease in GAT1 surface expression is related to increases in the serine phosphorylation on surface-resident GAT1.
Fig. 5.

Long-term GABA treatment alters the serine phosphorylation of GAT1. A, representative immunoblots of total GAT1, and serine and tyrosine phosphorylated GAT1, in the basal state or treated with 10 μM GABA for 24 hr. After drug treatment, cells were precipitated by GAT1 antibody and immunoblotted using either phosphoserine or phosphotyrosine antibodies. Quantitative results from eight experiments are shown in the histogram. The amount of serine-phosphorylated (gray bar) and tyrosine-phosphorylated GAT1 (black bar) in the basal state is compared to total cellular GAT1 and normalized to 100%. B, serine-phosphorylation of surface GAT1. Surface GAT1 was isolated by surface biotinylation assay, immunoprecipitated by anti-GAT1 antibodies and probed by phosphoserine antibodies. The immunoblot shows the phosphoserine immunoreactivity of precipitated GAT1 by GAT1 antibodies. The quantified results from six experiments are shown in the histogram. The amount of serine phosphorylated GAT1 in the basal state is compared to total cellular GAT1 and normalized to 100%. The difference between surface serine phosphorylation and total serine phosphorylation of GAT1 was significant (denoted by one asterisk; p < 0.05). When treated with 10 μM GABA for 24 hr, both total serine phosphorylation and surface serine phosphorylation of GAT1 increased significantly from the basal state (denoted by two asterisks; p < 0.05).
4. DISCUSSION
The reliability of synaptic transmission is maintained by the precise coordination of presynaptic and postsynaptic components. By removing transmitter from the synaptic cleft, neurotransmitter transporters contribute to synaptic signaling by fine-tuning its intensity and duration. Neurotransmitter transporter expression is not static. Instead, transporters cycle to and from the plasma membrane on the time scale of seconds to minutes and this cycling is regulated by factors which include extracellular signals (Sattler and Rothstein, 2006; Zahniser and Sorkin, 2004), protein phosphorylation (Foster et al., 2006) and interactions with accessory proteins (Quick, 2006). In the present study, we show that the substrate-mediated regulation of GAT1 trafficking occurs in a time-dependent manner. This regulation of GAT1 trafficking is reflected by changes in exocytosis and endocytosis rates, and in the size of the GAT1 recycling pool. We detail two separate GABA-mediated mechanisms responsible for these changes: (i) GABA transporter-mediated up-regulation of GAT1 surface expression, and (ii) GABA receptor-mediated down-regulation of GAT1 surface expression. On the time scale of minutes, the transporter-mediated up-regulation of GAT1 expression predominates, while on the time scale of hours, the receptor-mediated down-regulation of GAT1 expression predominates. Our current and previous data suggest that these processes trigger changes in the phosphorylation states of GAT1 to control the number of GAT1 molecules at cell surface.
Based upon our current results and previous studies, we propose the following model. In the basal state, GAT1 is inserted into the membrane in a calcium-dependent manner and internalized via a clathrin-dependent process (Deken et al., 2003). With short duration and lower concentrations of GABA, a transporter-mediated process leads to an increase in tyrosine phosphorylation of GAT1 that places transporters in a conformation favoring retention of GAT1 on the plasma membrane (Whitworth and Quick, 2001). This results in a net increase in the surface expression of GAT1 via alterations to the rates of recycling and not to changes in the numbers of recycling GAT1 molecules. In contrast, with longer duration and/or higher GABA concentrations, a receptor-mediated process leads to an increase in serine phosphorylation of GAT1 present on the cell surface. The changes in the phosphorylation state alter GAT1 endocytosis and exocytosis rates, and importantly, reduce the size of the GAT1 recycling pool, leading to a decrease in GAT1 surface expression. This model suggests a feedback mechanism in which synaptic activity affects the trafficking states of GAT1.
The average number of GAT1 molecules in a cultured cortical inhibitory bouton is approximately 3000 – 4000 (Chiu et al., 2002), and the size of the GAT1 recycling pool is approximately 30% of total GAT1 (Wang and Quick, 2005). Therefore, our estimates suggest that about 1000 GAT1 molecules traffic to and from the plasma membrane. Our study of GAT1 trafficking kinetics indicates that in the basal state, GAT1 constitutively traffics with an exocytosis rate slower than the endocytosis rate; this results in two intracellularly-localized transporters for every transporter present on the surface. Using 1000 as an estimate of the recycling pool size, this means there are approximately 330 transporters on the surface and 670 on intracellular recycling vesicles. The current data further show that GABA, acting through a transporter-dependent mechanism, up-regulates surface expression by 45%, meaning that the surface number of GAT1 molecules could be as high as 475 – 500. On the other hand, our data suggest that with regulation of GAT1 trafficking via GABA receptor activation, the size of the recycling pool goes from 1000 molecules in the basal state to 800 molecules. In addition, the change in the trafficking rates results in one transporter on the cell surface for every three that are located intracellularly; that is, approximately 200 GAT1 molecules would be on the surface and 600 on intracellular vesicles. Thus, these two forms of regulation can result in a greater than 2.5-fold change in surface transporter expression.
Receptor-mediated transporter trafficking and subsequent protein phosphorylation are likely to be a common regulatory mechanism. For the insulin-responsive glucose transporter 4 (GLUT4), insulin activates insulin receptor tyrosine kinase to recruit several intracellular effectors, including phosphatidylinositol (PI) 3-kinase (PI3K) to produce phosphoinositide intermediates. They in turn serve as substrates to activate a serine/threonine kinase cascade that alters GLUT4 trafficking (Simpson et al., 2001; Thurmond and Pessin, 2001; Watson and Pessin, 2006). As a result, insulin induces a dramatic increase in GLUT4 exocytosis and a slight decrease in GLUT4 endocytosis, leading to an approximately 10- to 20-fold increase in transporter levels at the plasma membrane (Bryant et al., 2002). Similarly, in response to external signals, GAT1 can be phosphorylated by different protein kinases, which may serve as the sorting cues for different trafficking pathways. For example, protein kinase C activation induces serine/threonine phosphorylation of GAT1 that in turn promotes GAT1 internalization, whereas in the presence of tyrosine kinase activators, the insertion of GAT1 to the plasma membrane is accelerated (Quick et al., 2004). The correlation between changes in phosphorylation states and synaptic localization has also been found in other neurotransmitter transporters, including those for serotonin (Blakely et al., 1998), dopamine (Foster et al., 2006; Mortensen and Amara, 2003), and glutamate (Robinson, 2006; Sattler and Rothstein, 2006). These phosphorylation events may serve as a final common pathway for the multiple signals that have been found to regulate trafficking.
Direct links between the GABAA receptor and serine phosphorylation of GAT1 have yet to be established. Indeed, the specific GABAA receptor inhibitor bicuculline, applied alone, did not significantly change the serine phosphorylation level of GAT1; however, it prevented the GABA-mediated increase in GAT1 serine phosphorylation. One potential mediator between the GABAA receptor and GAT1 serine phosphorylation is PICK1 (protein interacting with C kinase), a member of the PDZ (PSD-95, Discs large, Z0-1) family of binding proteins (Staudinger et al., 1995). PICK1 binds via its single PDZ domain to PKC in its activated form to cluster the kinase in spines where it may phosphorylate α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA)-type glutamate receptor subunit 2 (GluR2) (Perez et al., 2001). This PICK1-mediated phosphorylation of AMPA receptors is one of the principal mechanisms in the induction of LTD, a specific form of synaptic inhibition (Wang and Linden, 2000; Xia et al., 2000). The binding between PICK1 and neurotransmitter transporters is also found in members of the SLC6 transporter family, including transporters for dopamine, serotonin, and norepinephrine (Torres et al., 2001). The PDZ domain of PICK1 recognizes type II PDZ-binding motifs with a sequence pattern of X-φ-X-φ, where X designates any amino acid and φ a hydrophobic residue (Songyang et al., 1997; Madsen et al., 2005). GAT1 possesses this motif at its C-terminus (McHugh et al., 2004). Whether GAT1 directly binds to PICK1 is currently under investigation.
The lack of receptor-mediated regulation of GAT1 surface expression during short-term GABA treatment suggests that the second messenger pathways that regulate this effect operate on the time scale of hours. The evidence that the size of the GAT1 recycling pool is regulated on this time scale, and not at a shorter time scale, suggests that a major function of the receptor-mediated regulation is to determine the available pool of transporters for subsequent short-term trafficking to and from the plasma membrane. We are presently investigating the pathways that operate on this longer time scale. Our data suggest that protein synthesis is not required because the protein level of GAT1 in neurons remains unchanged during 24 hr GABA treatment. We are also investigating the conditions under which the receptor-mediated transporter regulation is likely to occur. Our data suggest that receptor-mediated regulation is likely to be seen with chronic elevations in extracellular GABA. For example, it is known that chronic administration of the anti-epileptic nipecotic acid derivative tiagabine (15 mg/kg, p.o. twice daily for 21 days) to mice significantly decreases [3H]tiagabine binding in various brain areas (Thomsen and Suzdak, 1995). The results of the present study suggest that this decrease in GAT1 surface expression by transporter antagonists may be the result of elevated extracellular GABA concentrations and the subsequent activation of GABAA receptors.
The abbreviations used are
- GABA
γ-aminobutyric acid
- GAT1
γ-aminobutyric acid transporter 1
- PKC
protein kinase C
- SNARE
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- TTX
tetrodotoxin
- GLUT4
glucose transporter 4
- PI3K
phosphatidylinositol 3-kinase
- AMPA
α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bernstein EM, Quick MW. Regulation of gamma-aminobutyric acid (GABA) transporters by extracellular GABA. J Biol Chem. 1999;274:889–895. doi: 10.1074/jbc.274.2.889. [DOI] [PubMed] [Google Scholar]
- Blakely RD, Ramamoorthy S, Schroeter S, Qian Y, Apparsundaram S, Galli A, DeFelice LJ. Regulated phosphorylation and trafficking of antidepressant-sensitive serotonin transporter proteins. Biol Psychiatry. 1998;44:169–178. doi: 10.1016/s0006-3223(98)00124-3. [DOI] [PubMed] [Google Scholar]
- Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol. 2002;3:267–277. doi: 10.1038/nrm782. [DOI] [PubMed] [Google Scholar]
- Chiu CS, Brickley S, Jensen K, Southwell A, Mckinney S, Cull-Candy S, Mody I, Lester HA. GABA transporter deficiency causes tremor, ataxia, nervousness, and increased GABA-induced tonic conductance in cerebellum. J Neurosci. 2005;25:3234–3245. doi: 10.1523/JNEUROSCI.3364-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CS, Jensen K, Sokolova I, Wang D, Li M, Deshpande P, Davidson N, Mody I, Quick MW, Quake SR, Lester HA. Number, density, and surface/cytoplasmic distribution of GABA transporters at presynaptic structures of knock-in mice carrying GABA transporter subtype 1-green fluorescent protein fusions. J Neurosci. 2002;22:10251–10266. doi: 10.1523/JNEUROSCI.22-23-10251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deken SL, Beckman ML, Boos L, Quick MW. Transport rates of GABA transporters: regulation by the N-terminal domain and syntaxin 1A. Nat Neurosci. 2000;3:998–1003. doi: 10.1038/79939. [DOI] [PubMed] [Google Scholar]
- Deken SL, Wang D, Quick MW. Plasma membrane GABA transporters reside on distinct vesicles and undergo rapid regulated recycling. J Neurosci. 2003;23:1563–1568. doi: 10.1523/JNEUROSCI.23-05-01563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel D, Schmitz D, Gloveli T, Frahm C, Heinemann U, Draguhn A. Laminar difference in GABA uptake and GAT-1 expression in rat CA1. J Physiol. 1998;512:643–649. doi: 10.1111/j.1469-7793.1998.643bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JD, Cervinski MA, Gorentla BK, Vaughan RA. Regulation of the dopamine transporter by phosphorylation. Handb Exp Pharmacol. 2006:197–214. doi: 10.1007/3-540-29784-7_10. [DOI] [PubMed] [Google Scholar]
- Guastella J, Nelson N, Nelson H, Czyzyk L, Keynan S, Miedel MC, Davidson N, Lester HA, Kanner BI. Cloning and expression of a rat brain GABA transporter. Science. 1990;249:1303–1306. doi: 10.1126/science.1975955. [DOI] [PubMed] [Google Scholar]
- Jensen K, Chiu CS, Sokolova I, Lester HA, Mody I. GABA transporter-1 (GAT1)-deficient mice: differential tonic activation of GABAA versus GABAB receptors in the hippocampus. J Neurophysiol. 2003:2690–2701. doi: 10.1152/jn.00240.2003. [DOI] [PubMed] [Google Scholar]
- Larsson OM, Falch E, Krogsgaard-Larsen P, Schousboe A. Kinetic characterization of inhibition of gamma-aminobutyric acid uptake into cultured neurons and astrocytes by 4,4-diphenyl-3-butenyl derivatives of nipecotic acid and guvacine. J Neurochem. 1988;50:818–823. doi: 10.1111/j.1471-4159.1988.tb02986.x. [DOI] [PubMed] [Google Scholar]
- Linden DJ, Connor JA. Participation of postsynaptic PKC in cerebellar long-term depression in culture. Science. 1991;254:1656–1659. doi: 10.1126/science.1721243. [DOI] [PubMed] [Google Scholar]
- Madsen KL, Beuming T, Niv MY, Chang CW, Dev KK, Weinstein H, Gether U. Molecular determinants for the complex binding specificity of the PDZ domain in PICK1. J Biol Chem. 2005;280:20539–20548. doi: 10.1074/jbc.M500577200. [DOI] [PubMed] [Google Scholar]
- Mager S, Naeve J, Quick M, Labarca C, Davidson N, Lester HA. Steady states, charge movements, and rates for a cloned GABA transporter expressed in Xenopus oocytes. Neuron. 1993;10:177–188. doi: 10.1016/0896-6273(93)90309-f. [DOI] [PubMed] [Google Scholar]
- Metzger F, Kapfhammer JP. Protein kinase C: its role in activity-dependent Purkinje cell dendritic development and plasticity. Cerebellum. 2003;2:206–214. doi: 10.1080/14734220310016150. [DOI] [PubMed] [Google Scholar]
- Mortensen OV, Amara SG. Dynamic regulation of the dopamine transporter. Eur J Pharmacol. 2003;479:159–170. doi: 10.1016/j.ejphar.2003.08.066. [DOI] [PubMed] [Google Scholar]
- Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB. PICK1 targets activated protein kinase Calpha to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA-type glutamate receptor subunit 2. J Neurosci. 2001;21:5417–5428. doi: 10.1523/JNEUROSCI.21-15-05417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW. The role of SNARE proteins in trafficking and function of neurotransmitter transporters. Handb Exp Pharmacol. 2006:181–196. doi: 10.1007/3-540-29784-7_9. [DOI] [PubMed] [Google Scholar]
- Quick MW, Hu J, Wang D, Zhang HY. Regulation of a gamma-aminobutyric acid transporter by reciprocal tyrosine and serine phosphorylation. J Biol Chem. 2004;279:15961–15967. doi: 10.1074/jbc.M306924200. [DOI] [PubMed] [Google Scholar]
- Robinson MB. Acute regulation of sodium-dependent glutamate transporters: A focus on constitutive and regulated trafficking. Handb Exp Pharmacol. 2006:251–275. doi: 10.1007/3-540-29784-7_13. [DOI] [PubMed] [Google Scholar]
- Roepstorff A, Lambert JD. Factors contributing to the decay of the stimulus-evoked IPSC in rat hippocampal CA1 neurons. J Neurophysiol. 1994;72:2911–2926. doi: 10.1152/jn.1994.72.6.2911. [DOI] [PubMed] [Google Scholar]
- Sattler R, Rothstein JD. Regulation and dysregulation of glutamate transporters. Handb Exp Pharmacol. 2006:277–303. doi: 10.1007/3-540-29784-7_14. [DOI] [PubMed] [Google Scholar]
- Simpson F, Whitehead JP, James DE. GLUT4--at the cross roads between membrane trafficking and signal transduction. Traffic. 2001;2:2–11. doi: 10.1034/j.1600-0854.2001.020102.x. [DOI] [PubMed] [Google Scholar]
- Solis JM, Nicoll RA. Postsynaptic action of endogenous GABA released by nipecotic acid in the hippocampus. Neurosci Lett. 1992;147:16–20. doi: 10.1016/0304-3940(92)90764-x. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- Staudinger J, Zhou J, Burgess R, Elledge SJ, Olson EN. PICK1: a perinuclear binding protein and substrate for protein kinase C isolated by the yeast two-hybrid system. J Cell Biol. 1995;128:263–271. doi: 10.1083/jcb.128.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen C, Suzdak PD. Effects of chronic tiagabine treatment on [3H]GABAA, [3H]GABAB and [3H]tiagabine binding to sections from mice brain. Epilepsy Res. 1995;21:79–88. doi: 10.1016/0920-1211(95)00010-8. [DOI] [PubMed] [Google Scholar]
- Thurmond DC, Pessin JE. Molecular machinery involved in the insulin-regulated fusion of GLUT4-containing vesicles with the plasma membrane (review) Mol Membr Biol. 2001;18:237–245. doi: 10.1080/09687680110082400. [DOI] [PubMed] [Google Scholar]
- Torres GE, Yao WD, Mohn AR, Quan H, Kim KM, Levey AI, Staudinger J, Caron MG. Functional interaction between monoamine plasma membrane transporters and the synaptic PDZ domain-containing protein PICK1. Neuron. 2001;30:121–134. doi: 10.1016/s0896-6273(01)00267-7. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Arriza JL, Amara SG, Kavanaugh MP. Kinetics of a human glutamate transporter. Neuron. 1995;14:1019–1027. doi: 10.1016/0896-6273(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Wang D, Quick MW. Trafficking of the plasma membrane gamma-aminobutyric acid transporter GAT1. Size and rates of an acutely recycling pool. J Biol Chem. 2005;280:18703–18709. doi: 10.1074/jbc.M500381200. [DOI] [PubMed] [Google Scholar]
- Wang YT, Linden DJ. Expression of cerebellar long-term depression requires postsynaptic clathrin-mediated endocytosis. Neuron. 2000;25:635–647. doi: 10.1016/s0896-6273(00)81066-1. [DOI] [PubMed] [Google Scholar]
- Watson RT, Pessin JE. Bridging the GAP between insulin signaling and GLUT4 translocation. Trends Biochem Sci. 2006;31:215–222. doi: 10.1016/j.tibs.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Whitworth TL, Quick MW. Substrate-induced regulation of gamma-aminobutyric acid transporter trafficking requires tyrosine phosphorylation. J Biol Chem. 2001;276:42932–42937. doi: 10.1074/jbc.M107638200. [DOI] [PubMed] [Google Scholar]
- Xia J, Chung HJ, Wihler C, Huganir RL, Linden DJ. Cerebellar long-term depression requires PKC-regulated interactions between GluR2/3 and PDZ domain-containing proteins. Neuron. 2000;28:499–510. doi: 10.1016/s0896-6273(00)00128-8. [DOI] [PubMed] [Google Scholar]
- Zahniser NR, Sorkin A. Rapid regulation of the dopamine transporter: role in stimulant addiction? Neuropharmacology. 2004;47(Suppl 1):80–91. doi: 10.1016/j.neuropharm.2004.07.010. [DOI] [PubMed] [Google Scholar]