

Abstract
Background
We previously showed that mice lacking the high mobility group A1 gene (Hmga1-knockout mice) developed a type 2-like diabetic phenotype, in which cell-surface insulin receptors were dramatically reduced (below 10% of those in the controls) in the major targets of insulin action, and glucose intolerance was associated with increased peripheral insulin sensitivity. This particular phenotype supports the existence of compensatory mechanisms of insulin resistance that promote glucose uptake and disposal in peripheral tissues by either insulin-dependent or insulin-independent mechanisms. We explored the role of these mechanisms in the regulation of glucose homeostasis by studying the Hmga1-knockout mouse model. Also, the hypothesis that increased insulin sensitivity in Hmga1-deficient mice could be related to the deficit of an insulin resistance factor is discussed.
Results
We first show that HMGA1 is needed for basal and cAMP-induced retinol-binding protein 4 (RBP4) gene and protein expression in living cells of both human and mouse origin. Then, by employing the Hmga1-knockout mouse model, we provide evidence for the identification of a novel biochemical pathway involving HMGA1 and the RBP4, whose activation by the cAMP-signaling pathway may play an essential role for maintaining glucose metabolism homeostasis in vivo, in certain adverse metabolic conditions in which insulin action is precluded. In comparative studies of normal and mutant mice, glucagon administration caused a considerable upregulation of HMGA1 and RBP4 expression both at the mRNA and protein level in wild-type animals. Conversely, in Hmga1-knockout mice, basal and glucagon-mediated expression of RBP4 was severely attenuated and correlated inversely with increased Glut4 mRNA and protein abundance in skeletal muscle and fat, in which the activation state of the protein kinase Akt, an important downstream mediator of the metabolic effects of insulin on Glut4 translocation and carbohydrate metabolism, was simultaneously increased.
Conclusion
These results indicate that HMGA1 is an important modulator of RBP4 gene expression in vivo. Further, they provide evidence for the identification of a novel biochemical pathway involving the cAMP-HMGA1-RBP4 system, whose activation may play a role in glucose homeostasis in both rodents and humans. Elucidating these mechanisms has importance for both fundamental biology and therapeutic implications.
Background
Insulin resistance is a metabolic condition found relatively frequently among humans with chronic hyperinsulinemia and in experimental animal models with defective insulin signaling [1-3]. Recently, a link has been established between peripheral insulin sensitivity and the retinol (vitamin A) metabolism, and insulin resistance in rodents and humans has been linked to abnormalities of the vitamin A signaling pathway [4-6]. According to these studies, impaired glucose uptake in adipose tissue results in secondary systemic insulin resistance through release of the adipose-derived serum RBP4 [4,5]. However, it is unknown whether RBP4 effects on insulin sensitivity are vitamin A-dependent or vitamin A-independent. RBP4 (also called RBP) is mainly produced by the liver but also by adipocytes [7]. In plasma, retinol-RBP4 is found in an equimolar complex with transthyretin (TTR), which is a thyroid hormone transport protein that is synthesized in and secreted from the liver. This ternary complex prevents retinol-RBP4 excretion by the kidney [8]. By impairing insulin signaling in muscle, RBP4 inhibits glucose uptake and interferes with insulin-mediated suppression of glucose production in the liver, causing blood glucose levels to rise [4]. Conversely, mice lacking the RBP4 gene show increased insulin sensitivity, and normalizing increased RBP4 serum levels improves insulin resistance and glucose intolerance [4].
HMGA1 is a small basic protein that binds to adenine-thymine (A-T) rich regions of DNA and functions mainly as a specific cofactor for gene activation [9]. HMGA1 by itself has no intrinsic transcriptional activity; rather, it can transactivate promoters through mechanisms that facilitate the assembly and stability of a multicomponent enhancer complex, the so-called enhanceosome, that drives gene transcription [9,10].
As part of an investigation into the molecular basis regulating the human insulin receptor gene, we previously showed that HMGA1 is required for proper insulin receptor gene transcription [11,12]. More recently, we showed that loss of HMGA1 expression, induced in mice by disrupting the HMGA1 gene, caused a type 2-like diabetic phenotype, in which, however, impaired glucose tolerance and overt diabetes coexisted with a condition of peripheral insulin hypersensitivity [13]. Concomitant insulin resistance and insulin hypersensitivity in peripheral tissues may paradoxically coexist as observed in livers of lipodystrophic and ob/ob mice [14], as well as in Cdk4 knockout mice with defective pancreatic beta cell development and blunted insulin secretion [15]. The hypothesis that the paradoxical insulin hypersensitivity of Hmga1-deficient mice could be due to a deficit, in these animals, of RBP4 is supported by our data. Herein, by employing the Hmga1-knockout mouse model, we provide compelling evidence for the identification of a novel biochemical pathway involving HMGA1 and RBP4, whose activation by the cAMP pathway may play an important role in maintaining glucose metabolism homeostasis in vivo, in both rodents and humans. The importance of HMGA1 in RBP4 gene transcription was substantiated in Hmga1-deficient mice, in which loss of HMGA1 expression considerably decreased RBP4 mRNA abundance and RBP4 protein production.
Results
RBP4 gene transcription is induced by HMGA1 and cAMP
We first performed experiments to see whether HMGA1 had a role in activating the mouse RBP4 gene promoter at the transcriptional level. To test this possibility, HepG2 human hepatoma cells and mouse Hepa1 hepatoma cells were cotransfected transiently with mouse RBP4-Luc reporter plasmid plus increasing amounts of the HMGA1 expression vector. As shown in Figure 1, overexpression of HMGA1 considerably increased RBP4-Luc activity in both cell types and this effect occurred in a dose-dependent manner. Consistent with these results, RBP4 mRNA abundance was increased in cells overexpressing HMGA1 and was reduced in cells pretreated with siRNA targeting HMGA1 (Figure 1), indicating that activation of the RBP4 gene requires HMGA1. These data were substantiated by chromatin immunoprecipitation (ChIP) assay, showing that binding of HMGA1 to the endogenous RBP4 locus was increased in whole, intact HepG2 and Hepa1 cells naturally expressing HMGA1, and was decreased in cells exposed to siRNA against HMGA1 (Figure 1). Based on these results, in addition to previous observations indicating that cAMP, or agents which elevate intracellular cAMP, increase RBP transcript levels [16], we were interested to see whether a functional link could be established between cAMP, HMGA1, and RBP4. To this end, we first confirmed and extended the observation made by Jessen and Satre [16] that RBP is induced by cAMP in Hepa1 cells. As measured by Northern blot analysis of total RNA (Figure 2), RBP4 mRNA increased ≈ 5-fold over the basal level in Hepa1 cells treated with 0.5 mM 8-bromo cAMP (Br-cAMP), a standard concentration for c-AMP induction experiments [16]. As shown in Figure 2, RBP4 mRNA levels increased starting at 3 h, peaking at 24 h and then declining, suggesting a transient transcriptional stimulation. To establish whether HMGA1 was required for basal and cAMP-dependent RBP4 transcription, we transfected the HMGA1 expression vector in Hepa1 cells treated or not with Br-cAMP and RBP4 protein levels were analyzed 48 h later by Western blot. As shown in Figure 2, RBP4 protein expression was enhanced in cells overexpressing HMGA1 and even further in cells treated with cAMP, in which an increase in HMGA1 protein expression was simultaneously observed, suggesting that induction of RBP4 by cAMP may occur, at least in part, through activation of endogenous HMGA1 expression. This hypothesis was supported by the fact that RBP4 was reduced in cAMP treated cells in which endogenous levels of HMGA1 were specifically lowered by transfecting cells with HMGA1 antisense expression plasmid (Figure 2). However, further experiments are needed to fully explain the role of cAMP on HMGA1 expression. The functional significance of HMGA1 in RBP4 gene expression was confirmed in transient transcription assays in Hepa1 (and differentiated 3T3-L1, data not shown) cells, in which overexpression of HMGA1 caused an increase in both basal and cAMP-induced Luc activity from the mouse RBP-Luc reporter plasmid (Figure 3). This effect was substantiated in HEK-293 cAMP-responsive cells, a cell line ideally suited for studying the effects of HMGA1 on transcription since it does not express appreciable levels of this protein. As shown in Figure 3, in support of the role that HMGA1 plays in the context of RBP4 gene, the direct effect of cAMP was less effective in promoting RBP4 transcription in HEK-293 cells expressing low levels of HMGA1, becoming considerably higher in cells with forced expression of HMGA1.
Figure 1.

RBP4 gene expression is induced by HMGA1. (Top) Mouse RBP4-Luc reporter vector (2 μg) was transfected into HepG2 and Hepa1 cells plus increasing amounts (0, 0.5, or 1 μg) of HMGA1 expression plasmid. Data represent the means ± standard errors for three separate experiments; values are expressed as factors by which induced activity increased above the level of Luc activity obtained in transfections with RBP4-Luc reporter vector plus the empty expression vector, which is assigned an arbitrary value of 1. (Middle) HMGA1 expression plasmid was transfected into HepG2 and Hepa1cells. After 6 h of transfection, the cells were treated with anti-HMGA1 (100 pmol), siRNA, or a non-targeting control siRNA, and endogenous RBP4 mRNA expression was measured 48 to 96 h later. Western blots of HMGA1 in each condition are shown in the autoradiograms. (Bottom) ChIP of the RBP4 promoter gene in HepG2 and Hepa1 cells, either untreated or pretreated with HMGA1 siRNA. ChIP was done using an anti-HMGA1 specific antibody (Ab).
Figure 2.

Stimulation of RBP4 mRNA and protein expression by cAMP and HMGA1. (Upper left) 20 μg of total RNA from Hepa1 cells treated with the indicated concentrations of Br-cAMP for 24 h (lanes 1–7) were analysed by Northern blot. Hybridization was carried out with an RBP4 cDNA or an 18S RNA probe as a control of the RNA loaded on each lane. (Lower left) 20 μg of total RNA from Hepa1 cells treated with 0.5 mM Br-cAMP for the indicated times were loaded on each lane (lanes 1–8) and analysed as above. (Right) Hepa1 cells, in the absence or presence of an expression plasmid (1 μg) containing the HMGA1 cDNA in either the sense (s) or antisense (as) orientation, were left untreated or treated with Br-cAMP (0.5 mM), total protein extracts were prepared 48 h later and HMGA1 and RBP4 protein expression levels were detected by Western blot (WB) with anti-HMGA1 and anti-RBP4 antibodies, respectively. β-actin, control of cellular protein loading. Densitometric analyses of three to five independent blots are shown.
Figure 3.

Role of HMGA1 in basal and cAMP-induced RBP4 expression. Rbp4-Luc reporter vector and HMGA1 expression plasmid (sense or antisense) were cotransfected into Hepa1 and HEK-293 cells, either untreated or treated with Br-cAMP. Data represent the means ± standard errors for three separate experiments. Transcriptional activity of the RBP4 gene promoter is shown as the ratio of luciferase activity to Renilla activity (Luc/Ren) as described in the experimental procedures. Values are expressed as the factors by which induced activity increased above the level of Luc activity obtained in transfections with the reporter vector alone, which is assigned an arbitrary value of 1. Open bar, mock (no DNA); black bar, pGL3-basic (vector without an insert). Western blots of HMGA1 and β-actin in each condition are shown in the autoradiograms.
Thus, these data together demonstrate that HMGA1 is of major importance for transcriptional regulation of the RBP4 gene, and indicate that a functional link exists between cAMP, HMGA1, and RBP4.
Hmga1-deficient mice have reduced expression of RBP4 in liver and fat tissue and reduced serum RBP4 levels
In the light of the above experimental results, indicating that HMGA1 plays a positive role in RBP4 gene transcription in living cultured cells, it was interesting to analyze the functional consequences of genetic ablation of HMGA1 on RBP4 in vivo, in Hmga1-knockout mice. To this end, we performed studies aimed at investigating the expression of RBP4 mRNA and protein in Hmga1-deficient mice and wild-type controls. As shown in Figure 4, RBP4 mRNA was severely attenuated in both liver and fat from Hmga1-null mice, and reduced by 50% in Hmga1 heterozygous mutants, as assessed by real-time quantitative polymerase chain reaction (qRT-PCR). Reduced RBP4 mRNA levels in liver and adipose tissue paralleled the decrease in RBP4 serum levels as detected by Western blot analysis of serum samples from age- and body weight-matched mice with diverse genotypes (Figure 4), thereby showing the requirement of HMGA1 for full RBP4 expression in whole animals.
Figure 4.
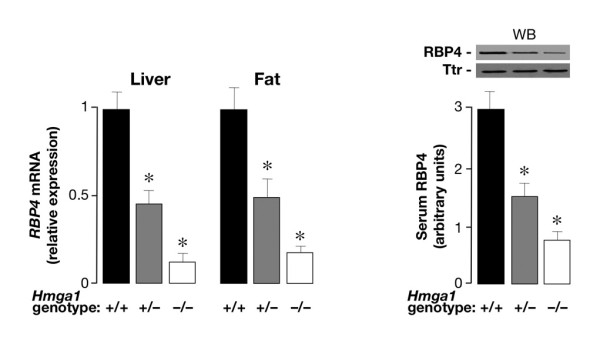
RBP4 expression in wild-type and Hmga1-deficient mice. RBP4 mRNA in liver and fat from control and Hmga1-deficient mice, as measured by qRT-PCR (left), and densitometric quantification of RBP4 serum levels as detected by Western blot (WB) of serum samples (2 μl) from mice with diverse genotypes (right). In WB analysis, an anti-transthyretin (TTR) antibody was used to confirm similar amounts of protein on each lane. Results are from 4–6 mice in each group. *P < 0.01 versus control mice.
HMGA1 and RBP4 expression increase in liver and fat of normal mice after intraperitoneal glucagon injection
Based on our observations in intact cultured cells, indicating a role for the cAMP signaling pathway in HMGA1 and RBP4 gene expression, cAMP-inducible transcriptional activation of the Hmga1 and RBP4 genes was investigated in vivo, in whole animals, by systemic administration of the intracellular cAMP-elevating hormone glucagon. Under these conditions, glucagon-stimulated cAMP responses in terms of both Hmga1 and RBP4 mRNA expression were first analyzed in wild-type control mice. Consistent with our data in Hepa1 cells, Hmga1 and RBP4 mRNA levels significantly increased in liver and fat of normal mice after intraperitoneal injection of glucagon (Figure 5). Time course analyses revealed that the induction and accumulation of Hmga1 mRNA preceded the expression of RBP4 mRNA in both tissues. In liver, RBP4 mRNA appeared after that for Hmga1, peaked after 6 h following glucagon injection, and then remained at a plateau (Figure 5). In fat, RBP4 mRNA appeared at 1 h after Hmga1 mRNA, peaked after 6 h of glucagon stimulation, and decreased smoothly thereafter (Figure 5). Increased levels of Hmga1 and RBP4 mRNAs were paralleled by the increase of Hmga1 and RBP4 protein expression, as measured by Western blot analysis of proteins from liver and fat of glucagon-injected animals (Figure 5). Interestingly, when similar experiments were carried out in glucagon injected Hmga1-deficient mice, tissue expression of RBP4 mRNA was severely attenuated in liver and fat from heterozygous (Hmga1+/-) and Hmga1-null (Hmga1-/-) animals (Figure 6), thereby indicating that HMGA1 is indeed required for maximal induction of the RBP4 gene in vivo, in the whole organism, and that the glucagon/adenylate cyclase system regulates both HMGA1 and RBP4 gene and protein expression. Consistent with this conclusion, liver RBP4 mRNA and protein expression levels were lower in fed wild-type mice, becoming higher during fasting, when circulating glucagon increases (Figure 6).
Figure 5.

Hmga1 and RBP4 mRNA and protein expression in vivo, in glucagon-injected wild-type mice. Total RNA was isolated from liver (upper left) and fat (upper right) of 3-h-fasted mice, before and after intraperitoneal injection of glucagon. Levels of Hmga1 and RBP4 mRNA were measured at the indicated time intervals by qRT-PCR and normalized to RPS9 mRNA abundance, as described in Methods. Results are the mean values ± s.e.m. from 4–6 animals per group. Black bars, Hmga1 mRNA; gray bars, RBP4 mRNA. Representative Western blots from liver (lower left) and fat (lower right) of mice before and after glucagon injection are shown. Densitometric analyses of immunoblots are shown in bar graphs as the mean ± s.e.m. of data from 3–5 mice per each time point. Black bars, HMGA1; gray bars, RBP4.*P < 0.05 versus control mice (time 0).
Figure 6.
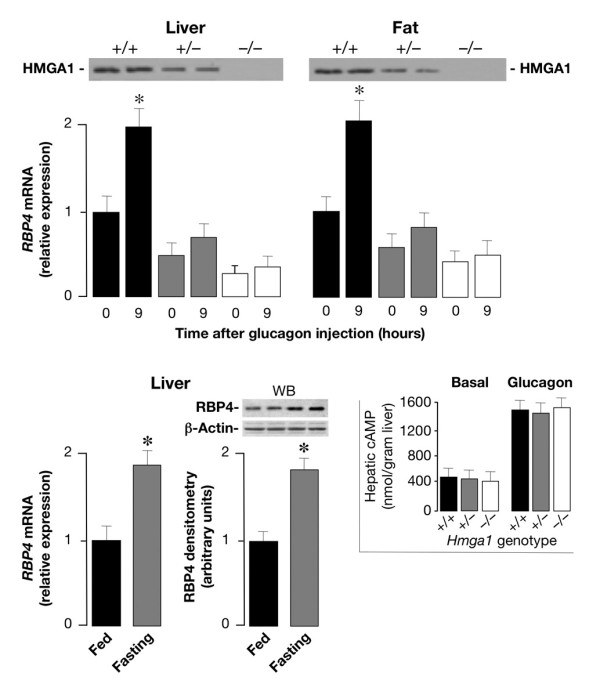
Comparison of RBP4 mRNA levels in glucagon-injected wild-type and Hmga1-deficient mice, and liver RBP4 expression in wild-type mice during fasting and fed. Total RNA was isolated from liver and fat of 3-h-fasted mice, before (time 0) and after 9 h of intraperitoneal injection of glucagon, and RBP4 mRNA was measured by qRT-PCR and normalized to RPS9 mRNA abundance. Results are the mean values ± s.e.m. from 6–8 animals per group. Black bars, Hmga1+/+, n = 8; gray bars, Hmga1+/-, n = 6; white bars, Hmga1-/-, n = 6. *P < 0.05 versus each control (time 0). Western blots for HMGA1 protein expression are shown in liver and fat from all three genotypes (top). The levels of RBP4 mRNA and protein (shown at the bottom of the figure) were measured in liver of fed and 6-h-fasted wild-type mice (6 animals per group), using qRT-PCR and Western blot (WB), respectively. *P < 0.05 versus fed mice. Inset, cAMP was measured in liver from control and Hmga1-deficient mice, in both basal conditions and 3 h after the intraperitoneal injection of glucagon (1 mg/kg body weight), as described in the Methods section. The data are mean ± s.e.m. for 4–6 animals per group.
As a measure of the glucagon efficacy in glucagon-injected mice, a liver biopsy was taken before and after glucagon injection, and cAMP levels in liver were determined for both control and Hmga1-deficient mice (Figure 6, inset). No substantial difference was found in basal levels of cAMP (0.45 and 0.50 in Hmga1-/- and Hmga1+/-, respectively, versus 0.52 μmol/g tissue in controls). After glucagon injection, hepatic levels of cAMP increased to 1.50 μmol/g tissue in control mice, compared with 1.48 and 1.52 in Hmga1-/- and Hmga1+/- mice, respectively. Results similar to those shown in the inset of Figure 6 were also obtained in epididymal and subcutaneous fat pads from control and mutant animals (data not shown), thus indicating that the glucagon-stimulated cAMP synthesis did not differ among mice with diverse genotypes.
Hmga1-deficient mice have increased Glut4 expression and insulin signaling activity in skeletal muscle and fat
Systemic insulin resistance has been associated with elevation of serum RBP4, whereas genetic and pharmacological interventions aimed at decreasing serum RBP4 levels enhance insulin action and improve insulin sensitivity [4]. Increased peripheral insulin sensitivity during insulin-tolerance test was previously observed by us in Hmga1-knockout mice [13]. To verify whether a functional link indeed existed between HMGA1 and RBP4, and whether insulin hypersensitivity in Hmga1-deficient mice could be mediated by the HMGA1-RBP4 system, we carried out quantitative measurements of Glut4 mRNA transcript abundance. Examination by qRT-PCR showed a significant increase of Glut4 transcripts in both skeletal muscle and adipose tissues from Hmga1-deficient mice compared with controls (Figure 7). Accordingly, immunoblotting of muscle and fat tissue showed a 2- to 3-fold increase of Glut4 in the insulin hypersensitive Hmga1-knockout mice compared with controls (Figure 7), clearly indicating that an inverse correlation between RBP4 and Glut4 indeed exists in vivo, in this animal model of diabetes, in which reduced RBP4 may contribute to the maintenance of glucose homeostasis by increasing insulin signaling and peripheral insulin sensitivity. In agreement with this interpretation, the activation state of the protein kinase Akt, an important downstream target of PI 3-kinase regulating insulin serum effects on Glut4 translocation and carbohydrate metabolism [17], was increased in mutant animals. As shown in Figure 7, basal phospho-Akt immunoreactivity was higher in skeletal muscle and adipose tissues from Hmga1-deficient mice compared with wild-type controls, and this increase paralleled closely the increase of Glut4 protein in adipose and muscle plasma membranes from heterozygous and homozygous Hmga1 mutants. In line with previous observations on transcriptional repression of the mouse Glut4 gene by cAMP [18], endocrine upregulation of Glut4 in Hmga1-deficient mice was substantiated further by in vitro experiments (not shown), indicating that in isolated adipocytes treated with Br-cAMP, Glut4 mRNA was decreased in all three genotypes. A positive correlation of RBP4 levels with markers of lipid metabolism adversely affecting insulin sensitivity has been reported recently in both clinical and experimental studies [19,20]. Hmga1-knockout mice had lower levels of serum free fatty acids (0.45 ± 0.13 and 0.34 ± 0.07 in Hmga1+/+ and Hmga1-/-, respectively; P < 0.05), which might contribute to their improved insulin sensitivity.
Figure 7.
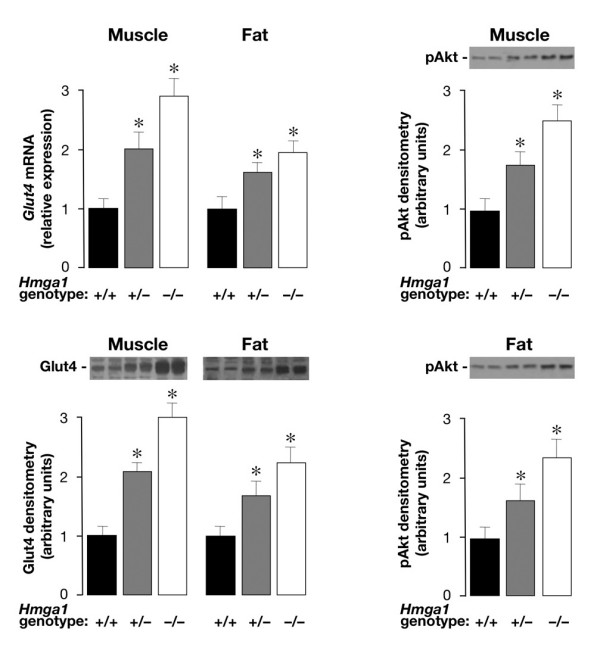
Glut4 and pAkt expression in wild-type and Hmga1-deficient mice. Glut4 mRNA (upper left) and protein content (lower left) in muscle and fat parallel pAkt protein abundance in skeletal muscle (upper right) and adipose tissue (lower right) from control and Hmga1-deficient mice. Representative Western blots of Glut4 and pAkt proteins are shown, together with the densitometric analyses of six to eight independent blots. Black bars, Hmga1+/+, n = 8; gray bars, Hmga1+/-, n = 6; white bars, Hmga1-/-, n = 6. *P < 0.05 versus Hmga1+/+.
Recombinant RBP4 injection reduces Glut4 and insulin signaling activity in muscle and fat tissue of Hmga1-deficient mice, and attenuates insulin hypersensitivity of these animals
To demonstrate that increased insulin sensitivity in mutant mice was directly due to HMGA1 regulation of RBP4, we determined the effect of recombinant RBP4 administration on Akt phosphorylation and Glut4 protein expression in skeletal muscle from Hmga1-deficient mice. As shown in Figure 8, Akt phosphorylation was reduced in muscle from RBP4-injected mutant animals compared with saline-injected Hmga1 mutants. The reduction in Akt phosphorylation in these genotypes correlated inversely with RBP4 serum levels in the same animals (Figure 8) and paralleled the reduction of Glut4 in skeletal muscle and adipose (not shown) plasma membranes (Figure 8), indicating that, in these conditions, activation of the Akt-Glut4 pathway is regulated, at least in part, by circulating RBP4. Plasma insulin levels were slightly higher in RBP4-injected mice, but no significant difference was found (1.6 ± 0.2 and 1.2 ± 0.2 in RBP4-injected Hmga1+/- and Hmga1-/- respectively, versus 1.4 ± 0.1 and 0.9 ± 0.1 ng/ml in saline-injected Hmga1+/- and Hmga1-/- mice).
Figure 8.
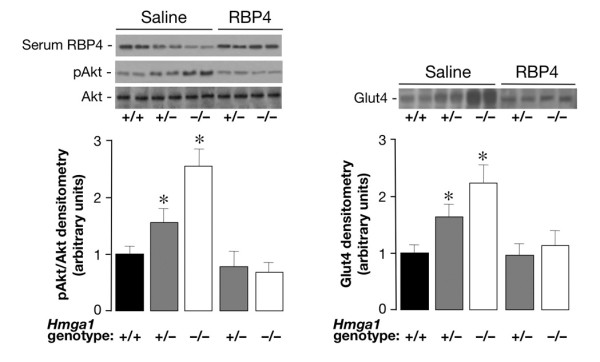
Effects of recombinant RBP4 administration on Akt phosphorylation and Glut4 protein expression in Hmga1-deficient mice. Basal (saline) levels of pAkt (left) and Glut4 (right) were increased in skeletal muscle of saline-injected Hmga1-deficient mice compared with controls, and were reduced following RBP4-injection (n = 6 per genotype). Densitometric quantifications of three independent experiments from 3 animals per genotype are shown, together with representative Western blots of pAkt, Glut4, and serum RBP4 of saline and RBP4-injected mice. *P < 0.05 versus Hmga1+/+.
Consistent with the condition of insulin hypersensitivity, we previously reported that the glucose-lowering effect of exogenous insulin was enhanced in Hmga1-deficient mice during insulin-tolerance test (ITT) [13]. To support further the role of RBP4 in insulin hypersensitivity in Hmga1 mutants, we have determined the effect of RBP4 administration on the glucose fall induced by insulin in these genotypes during ITT. As shown in Figure 9, injection of human RBP4 in heterozygous and homozygous Hmga1 mutants caused a less dramatic fall in blood glucose levels, lessening the hypoglycemic response to intraperitoneal insulin observed in the saline-injected animals. Thus, taken together, our findings consistently support the role of HMGA1 as a key element in the transcriptional regulation of genes involved in glucose metabolism and add new insights into the compensatory mechanisms that may contribute to counteract insulin resistance in vivo. By directly regulating RBP4 gene transcription, HMGA1 enhances peripheral insulin sensitivity, ensuring glucose uptake in skeletal muscle. This, if on one hand might represent an adaptive mechanism to ameliorate insulin resistance in animals with a disadvantageous metabolic risk profile, on the other might indicate that the cAMP/HMGA1-mediated RBP4 expression during fasting (when glucagon peaks) may act physiologically to reduce insulin sensitivity in peripheral tissues, thereby contributing to the maintenance of euglycemia under this condition. This was supported by the observation that after an overnight fasting period (12–16 h) plasma glucose concentration in wild-type mice was higher than that of Hmga1-deficient mice (89 ± 5 in Hmga1+/+ mice, versus 72 ± 6 and 62 ± 5 mg/dl in Hmga1+/- and Hmga1-/- mice, respectively; P < 0.05).
Figure 9.

Effects of RBP4 on insulin sensitivity. Insulin-tolerance test (ITT) was assessed in Hmga1-deficient mice injected with saline alone (left), and in Hmga1 mutants injected chronically with purified RBP4 (right) (n = 6–8 per genotype in each condition). ITT was performed by measuring blood glucose levels in 12-h-fasted conscious mice injected intraperitoneally with human insulin (Human Actrapid, Novo Nordisk), 1 U/kg body weight. Open squares, Hmga1+/+; open circles, Hmga1+/-; open diamonds, Hmga1-/-. The degree of statistical significance was less in RBP4-injected Hmga1-deficient mice compared with the significance for saline-injected Hmga1 mutants. *P < 0.0001, saline-injected Hmga1-deficient mice versus Hmga1+/+; # P < 0.05, RBP4-injected Hmga1-/- mice versus Hmga1+/+.
Discussion
We have previously shown that loss of HMGA1 protein expression, induced in mice by disrupting the HMGA1 gene, severely decreased insulin receptor expression (below 10% of control animals) and phosphorylation in the major targets of insulin action, largely impaired insulin signaling, and reduced insulin secretion, producing a type 2-like diabetic phenotype in which defects in both peripheral insulin sensitivity and pancreatic beta-cell insulin secretion were coexpressed simultaneously [13]. However, despite the severe decrease in insulin receptor signaling and insulin receptor production, the glucose-lowering effect of exogenous insulin was enhanced in Hmga1-deficient mice during ITT, and the glucose infusion rate necessary to maintain euglycemia was higher in mutant mice during hyperinsulinemic-euglycemic clamp [13], supporting the existence of alternative pathways of insulin signaling promoting glucose uptake and disposal in certain adverse metabolic conditions such as those found in the Hmga1-knockout mouse. The existence of signaling pathways promoting glucose uptake and utilization in peripheral tissues through mechanisms that are independent of insulin has been postulated before, on the basis of experimental observations supporting the existence of molecular circuits/pathways that can compensate for the decrease in insulin-stimulated glucose uptake in vivo, in both animal models and human patients with type 2 diabetes [21-23]. However, how these compensatory mechanisms are activated has remained hitherto largely undefined. As previously shown, consistent with the ubiquitous distribution of HMGA1, insulin receptor expression was also reduced in pancreatic tissue from Hmga1-deficient mice [13]. Loss of insulin secretion in response to glucose has been reported in IRβ knockout mice with tissue-specific knockout of the insulin receptor in pancreatic beta cells [24]. As in the IRβ knockout mice, plasma insulin after glucose challenge was considerably reduced in Hmga1-mutant animals, in which the acute first-phase insulin secretory response was severely blunted [13], indicating a glucose-induced insulin secretory defect. In addition, substantial abnormalities in pancreatic islet morphology and size have been described in Hmga1-knockout mice [13], indicating that decreased insulin secretion in this genotype may also depend on reduced beta-cell mass. Thus, defects in both pancreatic beta-cell insulin secretion and peripheral insulin action coexist simultaneously in this knockout mouse model of diabetes, in which activation of compensatory mechanisms to efficiently overcome these metabolic abnormalities may be of vital importance.
Downregulation of Glut4 in adipose tissue is a typical feature of insulin-resistant states, such as obesity and type 2 diabetes [25]. It has been found that the decrease in Glut4 expression that occurs in the fatty tissue of obese animals and humans is accompanied by increased expression and secretion of the adipocyte-derived RBP4 fraction [4,5], suggesting that RBP4 production is tightly regulated by adipose tissue glucose uptake. RBP4 has been recently implicated in systemic insulin sensitivity in rodents and humans, in which elevated serum RBP4 levels were associated with reduced expression of Glut4 in adipocytes, and correlated inversely with peripheral insulin sensitivity. However, based on current data, the role of RBP4 in insulin sensitivity in humans is still controversial and might be restricted to rodent models only. Interspecies differences are known to exist and discrepancies between humans and mice might emphasize the role of non-genetic environmental factors and genetic modifiers in determining the phenotypic variations in RBP4 and insulin sensitivity between humans and animal models. Our results in the present study clearly indicate that in Hmga1-knockout mice RBP4 levels are considerably decreased in serum and in whole liver and adipose tissue extracts, strictly linking HMGA1 and RBP4 expression. We propose that HMGA1 deficiency adversely affects RBP4 expression and this, in animals with a disadvantageous metabolic risk profile like that observed in the Hmga1-knockout mouse model, might reflect an adaptive mechanism to increase glucose uptake and glucose disposal. Consistent with the results obtained in Hmga1-deficient mice, RBP4 was considerably reduced in cells of both human (HepG2) and mouse (Hepa1) origin readily expressing RBP4, following perturbation of endogenous HMGA1 protein expression in cells treated with siRNA against HMGA1. Conversely, an increase in RBP4 mRNA abundance was observed in both cell lines following forced expression of HMGA1, consistently supporting a role for HMGA1 in the transcriptional activation of the RBP4 gene. These findings were substantiated further by ChIP analysis, showing that HMGA1 indeed binds to the RBP4 locus in intact living cells.
Signal transduction pathways which raise intracellular cAMP have been reported to have a potential role in the regulation of RBP4 gene expression [16]. Although the molecular mechanisms underlying this effect remain poorly understood, evidence exists supporting the notion that the regulation of RBP4 gene transcription via the cAMP signaling pathway may be physiologically relevant. One important physiological condition in which intracellular cAMP increases is in response to low glucose availability. In this metabolic setting, a concomitant predominance of circulating counter-regulatory hormones, in particular pancreatic glucagon acting via the cAMP pathway, induces glycogenolysis and gluconeogenesis in the liver, which produce and release hepatic glucose in the blood. In this regard, the cAMP-element-binding protein (CREB) has been identified as a critical transcriptional checkpoint which, in response to cAMP, promotes hepatic glucose output through the synergistic activation of distinct transcriptional effector pathways, which include the PPAR gamma coactivator 1 (PGC1) and the NR4A orphan nuclear receptors [26].
In this paper, we report that systemic injection of glucagon to wild-type control mice caused an increase in RBP4 mRNA and protein expression, along with an increase of both intracellular cAMP and HMGA1 levels. Glucagon effects were attenuated in Hmga1-deficient mice, supporting a distinct role for HMGA1 in the regulation of RBP4 gene expression and functionally linking this two genes. As a consequence of the functional link between HMGA1 and RBP4, a significant increase in Glut4 mRNA and protein was observed in both skeletal muscle and adipose tissues from Hmga1-deficient mice compared with controls. An inverse relationship between RBP4 and Glut4 has been described previously, in the adipose-Glut4-/- mouse, in which the decrease in Glut4 expression that occurs in the fatty tissue of this mutant genotype is accompanied by increased expression and secretion of the fat-derived RBP4 [4]. In our model, instead, RBP4 expression is genetically impaired due to the lack of HMGA1 and Glut4 is increased in both muscle and fat, suggesting that abnormalities in RBP4 and/or metabolites of the vitamin A metabolism may directly affect whole-body insulin action and peripheral insulin sensitivity. In support of this possibility, identification of regulatory single nucleotide polymorphisms in the RBP4 gene associated with type 2 diabetes has been recently reported [27,28], while correlations of RBP4 with insulin resistance have been confirmed in experimental clinical approaches in humans [7]. Although conflicting results have been reported, raising doubt about the postulated relationship of RBP4 with insulin sensitivity in humans, our results in Hmga1-deficient mice confirm that an inverse correlation indeed exists between RBP4 and insulin sensitivity in vivo, in this animal model of diabetes, lending support to previous hypotheses that lowering RBP4 levels would be helpful in ameliorating insulin resistance, at least in mice.
Overall, our findings provide mechanistic insight into the regulation of glucose uptake and disposal in peripheral tissues, and support further the role of HMGA1 as a molecule that is likely to be an important emerging factor in the transcriptional activity of genes implicated in the maintenance of glucose homeostasis and metabolic control, such as the insulin receptor gene [11-13], the leptin gene [29], and, as shown here, the RBP4 gene. Apart from the intrinsic biological interest in elucidating the mechanisms leading to improvement in insulin sensitivity, a clear understanding of the molecular process involved is of potential importance in the development of new therapeutic strategies for patients with metabolic disorders such as obesity, diabetes, and other insulin resistant states.
Conclusion
We propose that HMGA1 can serve as a modulator of both RBP4 gene expression and protein function and represents an important novel mediator of glucose homeostasis in vivo.
Methods
Plasmids, transfections, and ChIP
The RBP4-Luc reporter plasmid was obtained by cloning the NheI/XhoI 1427-bp sequence of the mouse RBP4 promoter (-1417 to +10) into pGL3 (Promega). This fragment was amplified from genomic DNA using the following modified primers: 5'-TTGCTAGCATGGCTAAGGTGCTTGTTGAAA-3', 5'-TTCTCGAGCACACCCACTCCATCTCACC-3' and the integrity of this construct was checked by DNA sequencing. RBP4-Luc reporter plasmid, together with either the control vector plasmid or expression plasmid encoding HMGA1 [11], was transiently transfected into cultured cells using LipofectAMINE 2000 reagent (Invitrogen), and Luc activity was assayed 48 h later, as previously described [30]. Renilla control vector served as an internal control of transfection efficiency, together with measurements of protein expression levels. For antisense HMGA1 experiments, RBP4-containing vector was cotransfected into Hepa1 cells with the expression plasmid pcDNA1 containing the HMGA1 cDNA in the antisense orientation [12]. Small interfering RNA (siRNA) targeted to HMGA1 [30] was transfected into cells at 50% to 60% confluency and cells were analyzed 48 to 96 h later. ChIP assay was performed in HepG2 and Hepa1 cells, either untreated or pretreated with HMGA1 siRNA as described previously [31]. Formaldehyde-fixed DNA-protein complex was immunoprecipitated with anti-HMGA1 antibody. Primers for the RBP4 sequence were used for PCR amplification of immunoprecipitated DNA (30 cycles), using PCR ready-to-go beads (Amersham Pharmacia Biotech). PCR products were electrophoretically resolved on 1.5% agarose gel and visualized by ethidium bromide staining.
Animals
Male Hmga1-deficient and wild-type mice aged 6–9 months were studied. The generation of these animals and many of the physiological characteristics of the mice have been described in detail [13]. All animal work was carried out at the Animal Facility at the 'Istituto dei Tumori di Napoli', and at the Faculty of Pharmacy, Roccelletta di Borgia, Catanzaro, using approved animal protocols and in accordance with institutional guidelines. Serum free fatty acid levels were measured in wild-type and Hmga1-knockout mice (n = 12–16 per genotype) using the NEFA C kit (Wako).
Real-time PCR and Western blot
For qRT-PCR, total cellular RNA was extracted from tissues using the RNAqueous-4PCR kit and subjected to DNase treatment (Ambion). RNA levels were normalized against 18S ribosomal RNA in each sample, and cDNAs were synthesized from 2 μg of total RNA using the RETROscript first strand synthesis kit (Ambion). Primers for mouse HMGA1 (NM_016660.2) (5'-GCAGGAAAAGGATGGGACTG-3'; 5'-AGCAGGGCTTCCAGTCCCAG-3'), RBP4 (NM_011255.2) (5'-AGGAGAACTTCGACAAGGCT-3'; 5'-TTCCCAGTTGCTCAGAAGAC-3'), Glut4 (NM_009204) (5'-TCATTGTCGGCATGGGTTT-3'; 5'-CGGCAAATAGAAGGAAGACGTA-3'), and RPS9 (NM_029767.2) (5'-CTGGACGAGGGCAAGATGAAGC-3'; 5'-TGACGTTGGCGGATGAGCACA-3') were designed according to sequences from the GenBank database. A real-time thermocycler (Eppendorf Mastercycler ep realplex ES) was used to perform quantitative PCR. In a 20-μl final volume, 0.5 μl of the cDNA solution was mixed with SYBR Green RealMasterMix (Eppendorf), and 0.3 μM each of sense and antisense primers. The mixture was used as a template for the amplification by the following protocol: a denaturing step at 95°C for 2 min, then an amplification and quantification program repeated for 45 cycles of 95°C for 15 s, 55°C for 25 s, and 68°C for 25 s, followed by the melting curve step. SYBR Green fluorescence was measured, and relative quantification was made against the RPS9 cDNA used as an internal standard. All PCR reactions were done in triplicate.
Western blot analysis was performed to analyze HMGA1 and RBP4 protein expression in whole-cell liver and fat extracts from normal and mutant mice, using polyclonal specific antibodies raised against HMGA1 [11] and RBP4 (AdipoGen, Inc.). For the measurement of serum RBP4, blood was collected from the retro-orbital sinus, plasma protein extracts were resolved on 12% SDS-PAGE, blotted onto nitrocellulose membranes and RBP4 was detected using rabbit polyclonal antisera at 1:2000 dilution, as suggested by the manufacturer. TTR was detected using a goat anti-TTR polyclonal antibody (Santa Cruz Biotechnology). Rabbit anti-Glut4 polyclonal antibody was used as previously described [13].
In vivo studies with the peptide hormone glucagon
For systemic administration of exogenous glucagon, mice were injected in the peritoneal cavity with human glucagon (1 mg/kg body weight) or saline after 3 h of fasting. At this dose, the peak increase of plasma glucagon in all genotypes was ~96% ± 10% above pre-injection levels, reflecting similar previous observations in rodents [32]. At different times after the injection the mice were killed by cervical dislocation, the liver and fat were rapidly removed, frozen into liquid nitrogen and stored at -80°C until processed. For cAMP determination, frozen samples were first homogenized in ice-cold trichloroacetic acid (TCA) (6% wt/vol), and cAMP was determined using the cAMP enzyme immunoassay kit (Amersham Pharmacia Biotech), according to the instructions specified by the manufacturer.
RBP4 purification and injection
Human RBP4 cDNA cloned into a pET3a expression vector was a kind gift from JW Kelly (The Scripps Research Institute). Based on previously published methodology [33], RBP4 protein expression vector was transformed into the BL21 strain of Escherichia coli (Stratagene), expanded in suspension culture and induced for 6 h with 1 mM isopropyl-D-thiogalactopyranoside to stimulate protein expression. Bacteria were pelleted and lysed by osmotic shock [34]. From this point on, all steps, including denaturation, refolding, and RBP4 purification, were performed essentially as described elsewhere [35]. Protein fractions were examined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting, and desired fractions were pooled together, concentrated with an Amicon Centriprep-10 concentrator (Millipore), and stored at -80°C.
To determine whether elevation of RBP4 affected insulin hypersensitivity in vivo, in Hmga1-deficient mice, heterozygous and homozygous Hmga1 mutants, were intraperitoneally injected twice daily (at 12-h intervals) with 200 μg of purified human RBP4 (13 μg/g body weight per mouse) for 7 days. This resulted in a daily average serum level of human RBP4 similar to that of control mice (see Figure 8), which received physiological saline solution according to the same schedule above.
Statistical analysis
The ANOVA test was used to evaluate the differences between the groups of mice. For all analyses, P < 0.05 was considered significant.
Abbreviations
Akt: protein kinase B; Br-cAMP: 8-bromo cAMP; cAMP: cyclic adenosine monophosphate; CREB: cAMP-element-binding protein; Glut4: glucose transporter-4; HEK-293: human embryonic kidney-293; Hepa1: mouse hepatoma; HMGA1: high mobility group A1; ITT: insulin-tolerance test; PGC1: PPAR gamma coactivator 1; qRT-PCR: quantitative Real-Time PCR; RBP4: retinol-binding protein 4; siRNA: small interfering RNA; TCA: trichloroacetic acid; TTR: transthyretin.
Authors' contributions
EC and FP performed qRT-PCR studies as well as transient transfections with reporter and expression vectors, and participated in the design of the study. SI was involved in Western blotting studies and assisted FP in performing transfections with siRNA. ILP participated in Western blotting and performed cloning studies. DP, EG, GDS and AF participated in the analysis and discussion of the in vivo data from normal and mutant mice. GB, AL, and VC performed certain aspects of the assays detailed in Figures 1, 2 and 3 and contributed with Northern blotting studies. DF provided helpful discussion on this manuscript and participated in ChIP analysis. AB conceived, coordinated, and supervised the project, analyzed data, and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
We remain extraordinarily indebted to Drs R Citraro and N Costa and the entire staff of the animal facilities for animal care. We are most grateful to Dr JW Kelly for his generous gift of RBP4 expression vector, pET3a. We would also like to thank Mrs A Malta and Dr G Ceravolo for secretarial help. This work was supported by Telethon-Italy, grant GGP04245, and MIUR, protocol 2004062059-002 Italy (AB).
Contributor Information
Eusebio Chiefari, Email: echiefari@libero.it.
Francesco Paonessa, Email: fpao@hotmail.it.
Stefania Iiritano, Email: iiritanostefania@yahoo.it.
Ilaria Le Pera, Email: lepera@unicz.it.
Dario Palmieri, Email: dario.palmieri@unina.it.
Giuseppe Brunetti, Email: giuseppe.brunetti@unimi.it.
Angelo Lupo, Email: lupo@unisannio.it.
Vittorio Colantuoni, Email: colantuoni@unisannio.it.
Daniela Foti, Email: foti@unicz.it.
Elio Gulletta, Email: gulletta@unicz.it.
Giovambattista De Sarro, Email: desarro@unicz.it.
Alfredo Fusco, Email: afusco@napoli.com.
Antonio Brunetti, Email: brunetti@unicz.it.
References
- Kahn CR. Insulin action, diabetogenes, and the cause of type II diabetes (Banting Lecture) Diabetes. 1994;43:1066–1084. doi: 10.2337/diab.43.8.1066. [DOI] [PubMed] [Google Scholar]
- Polonsky KS, Sturis J, Bell GI. Non-insulin-dependent diabetes mellitus – a genetically programmed failure of the beta cell to compensate for insulin resistance. New Engl J Med. 1996;334:777–783. doi: 10.1056/NEJM199603213341207. [DOI] [PubMed] [Google Scholar]
- Taylor S. Insulin resistance or insulin deficiency: which is the primary cause of NIDDM? Diabetes. 1994;43:735–740. doi: 10.2337/diab.43.6.735. [DOI] [PubMed] [Google Scholar]
- Yang Q, Grahm TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, Kotani K, Quadro L, Kahn BB. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. 2005;436:356–362. doi: 10.1038/nature03711. [DOI] [PubMed] [Google Scholar]
- Graham TE, Yang Q, Blüher M, Hammarstedt A, Ciaraldi TP, Henry RR, Wason CJ, Oberbach A, Jansson P-A, Smith U, Kahn BB. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. New Engl J Med. 2006;354:2552–2563. doi: 10.1056/NEJMoa054862. [DOI] [PubMed] [Google Scholar]
- Ziouzenkova O, Orasanu G, Sharlach M, Akiyama TE, Berger JP, Viereck J, Hamilton JA, Tang G, Dolnikowski GG, Vogel S, Duester G, Plutzky J. Retinaldehyde represses adipogenesis and diet-induced obesity. Nat Med. 2007;13:695–702. doi: 10.1038/nm1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Eynatten M, Humpert PM. Retinol-binding protein-4 in experimental and clinical metabolic disease. Expert Rev Mol Diagn. 2008;8:289–299. doi: 10.1586/14737159.8.3.289. [DOI] [PubMed] [Google Scholar]
- Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: transthyretin and retinol binding protein. Science. 1995;268:1039–1041. doi: 10.1126/science.7754382. [DOI] [PubMed] [Google Scholar]
- Bustin M, Reeves R. High-mobility group proteins: architectural components that facilitate chromatin function. Prog Nucleic Acid Res Mol Biol. 1996;54:35–100. doi: 10.1016/s0079-6603(08)60360-8. [DOI] [PubMed] [Google Scholar]
- Thanos D, Maniatis T. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell. 1995;83:1091–1100. doi: 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- Brunetti A, Manfioletti G, Chiefari E, Goldfine ID, Foti D. Transcriptional regulation of human insulin receptor gene by the high-mobility group protein HMGI(Y) FASEB J. 2001;15:492–500. doi: 10.1096/fj.00-0190com. [DOI] [PubMed] [Google Scholar]
- Foti D, Iuliano R, Chiefari E, Brunetti A. A nucleoprotein complex containing Sp1, C/EBP beta, and HMGI-Y controls human insulin receptor gene transcription. Mol Cell Biol. 2003;23:2720–2732. doi: 10.1128/MCB.23.8.2720-2732.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foti D, Chiefari E, Fedele M, Iuliano R, Brunetti L, Paonessa F, Barbetti F, Croce CM, Fusco A, Brunetti A. Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in humans and mice. Nat Med. 2005;11:765–773. doi: 10.1038/nm1254. [DOI] [PubMed] [Google Scholar]
- Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000;6:77–86. [PubMed] [Google Scholar]
- Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in β-islet cell hyperplasia. Nat Genet. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- Jessen KA, Satre MA. Induction of mouse retinol binding protein gene expression by cyclic AMP in Hepa 1–6 cells. Arch Biochem Biophys. 1998;357:126–130. doi: 10.1006/abbi.1998.0821. [DOI] [PubMed] [Google Scholar]
- Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, 3rd, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- Kaestner KH, Flores-Riveros JR, McLenithan JC, Janicot M, Lane MD. Transcriptional repression of the mouse insulin-responsive glucose transporter (GLUT4) gene by cAMP. Proc Natl Acad Sci USA. 1991;88:1933–1937. doi: 10.1073/pnas.88.5.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Eynatten M, Lepper PM, Liu D, Lang K, Baumann M, Nawroth PP, Bierhaus A, Dugi KA, Heemann U, Allolio B, Humpert PM. Retinol-binding protein 4 is associated with components of the metabolic syndrome, but not with insulin resistance, in men with type 2 diabetes or coronary artery disease. Diabetologia. 2007;50:1930–1937. doi: 10.1007/s00125-007-0743-8. [DOI] [PubMed] [Google Scholar]
- Stefan N, Hennige AM, Staiger H, Machann J, Schick F, Schleicher E, Fritsche A, Haring HU. High circulating retinol-binding protein 4 is associated with elevated liver fat but not with total, subcutaneous, visceral, or intramyocellular fat in humans. Diabetes Care. 2007;30:1173–1178. doi: 10.2337/dc06-2342. [DOI] [PubMed] [Google Scholar]
- Saltiel AR, Pessin JE. Insulin signaling pathways in time and space. Trends Cell Biol. 2002;12:65–71. doi: 10.1016/s0962-8924(01)02207-3. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Kahn CR, Accili D. Insulin receptor knockout mice. Annu Rev Physiol. 2003;65:313–332. doi: 10.1146/annurev.physiol.65.092101.142540. [DOI] [PubMed] [Google Scholar]
- Bouché C, Serdy S, Kahn CR, Goldfine AB. The cellular fate of glucose and its relevance in type 2 diabetes. Endocr Rev. 2004;25:807–30. doi: 10.1210/er.2003-0026. [DOI] [PubMed] [Google Scholar]
- Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic β cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96:329–339. doi: 10.1016/s0092-8674(00)80546-2. [DOI] [PubMed] [Google Scholar]
- Shepherd PR, Kahn BB. Glucose transporters and insulin action-implications for insulin resistance and diabetes mellitus. New Engl J Med. 1999;341:248–257. doi: 10.1056/NEJM199907223410406. [DOI] [PubMed] [Google Scholar]
- Desvergne B, Michalik L, Wahli W. Transcriptional regulation of metabolism. Physiol Rev. 2006;86:465–514. doi: 10.1152/physrev.00025.2005. [DOI] [PubMed] [Google Scholar]
- Munkhtulga L, Nakayama K, Utsumi N, Yanagisawa Y, Gotoh T, Omi T, Kumada M, Erdenebulgan B, Zolzaya K, Lkhagvasuren T, Iwamoto S. Identification of a regulatory SNP in the retinol binding protein 4 gene associated with type 2 diabetes in Mongolia. Hum Genet. 2007;120:879–888. doi: 10.1007/s00439-006-0264-4. [DOI] [PubMed] [Google Scholar]
- Craig RL, Chu WS, Elbein SC. Retinol binding protein 4 as a candidate gene for type 2 diabetes and prediabetic intermediate traits. Mol Genet Metab. 2007;90:338–344. doi: 10.1016/j.ymgme.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melillo RM, Pierantoni GM, Scala S, Battista B, Fedele M, Stella A, De Biasio MC, Chiappetta G, Fidanza V, Condorelli G, Santoro M, Croce CM, Viglietto G, Fusco A. Critical role of the HMGI(Y) proteins in adipocytic cell growth and differentiation. Mol Cell Biol. 2001;21:2485–2495. doi: 10.1128/MCB.21.7.2485-2495.2001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Paonessa F, Foti D, Costa V, Chiefari E, Brunetti G, Leone F, Luciano F, Wu F, Lee AS, Gulletta E, Fusco A, Brunetti A. Activator protein-2 overexpression accounts for increased insulin receptor expression in human breast cancer. Cancer Res. 2006;66:5085–5093. doi: 10.1158/0008-5472.CAN-05-3678. [DOI] [PubMed] [Google Scholar]
- Costa V, Paonessa F, Chiefari E, Palaia L, Brunetti G, Gulletta E, Fusco A, Brunetti A. The insulin receptor: a new anticancer target for peroxisome proliferator-activated receptor-g (PPARg) and thiazolidinedione-PPARg agonists. Endocr Relat Cancer. 2008;15:325–335. doi: 10.1677/ERC-07-0226. [DOI] [PubMed] [Google Scholar]
- Velliquette RA, Koletsky RJ, Ernsberger P. Plasma glucagon and free fatty acid responses to a glucose load in the obese spontaneous hypertensive rat (SHROB) model of matabolic syndrome X. Exp Biol Med (Maywood) 2002;227:164–170. doi: 10.1177/153537020222700303. [DOI] [PubMed] [Google Scholar]
- Xie Y, Lashuel HA, Miroy GJ, Dikler S, Kelly JW. Recombinant human retinol-binding protein refolding, native disulfide formation, and characterization. Protein Expr Purif. 1998;14:31–37. doi: 10.1006/prep.1998.0944. [DOI] [PubMed] [Google Scholar]
- Burger A, Berendes R, Voges D, Huber R, Demange P. A rapid and efficient purification method for recombinant annexin V for biophysical studies. FEBS Lett. 1993;329:25–28. doi: 10.1016/0014-5793(93)80185-w. [DOI] [PubMed] [Google Scholar]
- Isken A, Golczak M, Oberhauser V, Hunzelmann S, Driever W, Imanishi Y, Palczewski K, von Lintig J. RBP4 disrupts vitamin A uptake homeostasis in a STRA6-deficient animal model for Matthew-Wood syndrome. Cell Metab. 2008;7:258–268. doi: 10.1016/j.cmet.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]