

Abstract
During illumination, Ca2+ enters fly photoreceptor cells through light-activated channels that are located in the rhabdomere, the compartment specialized for phototransduction. From the rhabdomere, Ca2+ diffuses into the cell body. We visualize this process by rapidly imaging the fluorescence in a cross section of a photoreceptor cell injected with a fluorescent Ca2+ indicator in vivo. The free Ca2+ concentration in the rhabdomere shows a very fast and large transient shortly after light onset. The free Ca2+ concentration in the cell body rises more slowly and displays a much smaller transient. After ≈400 ms of light stimulation, the Ca2+ concentration in both compartments reaches a steady state, indicating that thereafter an amount of Ca2+, equivalent to the amount of Ca2+ flowing into the cell, is extruded. Quantitative analysis demonstrates that during the steady state, the free Ca2+ concentration in the rhabdomere and throughout the cell body is the same. This shows that Ca2+ extrusion takes place very close to the location of Ca2+ influx, the rhabdomere, because otherwise gradients in the steady-state distribution of Ca2+ should be measured. The close colocalization of Ca2+ influx and Ca2+ extrusion ensures that, after turning off the light, Ca2+ removal from the rhabdomere is faster than from the cell body. This is functionally significant because it ensures rapid dark adaptation.
Many neurons, including sensory neurons, localize Ca2+ influx channels to small, often diffusionally isolated compartments of the cell body (1). This localized Ca2+ influx into a small volume often results in sizeable but local changes in free Ca2+ concentration (Cai; refs. 2 and 3). Photoreceptor cells of flies exemplify this strategy. On light stimulation, channels that are highly permeable for Ca2+ (4, 5) and exclusively located in the membranes of the microvilli (6, 7) are activated. The microvilli, tube-like protrusions of the plasma membrane with exceptionally small dimensions, are regularly packed together to form a dense stack, the rhabdomere (8). Cai in the rhabdomere rises up to 600 μM when the cells are stimulated with bright light (9). Calculations indicate that similar or even higher concentrations also occur in one or a few microvilli already after the absorption of a single photon (10). From the rhabdomeric microvilli, Ca2+ diffuses into the cell body (11), where measured increases in Cai are more moderate but still exceed 10 μM (12, 13). These changes of Cai are functionally important, because they mediate light adaptation (14, 15).
Creating high local Cai values by confining the influx to specialized compartments potentially causes the problem that Cai in these compartments diminishes only slowly after the stimulus has ceased. This problem can be counteracted by colocalizing proteins that efficiently extrude Ca2+. The implementation of this design principle has been demonstrated, e.g., in stereocilia of haircells (16), in synaptic terminals of rods (17, 18), and in presynaptic boutons of hippocampal cells (19). In fly photoreceptor cells, highly active Na+/Ca2+ exchangers extrude Ca2+ (20–23). Two genes encoding two different types of Na+/Ca2+ exchangers have been shown to be expressed in the photoreceptor cells of Drosophila (24, 25), but the subcellular location of the proteins is unknown. In squid photoreceptor cells, however, the rhabdomeric membranes show Na+/Ca2+ exchange activity (26). Using a combination of Ca2+ imaging and modeling, we show in this report that Ca2+ extrusion in fly photoreceptors takes place close to the location of Ca2+ influx, the rhabdomere, suggesting that Na+/Ca2+ exchangers are located in or close to the rhabdomere. We show that this has profound physiological consequences, as it increases the speed of Ca2+ removal in the rhabdomere and avoids large gradients of Cai in the cells during continuous stimulation.
Materials and Methods
High-Speed Fluorescent Imaging and Data Analysis.
Single photoreceptor cells of female white-eyed mutant chalky blowflies (Calliphora vicina) were impaled with intracellular electrodes and iontophoretically injected with the fluorescent Ca2+ indicator dye Oregon green 5N (OG5N, Molecular Probes) as described previously (9, 13). The electrode was withdrawn, and the fly was placed on a goniometer that served as stage of a standard microscope (Nikon Optiphot-2, Nikon) connected to a confocal microscope (Odyssey XL, Noran Instruments, Middletown, WI), from which the confocal pinhole was removed to maximize light intensity. The fly was reoriented to ensure that the rhabdomere of the dye-filled cell was coaxial with the optical axis of the objective (×25 water immersion, NA 0.6; SW25, Leitz). Light came from the 488-nm line of a krypton–argon laser, and the emitted fluorescence light passed a triple-filter (Noran) or a 510-nm fluorescence cube (Nikon). An LS2 shutter (Uniblitz, Vincent Associates, Rochester, NY) was added in the light path of the confocal microscope before the beam expander to rapidly turn the light on or off. Several sequences of images were recorded at a rate of 480 images per s and averaged off line; care was taken to ensure that the preparation did not move between the recordings. All findings were confirmed in at least three different preparations.
Modeling the Steady-State Distribution of Ca2+.
For the calculations of the steady-state distribution of Cai, we assume that a constant influx of Ca2+ (see Results) occurs in the rhabdomere homogeneously along the entire length of a photoreceptor cell (l = 225 μm; ref. 8), reducing diffusion to two dimensions. This assumption seems justified because the light intensities used for measuring the fluorescence saturate the light response: each microvillus was hit at least once in a few milliseconds. The cross section through the modeled cell was assumed square with side lengths a = 7.1 μm. The diffusion coefficient for free Ca2+ ions (DCa) was assumed to be 220 μm2⋅s−1 (27). The diffusion coefficient of the mobile buffer, whether bound to Ca2+ or not, was equally assumed to be DB = 220 μm2⋅s−1 (28), as it is possible that most of the mobile buffer stems from the Ca2+ indicator. Accordingly, the dissociation constant of the buffer (Kd,B) was taken to be 20 μM, the value reported for OG5N (29, 30). As only the steady-state distribution of Ca2+ was investigated here, fixed (i.e., nondiffusing) Ca2+ buffers do not need to be considered (31). Equally, Ca2+ release (32) from or Ca2+ uptake (33) into organelles that might occur in fly photoreceptor cells do not influence the steady-state distribution of free Ca2+.
The square cross section was divided by a grid into 40 × 40 compartments of equal size. In the middle of one side, a 1.5 μm large region was designated to be the region of Ca2+ influx. The concentration of Ca2+ ions, either free or bound to a Ca2+ buffer, was calculated at the intersections of the grid lines. The volume assigned to each grid point was 1/1,600 of the total cell volume (Vtot = 11 pl), but the points lying on one of the cell borders were assigned half that value, and the points in the corners a quarter. Diffusion was calculated to occur along the grid lines. The change of either free or bound calcium at positions x and y (ΔCx,y) during a time step (Δt = 28 μs) caused by diffusion with coefficient D is then given by:
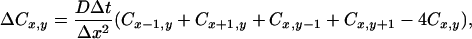 |
for grid points that do not lie on the border of the cell; for grid points on the border, the coefficients need to be changed appropriately. The distance between two grid points Δx = a/40 = 177 nm. After calculating ΔC for every grid point, the new total Ca2+ concentration was obtained. From the total Ca2+ concentration, the concentrations of free and bound Ca2+ were determined with the assumption that the buffer reactions are in equilibrium. The calculations continued until a stable distribution of Cai was reached, i.e., until the amount of Ca2+ extruded differed less than 0.1% from the amount of Ca2+ flowing in.
The activity of Na+/Ca2+ exchangers, the proteins that extrude Ca2+ from fly photoreceptor cells (20–23), depends on many parameters, including the membrane potential and the intra- and extracellular Na+ and Ca2+ concentrations (for review, see ref. 34). As we analyze only the steady state, all parameters can be assumed constant except Cai. Ca2+ binding to the transport site of the exchanger proteins is typically described with a Hill function (Hill coefficient h = 1), for which two parameters, the dissociation constant Kd,X and the maximal current Imax,X, need to be determined (34). A rough estimate of the Kd,X value for the Ca2+ extrusion can be inferred from data by Hardie (22), who showed that the Na+/Ca2+ exchanger is 10 times more active when Cai = 20 μM than when Cai = 1 μM. This yields 18 μM for Kd,X, which, however, is rather high compared with the values reported for other preparations (34). The maximum calcium current transported by the Ca2+ extrusion process was set to Imax,X = 1.3 nA, to ensure that the calculated Cai values at the location of extrusion lie between 12 and 40 μM, as found experimentally for Cai in strongly light-stimulated cells (13). The choice of parameters for the Ca2+ extrusion is rather uncritical for the conclusions drawn in Results, because changing Kd,X or Imax,X affects the average Ca2+ concentration strongly but the calculated gradients only very weakly.
Results
Imaging the Ca2+-Induced Change in Fluorescence in a Cross Section of a Photoreceptor Cell.
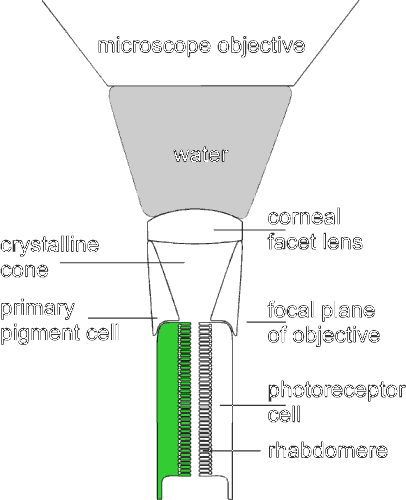
We imaged the distal cross section of a blowfly photoreceptor cell iontophoretically injected with the fluorescent Ca2+ indicator OG5N in vivo by optically neutralizing the natural optics of the cornea [ref. 35; the principle of this approach is outlined in a schematic drawing provided as supplementary material (see www.pnas.org)]. Fig. 1 shows false-color images representing raw intensity values taken from a time series recorded with high temporal resolution (480 images per s). The image at 2 ms is the first image recorded after the light was turned on. Because the photoreceptor cells have a latency period of ≈4 ms before an increase in Cai can be detected (9, 12), this first image represents the autofluorescence of the tissue together with the fluorescence of the Ca2+ indicator at low values of Cai. The intense light used for measuring the fluorescence strongly activates the phototransduction cascade, resulting in opening channels that are permeable for Ca2+ (4, 5). This leads to a Ca2+ influx, visible in the image taken 10 ms after light onset. The fluorescence intensity has increased in the area that corresponds to the rhabdomere but not in the rest of the cell. This observation confirms that the influx of Ca2+ through the light-activated channels is localized exclusively to the rhabdomere (6, 7, 11). The subsequent images show that the Ca2+ increase spreads into the cell body and concomitantly reduces in the rhabdomere. A movie showing the initial 200 ms of the measurement depicted in Fig. 1 is provided as supplementary material.
Figure 1.

A time series of images showing the Ca2+-induced fluorescence of the low-affinity dye OG5N during light stimulation. Raw intensity images are plotted by using false-color coding. Two milliseconds after the light was turned on, the first image was recorded. It represents the initial level of fluorescence during the latency period of the cell, because of autofluorescence of the tissue and the residual fluorescence of the Ca2+ indicator at low Cai. The other images are taken at times indicated by the numbers (in milliseconds). Ten milliseconds after the onset of illumination, a strong increase in fluorescence is visible in the region that corresponds to the rhabdomere. The fluorescence signal in the rhabdomere starts to decline after 100 ms, and it spreads into the cell body.
A Sizeable Ca2+ Current Flows into the Photoreceptor Cells During the Steady State.
The electrical response caused by bright illumination consists of a fast transient depolarization (on average 65 mV) that quickly decays to a steady-state depolarization of about 30 mV (e.g., ref. 13). During this steady state, a continuous current flows through the light-activated channels, which is counterbalanced by the strong noninactivating currents through voltage-dependent potassium channels (36). The voltage-dependent potassium current at 30 mV depolarization is ≈3 nA (36). This value can be used to estimate the Ca2+ current through the light-activated channels, as the current through the light-activated channels (Il) needs to balance the currents through the voltage-dependent K+ channels. Assuming the validity of the Goldman–Hodgkin–Katz theory (ref. 37; for application to fly photoreceptors, see refs. 10, 23, and 38), the current carried by Ca2+ ions (ICa) through the light-activated channels can be calculated as (10):
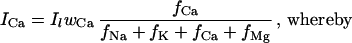 |
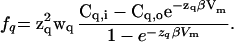 |
The index q denotes the four cations considered, zq the valence, Cq,i the intracellular, and Cq,o the extracellular concentration of ion sort q. β = F/(RT), with F the Faraday constant, R the molar gas constant, and T = 293 K. The membrane potential (Vm) is taken to be −30 mV, the steady-state value of strongly stimulated cells (13). The relative permeabilities (wq) of the light-activated channels have been measured in Drosophila photoreceptor cells (4, 5); here we use wNa = wK = 0.02, wCa = 0.85 and wMg = 0.11, the values described for wild-type flies (ref. 5; see also ref. 10). The following ion concentrations (in mM) are typically found in insect photoreceptor cells and retinas: CNa,i = 10, CNa,o = 140, CK,i = 120, CK,o = 4, CMg,i = 4, CMg,o = 2 (39, 40). In strongly stimulated photoreceptor cells, rather high values for the intracellular Ca2+ concentration (CCa,i) have been reported (12, 13), and the extracellular Ca2+ concentration (CCa,o) is probably reduced from its resting value of 1.4 mM (40). Here we use CCa,i = 20 μM and CCa,o = 1.2 mM.
By using these values, ICa works out to be 1.4 nA when Il is assumed to be 3 nA. This, however, neglects the contribution of Na+/Ca2+ exchange that can depolarize the cells because of its electrogenicity (21, 23). Furthermore, currents through the voltage-dependent K+ channels might have been overestimated, as we neglected possible light-induced accumulation of K+ in the extracellular space (39). In the following, we take ICa = 0.7 nA, in order not to overestimate the Ca2+ current through the light-activated channels.
Our previous measurements (13) and the data presented in Fig. 1 indicate that Cai does not increase substantially after ≈200–500 ms of light stimulation. This shows that Ca2+-extruding mechanisms must generate a Ca2+ current of (at least) 0.7 nA during the steady state to balance the influx.
The Steady-State Distribution of Cai Contains Information About the Localization of Ca2+-Extruding Proteins.
The Ca2+ ions, flowing in through the light-activated channels located in the rhabdomere, have to diffuse to the location of the Ca2+-extruding proteins to be extruded. The subcellular location of the Ca2+-extruding proteins therefore has a profound influence on the shape and size of the gradients that build up in the cytosol. This is illustrated in Fig. 2, where we calculated distributions of Cai in the cross section of a square model cell for four different locations of Ca2+-extruding proteins, as indicated in Fig. 2 (Top). It was assumed that a continuous Ca2+ current (ICa) of 0.7 nA flows into the model cell at the center of one side, corresponding to the place where the rhabdomeric microvilli are attached. The influence of mobile Ca2+ buffers was initially neglected. When assuming that the Ca2+ extrusion takes place on the basolateral sides (Fig. 2a), where the Na+/K+ pumps (41) are located, Cai in the rhabdomere should be as high as 97 μM, leveling off to reach 12–40 μM at the basolateral sides of the cell. Placing the Ca2+ extrusion to the apical side (except the rhabdomere) results in a much flatter distribution in the cell body (Fig. 2b). Still, Cai in the rhabdomere would be at 73 μM and on the apical side as low as 14 μM. Modeling extrusion on all sides except the rhabdomere (Fig. 2c) results in an intermediate between the situations shown in Fig. 2 a and b. Only when the Ca2+ extrusion is modeled to take place exclusively at the location of Ca2+ influx, the rhabdomere, a homogeneous distribution of Cai is found throughout the cell (Fig. 2d).
Figure 2.

Calculated distribution of Cai in a cross section of a square model cell. The continuous influx is assumed to occur at a 1.5-μm-wide region of one side (arrow in the diagrams). Extrusion is modeled at different regions of the plasma membrane, as indicated in the diagrams by the gray shading. (a) Ca2+ extrusion only at the basolateral sides. (b) Ca2+ extrusion only at the apical side, the Ca2+ influx region (rhabdomere) excluded. (c) Ca2+ extrusion at all sides, but the Ca2+ influx region excluded. (d) Ca2+ extrusion only at the Ca2+ influx region. In a–c, the resulting distribution of Cai shows large gradients, but not in d.
The gradients that can possibly be expected to be measured experimentally, however, are not as large as those depicted in Fig. 2. The addition of fluorescent Ca2+ indicators, highly mobile Ca2+ buffers that are necessary for the measurements, will reduce those gradients (31, 42). To estimate more realistically the measurable gradients, we repeated the calculations with varying concentrations of a highly mobile Ca2+ buffer (Fig. 3). Increasing the buffer concentration flattens the expected distribution of Cai. For the extreme case of 5.0 mM Ca2+ buffer (the concentration of the indicator in the recording electrode), barely any gradients are discernible, no matter where the extrusion is assumed to take place (Fig. 3 a and b). These calculated values of Cai can be used to derive the expected fluorescence intensity of a Ca2+ indicator with Kd,B = 20 μM. The expected difference in indicator fluorescence intensity between the rhabdomeric region and the side of the cell body opposite the rhabdomere is plotted as a function of the buffer concentration in Fig. 3c. It shows that even with buffer concentrations of 750 μM, the intensity difference should be 18% if the Ca2+ extrusion is located at the basolateral sides. When the Ca2+ extrusion takes place on the apical side, the intensity difference should amount to 9%. For lower buffer concentrations, the fluorescence difference is predicted to be even larger. Importantly, no matter how large the buffer concentration, no gradient will arise if Ca2+ extrusion is confined to the place of Ca2+ influx, the rhabdomere (not shown). These considerations show that measurements of Ca2+ gradients in the cell body can yield information about the location of Ca2+ extrusion, provided the concentration of the Ca2+ indicator is not too high.
Figure 3.

The concentration of the Ca2+ buffer strongly influences the modeled distribution of the free Ca2+ concentration. (a and b) The free Ca2+ concentration profile along the symmetry line through the cell body, from the Ca2+ influx region to the opposite side of the cell body, is plotted. The extrusion was assumed to take place only at basolateral sides (a; as in Fig. 2a) or only at the apical side (b; as in Fig. 2b). Increasing the buffer concentration (indicated by the numbers, in millimolars) reduces the size of the predicted gradients. (c) The relative fluorescence intensity difference of a Ca2+ indicator with Kd = 20 μM between the Ca2+ influx region (x = 0 μm in a and b) and the opposite end of the cell body (x = 7.1 μm in a and b) is plotted as a function of the buffer concentration, when assuming extrusion on the basolateral sides (○) or on the apical side (▪). Below a buffer concentration of 0.75 mM, the fluorescence intensity between the two points differs by more than 10% (dashed line).
The Steady-State Distribution of Cai in a Cross Section of the Photoreceptor Cells Is Homogeneous.
Fig. 4 shows the quantitative analysis of the data presented in Fig. 1. To obtain a reasonable signal-to-noise ratio, we defined three regions of interest in the cross section of the photoreceptor cell (Fig. 4a). The regions correspond to the rhabdomere (red), roughly the center of the cell body (green), and a part of the cell that lies still further away from the rhabdomere (blue). A fourth region was selected at least 10 μm away from the injected cell to measure the background signal (yellow); inspection of the background showed that the fluorescence intensity distribution is uniform (not shown). The raw intensity values recorded, therefore, are a superposition of a uniform background signal and the signal from the Ca2+ indicator. The signal from the Ca2+ indicator is shaped by the remaining optical properties of the tissue (waveguide properties of the rhabdomere and residual optical effects of the cornea and crystalline cone cells) and inhomogeneities in the distribution of the Ca2+ indicator. Cai in dark-adapted fly photoreceptor cells is below the working range of the indicator OG5N (12, 13), but OG5N has a significant residual fluorescence even in the absence of Ca2+. The distribution of intensity values during the latency period thus reflects these optical properties and inhomogeneities; it can therefore be used for normalization of the traces that have been background subtracted. Hence, the intensity values in the regions of interest were averaged, background subtracted, and subsequently normalized to the initial fluorescence level during the latency period (Fig. 4 b and c, arrows).
Figure 4.

The steady-state distribution of Cai in a cross section of a photoreceptor cell is homogeneous. (a) Raw intensity image (gray scale) of 100 successive images averaged during the steady state. The colored squares indicate the regions of interest quantitatively analyzed in b and c; red, rhabdomere; green, center of cell; blue, side of cell opposite the rhabdomere; yellow, background, outside the cell that was injected with the Ca2+ indicator. (b and c) Normalized fluorescence traces (the first value obtained after turning on the light was used for normalization; arrows). The normalized fluorescence rises sharply in the rhabdomere (red line) and displays a large transient. After the decay of the transient, the normalized fluorescence shows the same values, independent of location of the region of interest. The distribution of Cai during the steady state thus is homogeneous. The traces are averages of five experiments.
The intensity vs. time plots of the normalized fluorescence (Fig. 4 b and c) are fully consistent with our earlier measurements (9, 13). In the rhabdomere, very fast and large Ca2+ transients occur; 10 ms after the onset of the light stimulation, the normalized fluorescence in the rhabdomere has reached its maximum and stays at a plateau for about 100 ms before declining again. The plateau at the highest level of the normalized fluorescence is caused by the saturation of the Ca2+ indicator OG5N (9). The further away from the rhabdomere the region of interest is chosen, the slower the increase of the normalized fluorescence is found to be. Also, the transient at light onset is much reduced in the cell body as compared with the rhabdomere. About 400 ms after onset of light stimulation, however, the normalized fluorescence, and hence Cai, has reached the same value in all regions of interest, i.e., the distribution of Cai in the steady state is homogeneous. This observation argues that the Ca2+ extrusion is closely colocalized with the Ca2+ influx, assuming that the concentration of highly mobile Ca2+ buffers is below 750 μM (Fig. 3). Evidence presented below shows that this condition is likely to be met in our experiments.
Ca2+ Removal in the Rhabdomere Is Faster than in the Cell Body.
After turning off the light stimulation, Ca2+ influx through the light-activated channels ceases, and Cai diminishes because of the ongoing action of the Ca2+ extrusion mechanisms. When Ca2+ extrusion takes place in the rhabdomere, it can be expected that Cai reduces faster in the rhabdomere than in the rest of the cell. We therefore exposed a cell to bright light for 1 s, which allowed Cai to reach a homogeneous steady state (as in Fig. 4) and a subsequent dark period of 800 ms (Fig. 5a). At the end of the dark period, the normalized fluorescence, and hence Cai, in the rhabdomere (red trace) had reached lower values than in the other parts of the cell (green and blue traces, Fig. 5a). In a different experimental protocol (Fig. 5 b-d), we stimulated a dark-adapted cell for only 200 ms and then investigated how Cai developed during the subsequent dark period. Fig. 5b shows that after 200 ms of darkness Cai has diminished in the rhabdomere, whereas it has continued to rise in the cell body, consistent with Ca2+ being redistributed by diffusion. After 400 ms of darkness (Fig. 5c), Cai in the cell body has started to decline; by the same time, Cai in the rhabdomere has fallen to a value lower than that found in the cell body. The same is seen more clearly after 600 ms of darkness (Fig. 5d). Hence, the Ca2+ gradient in the photoreceptor cell reverses its direction: after 200 ms of darkness, Cai in the rhabdomere is still higher than in the cell body, whereas the opposite is found after 400–600 ms of dark adaptation. To analyze the extent of the Ca2+ gradient after 600 ms of dark adaptation, the normalized fluorescence intensity at the onset of the second light stimulation in Fig. 5d was plotted as a function of the distance to the rhabdomere (Fig. 5e). Converting these values to free calcium concentrations (as described in ref. 9) shows that in this particular cell the gradient is substantial, with Ca2+ concentrations ranging from ≈7 μM in the rhabdomere to ≈14 μM at the opposite side of the cell body (Fig. 5f). Together, these results show that Ca2+ removal from the rhabdomere is faster than from the cell body. Consequently, Ca2+ diffuses from the cell body into the rhabdomere during the recovery from light stimulation.
Figure 5.

Ca2+ extrusion is faster in the rhabdomere than in the rest of the cell. (a) After 1 s of light stimulation (of which the last 100 ms are shown), which allowed Cai to reach a homogeneous steady state (as in Fig. 4), the cell was dark adapted for 800 ms. After this dark period, the reduction of the Cai-induced fluorescence was stronger in the rhabdomere (red trace) than in other parts of the cell (green and blue traces). (b–d) A different cell was stimulated for 200 ms, and the light was then turned off for 200 ms (b), 400 ms (c), or 600 ms (d) and turned on again (for 200 ms) to probe the change of fluorescence, and hence Cai, during the dark period. Again, in the rhabdomere, Ca2+ declines much faster and to lower values than in the cell body. (e) The gradient of Cai at the onset of the second light stimulus in d plotted as normalized fluorescence intensity vs. distance from the rhabdomere. The error bars give the SEM obtained by averaging over the amount of pixels indicated by the numbers. (f) From the normalized fluorescence traces, the Cai values were calculated as described in ref. 9 by assuming Fmax = 6.3, Fmin = 1, and Kd = 20 μM (29, 30). The traces were averaged 10 (a), 5 (b), 2 (c), and 6 (d) times.
We cannot directly control or determine the indicator concentration used in the experiments. The measurements shown in Fig. 5 demonstrate that Ca2+ gradients can exist under our experimental conditions after the Ca2+ transients in the rhabdomere at the onset of illumination has ceased. Therefore, the concentration of the Ca2+ indicator used in the experiments was sufficiently low to enable the measurements of Ca2+ gradients. This argues that the homogeneous distribution of Cai in the steady state (Fig. 4) cannot be explained solely with a high concentration of Ca2+ buffer but must indeed be caused by colocalization of Ca2+ influx and extrusion in or close to the rhabdomere (Figs. 2, 3).
Discussion
An increase in Cai in fly photoreceptor cells is brought about by an influx of Ca2+ through the light-activated channels that are exclusively located in the rhabdomere (6, 7, 11). In this report, we show that also Ca2+ extrusion takes place in or close to the rhabdomere. Ca2+ influx and Ca2+ extrusion are therefore colocalized.
Two independent arguments indicate that the homogeneous distribution of Cai we observe in the steady state is not caused by a large mobile Ca2+ buffer capacity: (i) Ca2+ gradients exist in the cell body during periods of dark adaptation (Fig. 5). High concentrations of a Ca2+ buffer would also strongly reduce these gradients, probably to a degree that makes it impossible to measure them. (ii) Using blunt electrodes very easily overloads cells with the Ca2+ indicator. Recordings from those cells show profoundly modified kinetics of the membrane potential (15) and the fluorescence (data not shown) and hence were discarded. Consequently, in not-overfilled cells the concentration of the indicator must have been much lower than 5 mM (the concentration of the indicator in the electrode). Therefore, our measurements exclude the possibility that Ca2+ extrusion is located only on the basolateral membranes (Fig. 2a) and make it unlikely that Ca2+ extrusion takes place only in the apical membrane excluding the rhabdomere (Fig. 2b).
A Method for Locating Ca2+ Extrusion.
Imaging of cells injected with fluorescent Ca2+ indicator dyes is a well-established method for demonstrating the location of Ca2+ channels (e.g., refs. 11, 43, and 44). Here we show that measuring spatial Ca2+ gradients, in combination with modeling, can be used for demonstrating the location of Ca2+ extrusion in vivo. As this method characterizes the location of Ca2+ extrusion by its very function, it is not necessary to know which types of molecules extrude Ca2+. It contrasts in this respect with the traditional approach using antibodies. An intrinsic problem of the latter is that not every protein detected by an antibody might be functional, and proteins might be regulated differentially depending on where they are located. The approach used in this report therefore might provide a viable and direct alternative in systems where data on Ca2+-extruding proteins are absent and might complement traditional approaches in other systems.
The Identity of the Ca2+-Extruding Mechanism.
Ample evidence suggests that Na+/Ca2+ exchanger proteins are present in fly photoreceptor cells (20–25) and in photoreceptor cells of other invertebrates (bee, ref. 45; Limulus, refs. 46 and 47; squid, ref. 26). Manipulating the function of the Na+/Ca2+ exchanger can augment Cai in Limulus (46), which mimics the phenomena of light adaptation (14), generally believed to depend on an increase in Cai (14, 15). The Na+/Ca2+ exchanger thus appears to be the main mechanism of Ca2+ extrusion in invertebrate photoreceptor cells. Probably, therefore, the Na+/Ca2+ exchanger proteins contribute the largest part (or all) of the Ca2+ extrusion observed in our experiments. From the experiments presented here, it follows that Na+/Ca2+ exchanger proteins are located in or close to the rhabdomere of fly photoreceptor cells. This is in agreement with Bauer et al. (26), who demonstrated Na+/Ca2+ exchange in the rhabdomeres of squid photoreceptor cells. Because squids and flies do not belong to phylogenetically closely related groups, this raises the possibility that colocalization of Na+/Ca2+ exchangers and light-activated channels in rhabdomeres is a general feature of invertebrate photoreceptor cells.
The Close Colocalization of Ca2+ Influx and Ca2+ Extrusion Has Important Functional Implications.
Ca2+ directly regulates light-activated channels (22, 48) and either directly or via calmodulin regulates many other proteins involved in phototransduction (49). Because most of those proteins are located in or close to the rhabdomere (49), Cai in the rhabdomere rather than in the cell body may control the state of light adaptation. Rapid dark adaptation depends therefore on rapid removal of Ca2+ from the rhabdomere. Localizing Na+/Ca2+ exchanger proteins to the rhabdomere helps achieve this (Fig. 5). Similarly, the colocalization of Ca2+ influx channels and Ca2+ extrusion mechanisms in small compartments observed in other cell types (16–19) might have the same function, i.e., to increase the speed of Ca2+ removal.
Colocalization of Ca2+ influx and extrusion results in a homogeneous distribution of Cai in the steady state throughout the cell body (Fig. 4c). This ensures that structures located remotely from the Ca2+ influx are also exposed to the same levels of Cai. This is potentially important for the regulation of mitochondria that are located near the basolateral sides of fly photoreceptor cells (33) and are known to be regulated by Cai (50).
Supplementary Material
Acknowledgments
We thank J. Land and H. L. Leertouwer for expert technical assistance and M. Postma for helpful discussions concerning the numerical calculations.
Abbreviations
- Cai
free intracellular Ca2+ concentration
- OG5N
Oregon green 5N
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Koch C, Zador A. J Neurosci. 1993;13:413–422. doi: 10.1523/JNEUROSCI.13-02-00413.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lenzi D, Roberts W M. Curr Opin Neurobiol. 1994;4:496–502. doi: 10.1016/0959-4388(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 3.Augustine G J, Neher E. Curr Opin Neurobiol. 1992;2:302–307. doi: 10.1016/0959-4388(92)90119-6. [DOI] [PubMed] [Google Scholar]
- 4.Hardie R C, Minke B. Neuron. 1992;8:643–651. doi: 10.1016/0896-6273(92)90086-s. [DOI] [PubMed] [Google Scholar]
- 5.Reuss H, Mojet M H, Chyb S, Hardie R C. Neuron. 1997;19:1249–1259. doi: 10.1016/s0896-6273(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 6.Huber A, Sander P, Gobert A, Bähner M, Hermann R, Paulsen R. EMBO J. 1996;15:7036–7045. [PMC free article] [PubMed] [Google Scholar]
- 7.Niemeyer B A, Suzuki E, Scott K, Jalink K, Zuker C S. Cell. 1996;85:651–659. doi: 10.1016/s0092-8674(00)81232-5. [DOI] [PubMed] [Google Scholar]
- 8.Hardie R C. In: Progress in Sensory Physiology. Ottoson D, editor. Vol. 5. Heidelberg: Springer; 1985. pp. 1–79. [Google Scholar]
- 9.Oberwinkler J, Stavenga D G. J Neurosci. 2000;20:1701–1709. doi: 10.1523/JNEUROSCI.20-05-01701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Postma M, Oberwinkler J, Stavenga D G. Biophys J. 1999;77:1811–1823. doi: 10.1016/S0006-3495(99)77026-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ranganathan R, Bacskai B J, Tsien R Y, Zuker C S. Neuron. 1994;13:837–848. doi: 10.1016/0896-6273(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 12.Hardie R C. J Neurosci. 1996;16:2924–2933. doi: 10.1523/JNEUROSCI.16-09-02924.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oberwinkler J, Stavenga D G. J Gen Physiol. 1998;112:113–124. doi: 10.1085/jgp.112.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lisman J E, Brown J E. J Gen Physiol. 1972;59:701–719. doi: 10.1085/jgp.59.6.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muijser H. J Comp Physiol. 1979;132:87–95. [Google Scholar]
- 16.Yamoah E N, Lumpkin E A, Dumont R A, Smith P J, Hudspeth A J, Gillespie P G. J Neurosci. 1998;18:610–624. doi: 10.1523/JNEUROSCI.18-02-00610.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgans C W, El Far O, Berntson A, Wässle H, Taylor W R. J Neurosci. 1998;18:2467–2474. doi: 10.1523/JNEUROSCI.18-07-02467.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krizaj D, Copenhagen D R. Neuron. 1998;21:249–256. doi: 10.1016/s0896-6273(00)80531-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reuter H, Porzig H. Neuron. 1995;15:1077–1084. doi: 10.1016/0896-6273(95)90096-9. [DOI] [PubMed] [Google Scholar]
- 20.Armon E, Minke B. Biophys Struct Mech. 1983;9:349–357. [Google Scholar]
- 21.Hochstrate P. Z Naturforsch. 1991;46c:451–460. [Google Scholar]
- 22.Hardie R C. J Neurosci. 1995;15:889–902. doi: 10.1523/JNEUROSCI.15-01-00889.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerster U. Vision Res. 1997;37:2477–2485. doi: 10.1016/s0042-6989(97)00079-5. [DOI] [PubMed] [Google Scholar]
- 24.Schwarz E M, Benzer S. Proc Natl Acad Sci USA. 1997;94:10249–10254. doi: 10.1073/pnas.94.19.10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haug-Collet K, Pearson B, Webel R, Szerencsei R T, Winkfein R J, Schnetkamp P P, Colley N J. J Cell Biol. 1999;147:659–770. doi: 10.1083/jcb.147.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bauer P J, Schauf H, Schwarzer A, Brown J E. Am J Physiol. 1999;276:C558–C565. doi: 10.1152/ajpcell.1999.276.3.C558. [DOI] [PubMed] [Google Scholar]
- 27.Allbritton N L, Meyer T, Stryer L. Science. 1992;258:1812–1815. doi: 10.1126/science.1465619. [DOI] [PubMed] [Google Scholar]
- 28.Hall J D, Betarbet S, Jaramillo F. Biophys J. 1997;73:1243–1252. doi: 10.1016/S0006-3495(97)78157-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haugland R P. Handbook of Fluorescent Probes and Research Chemicals. Ed. 6. Eugene, OR: Molecular Probes; 1996. [Google Scholar]
- 30.Dabdoub A, Payne R. J Neurosci. 1999;19:10262–10269. doi: 10.1523/JNEUROSCI.19-23-10262.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roberts W M. J Neurosci. 1994;14:3246–3262. doi: 10.1523/JNEUROSCI.14-05-03246.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cook B, Minke B. Cell Calcium. 1999;25:161–171. doi: 10.1054/ceca.1998.0018. [DOI] [PubMed] [Google Scholar]
- 33.Walz B. J Ultrastruct Res. 1982;81:240–248. doi: 10.1016/s0022-5320(82)90079-x. [DOI] [PubMed] [Google Scholar]
- 34.Blaustein M P, Lederer W J. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- 35.Kirschfeld K, Franceschini N. Kybernetik. 1969;6:13–22. doi: 10.1007/BF00288624. [DOI] [PubMed] [Google Scholar]
- 36.Weckström M, Hardie R C, Laughlin S B. J Physiol (London) 1991;440:635–657. doi: 10.1113/jphysiol.1991.sp018729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hille B. Ionic Channels of Excitable Membranes. Ed. 2. Sunderland, MA: Sinauer; 1992. [Google Scholar]
- 38.Gerster U, Stavenga D G, Backhaus W. J Comp Physiol A. 1997;180:113–122. [Google Scholar]
- 39.Coles J A, Orkand R K, Yamate C L, Tsacopoulos M. Ann NY Acad Sci. 1985;481:303–317. doi: 10.1111/j.1749-6632.1986.tb27160.x. [DOI] [PubMed] [Google Scholar]
- 40.Sandler K, Kirschfeld K. J Comp Physiol A. 1991;169:299–311. [Google Scholar]
- 41.Baumann O, Lautenschläger B, Takeyasu K. Cell Tissue Res. 1994;275:225–234. doi: 10.1007/BF00319420. [DOI] [PubMed] [Google Scholar]
- 42.Neher E, Augustine G J. J Physiol (London) 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Denk W, Holt J R, Shepherd G M G, Corey D P. Neuron. 1995;15:1311–1321. doi: 10.1016/0896-6273(95)90010-1. [DOI] [PubMed] [Google Scholar]
- 44.Lumpkin E A, Hudspeth A J. Proc Natl Acad Sci USA. 1995;92:10297–10301. doi: 10.1073/pnas.92.22.10297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Minke B, Tsacopoulos M. Vision Res. 1986;26:679–690. doi: 10.1016/0042-6989(86)90082-9. [DOI] [PubMed] [Google Scholar]
- 46.O'Day P M, Gray-Keller M P. J Gen Physiol. 1989;93:473–492. doi: 10.1085/jgp.93.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deckert A, Stieve H. J Physiol (London) 1991;443:467–482. doi: 10.1113/jphysiol.1991.sp018438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hardie R C, Minke B. J Gen Physiol. 1994;103:409–427. doi: 10.1085/jgp.103.3.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Montell C. Annu Rev Cell Dev Biol. 1999;15:231–268. doi: 10.1146/annurev.cellbio.15.1.231. [DOI] [PubMed] [Google Scholar]
- 50.Fein A, Tsacopoulos M. Nature (London) 1988;331:437–440. doi: 10.1038/331437a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}