

Summary
It has been known for some time that the double-helix is not a uniform structure but rather exhibits sequence-specific variations that, combined with base-specific intermolecular interactions, offer the possibility of numerous modes of protein-DNA recognition. All-atom simulations have revealed mechanistic insights into the structural and energetic basis of various recognition mechanisms for a number of protein-DNA complexes while coarser grained simulations have begun to provide an understanding of the function of larger assemblies. Molecular simulations have also been applied to the prediction of transcription factor binding sites, while empirical approaches have been developed to predict nucleosome positioning. Studies that combine and integrate experimental, statistical and computational data offer the promise of rapid advances in our understanding of protein-DNA recognition mechanisms.
Introduction
The DNA double-helix is extremely flexible and its detailed structure is affected by its base sequence, environmental conditions, and interactions with other molecules. Sequence-dependent deviations from ideal B-DNA are the rule rather than the exception and, as will be discussed, appear to play a crucial role in protein-DNA readout mechanisms. Protein-DNA recognition is often described in terms of direct and indirect readout [1]. Direct readout involves the formation of specific contacts between amino acid side chains and DNA bases in such a way that a particular DNA sequence forms an energetically favorable interface with the residues on the surface of a given protein. Indirect readout describes the sequence-dependent bending of the overall DNA structure so as to optimize the protein-DNA interface geometry. Direct and indirect readout are somewhat ambiguous terms that provide conceptual simplifications of the complexities that underlie any binding process involving macromolecules. In this vein we have recently identified another mode of recognition involving sequence-specific local deviations from ideal B-DNA structure which are recognized by specific amino acids [2••].
A large and diverse set of computational approaches have been applied to the problem of predicting DNA structure and function. These include all-atom Molecular Dynamics (MD) [3•, 4•] and Monte Carlo (MC) simulations [2••, 5•, 6•], based on all-atom or coarser grained representations, as well as bioinformatics analysis [7•]. Such approaches generate a link between molecular structure and genome-wide analysis, a goal that has assumed increased importance as high-throughput methods continue to provide a wealth of data about sequence-dependent recognition. This article summarizes recent progress in molecular simulations of DNA structure and protein-DNA interactions.
Predicting DNA structure and dynamics
Experimental data provide an incomplete structural map of free DNA
Our current knowledge of sequence-dependent DNA conformation is often insufficient to determine whether a particular DNA deformation observed in a complex is induced by binding or is an inherent property of a particular nucleotide sequence. In fact, the effort and complexity of solving a structure with X-ray crystallography is significantly higher for free DNA than for complexes, due in part to the higher flexibility of the smaller molecules. Moreover, the paucity of NOE constraints complicates the use of NMR to solve free DNA structures although the use of dipolar coupling has significantly improved the situation [8]. The general problem is highlighted in Figure 1 which illustrates, for crystal structures, that the number of PDB releases has increased rapidly for protein-DNA complexes in comparison with free-DNA structures. Indeed, there has been only limited recent interest in studying the structure of free B-DNA, perhaps due in part to uncertainties associated with the biological relevance of the problem. Yet, it has become apparent that sequence-specific variations in the structure of the double-helix play a central role in protein-DNA recognition.
Figure 1.

Histogram of number of released crystal structures of Protein-DNA complexes and free DNA organized by Protein Data Bank release date
Despite the limited quantity of experimental data on free-DNA structure, much has been learned from the analysis of the data that are available. A particularly important set of observations concerns the structural properties of A-tracts, which are runs of at least three consecutive ApA, TpT, or ApT base pair steps [9,10]. A-tracts produce a characteristic narrowing of the minor groove which is due to favorable interactions between functional groups of adjacent base pairs in the major groove [11]. TpA steps are an exception to this behavior since base stacking is minimal for this step which results in fewer structural constraints and hence greater conformational variability. Olson and coworkers have carried out detailed analysis of the conformational properties of DNA at the base-pair level by describing the geometry of the ten unique dinucleotides, as seen in all available crystal structures [12,13]. Structural fluctuations observed among these dinucleotide steps were associated with conformational variability and have formed the basis of empirical force fields for DNA [13].
Dinucleotide geometry is affected by adjacent base pairs, which is why a description at the level of tetra-nucleotides is preferable. However, the relatively small number of available crystal structures of free B-DNA and their lack of diversity (some sequences are easier to crystallize than others) has the consequence that there is structural information for only 95 of the 136 possible unique tetra-nucleotides. This situation is compounded by the fact that flanking sequences and crystallization conditions can affect tetra-nucleotide geometry so that it is often difficult to draw meaningful conclusions from just a single structure. The Ascona B-DNA Consortium (ABC) [3•] and the Sarai lab [4•] have sought to complete structural knowledge of all tetra-nucleotides through all-atom MD simulations.
Another powerful approach that can provide information on sequence-dependent DNA structure is hydroxyl radical cleavage [14]. The Tullius lab recently used hydroxyl cleavage to screen, in a high-throughput manner, for sequence-dependent variations of the solvent-accessible surface area of DNA [15••]. This method provides information on local shape and structure effects of DNA without the need for atomic-resolution data.
Molecular dynamics simulations
AMBER is still the only force field that has been extensively tested for free-DNA simulations [16]. It produces structures in qualitative agreement with experiment for the Dickerson dodecamer, DNA bending, and for the A-DNA/B-DNA transition [17]. However, use of AMBER94 appears to result in systematic under-twisting [17], apparently due to artifactual α/γ-flips [3•]. Sarai and coworkers report a less serious under-twisting in MD simulations with AMBER99 [4•]. However, since a 50ns MD simulation based on AMBER99 reported the total corruption of a double-helix, it was concluded that force field artifacts become more prevalent in longer MD simulations [18•]. Related problems were reported in an MD study of DNA minicircles [19].
The Barcelona modification makes improvements in AMBER through a refinement of torsional parameters based on quantum-mechanical calculations [18•]. Long MD simulations of the Dickerson dodecamer suggest that the Barcelona force field produces more stable simulations [18•] and, indeed, the first microsecond MD simulation of DNA has been reported [20]. The CHARMM force field has been applied to MD simulations of free DNA as well but its performance has not yet been tested in long simulations [21]. Further advances in MD methods in recent years included the development of a polarizable force field for DNA [22] and the use of implicit solvent simulations [23–25]. However, a comprehensive validation of the Barcelona and other force fields through large-scale comparisons to experimentally determined structures is still required.
Monte Carlo simulations
MC simulations offer an alternate approach to CPU-expensive MD simulations. A new MC approach uses variables that are derived from the chemical topology of DNA [26], a slightly modified version of AMBER94, explicit ions, and a screened Coulomb potential [5•]. Although solvation/desolvation effects are not accounted for, the force field appears to capture many important features of sequence-dependent DNA structure. In addition, the MC algorithm is particularly effective in sampling thus allowing it to be applied to global conformational search and structure prediction problems. The method has been used to predict sequence-dependent DNA bending [5•] and local intrinsic deviations from B-DNA conformation [2••], and was employed for drug-DNA docking [27•]. Figure 2 illustrates the prediction of sequence-specific minor groove width of the Dickerson dodecamer. In addition to the excellent agreement with the experimentally determined minor groove width that is evident from the figure, the predicted average helix twist of 34.7° also agrees well with experiment (X-ray 35.4°; NMR 35.3°). The success of an approach based on a detailed description of the DNA but a crude solvent model indicates that intramolecular interactions dominate stacking geometry, and in turn, the detailed structure of the double-helix. This finding suggests that simplified approaches that do not require a full all-atom description of solvent molecules may be quite effective in the prediction of the sequence dependence of DNA conformation.
Figure 2. Sequence-dependence of minor groove shape for Dickerson dodecamer.

– Ideal B-DNA (A) and the Dickerson dodecamer, PDB code 1duf (B), differ in minor groove shape as shown by the color coding of the molecular surface (green for convex, black/grey for concave surfaces). (C) The intrinsically narrow minor groove in the center of the Dickerson dodecamer is predicted by MC simulations using ideal B-DNA as a starting conformation. The X-ray data represent an average of the 15 available crystal structures, symmetrized based on the palindromic sequence CGCGAATTCGCG in order to remove crystal packing effects. The NMR data represent an average of 10 structures that included dipolar coupling data in their structure determination. Minor groove width is calculated with the CURVES program [55].
Protein-DNA interactions
Molecular dynamics simulations
MD simulations have been increasingly employed to study the dynamics of protein-DNA binding and the interplay of direct and indirect readout. Valuable insights have been obtained although the balance between intra- and intermolecular interactions in force fields is somewhat uncertain and remains a challenge for the study of protein-DNA complexes [28]. Currently, CHARMM is predominantly used in MD simulations of complexes but AMBER has been the force field of choice for free DNA.
MD simulations were used to study the recognition of DNA by the tumor-suppressor protein p53 [29•,30,31]. The p53 consensus binding site is comprised of two decameric half-sites that are separated by a variable number of base pairs. Shakked and coworkers solved the first crystal structures of p53-tetramer binding to DNA [32••]. The DNA binding site in these structures was composed of two half-sites separated by a two-base pair linker. Nussinov and coworkers employed MD simulations to study the complex without base pair insertions between the half-sites and showed that bending of the full site enhances contacts between p53 core domains [29•]. In another MD study, it was demonstrated that alterations in the sequence of the p53 consensus site affect DNA shape and the strength of crucial p53-DNA contacts [30].
Monte Carlo simulations
MC simulations, based on a coarse-grained model that was derived from sequence-specific elastic properties of DNA, have been applied to study the binding of the Lac repressor to its operator. It was found that nucleotide sequence affects the conformation of the DNA loop in the Lac repressor-DNA complex [33]. Similarly, non-specific binding of the nucleotide protein HU to circular DNA was studied, and it was shown that HU proteins induce local DNA bending and untwisting [6•]. Interest in the mechanism of nucleosome packaging in chromatin has led to the development of a coarse-grained model that accounts for histone tail and inter-nucleosomal linker flexibility. This model treats the nucleosome as a rigid core with uniformly distributed charges, and stresses the role of the positively charged histone tails in packaging of oligonucleosomes [34].
Minor groove recognition exploits shape and electrostatic potential
Recent work on Hox proteins revealed that subtle sequence-dependent local variations in minor groove geometry provide a mechanism through which different proteins in the same family can recognize small differences in nucleotide sequence [2••]. Crystal structures were determined for ternary complexes involving the homeodomains of one of the eight Drosophila Hox proteins, Scr, and its Exd co-factor, and DNA. One complex contained a DNA binding site, fkh250, that was specific for Scr, while the other contained a consensus DNA site, fkh250con*, that binds other Hox proteins as well. Both complexes have the homeodomain recognition helices of the Scr protein and its Exd co-factor bound in the major groove, which is the main source for binding affinity. However, as shown in Figure 3, two side chains, His-12 and Arg3, are seen in the crystal structure of the complex with the fkh250 site whereas they are disordered when presented with the fkh250con* site. Binding studies suggest that both side chains play a key role in determining in vivo specificity [2••]. The specific recognition of the fkh250 sequence appears to be related to the narrow minor groove in the His-12/Arg3 binding site whereas the groove is much wider in the equivalent region of the fkh250con* sequence. MC simulations indicate that this difference in shape is a property of the free DNA.
Figure 3. Recognition of minor grove shape and electrostatic potential by a Hox homeodomain.
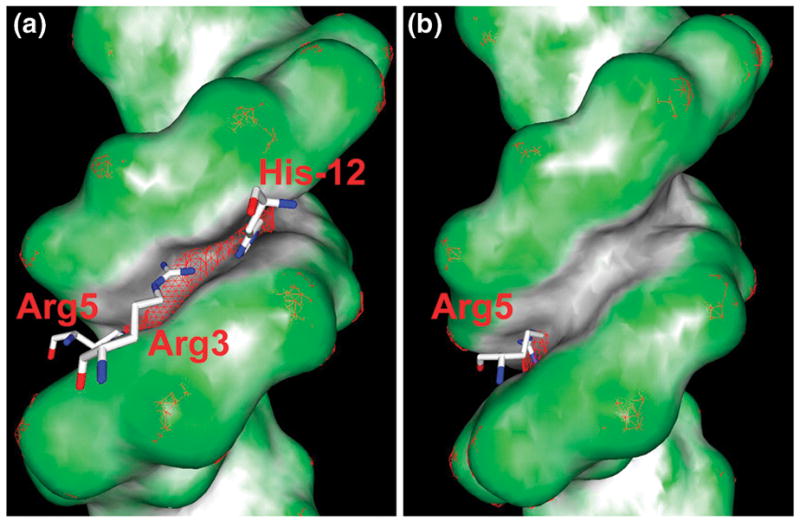
- Crystal structures of Scr bound to its specific fkh250 sequence, PDB code 2r5z (A), and the Hox consensus sequence fkh250con*, PDB code 2r5y (B). Color coding of the molecular surface reveals, for the fkh250 site, an extended region with a narrow minor groove, which binds His-12, Arg3, and Arg5. In contrast, the minor groove of the fkh250con* site is only narrow at the local region that binds Arg5. A mesh of the -8kT/e isosurface illustrates that the electrostatic potential, as calculated with DelPhi [35], is more negative in the minor groove of the fkh250 site than in the fkh250con* site. MC simulations of the free binding sites indicate the distinct minor groove shape to be an intrinsic feature of the free binding sites (see text). Molecular shape and electrostatic isosurface representations were generated with GRASP2 [56].
Calculations using the DelPhi program [35] indicated that the effect of minor groove width on binding can be traced to the electrostatic potential of the DNA. Narrow grooves produce enhanced electrostatic potentials due to electrostatic focusing effects originally discovered for enzyme active sites [36]. The effect of minor groove shape on electrostatic potential offers a new mode of protein-DNA recognition. Specifically, sequence-dependent variations in DNA shape can exploit corresponding variations in electrostatic potential to tune binding affinities, even among closely related members of the same protein family.
Prediction of protein-DNA binding sites
Transcription factor binding sites
All-atom calculations of protein-DNA binding energies have been used to predict the nucleotide sequence recognized by a particular protein. Most such studies start with a protein-DNA complex of known structure. Mutations are then computationally introduced into the base sequence and/or the protein and changes in binding affinity are calculated. In one such study, it was shown that either direct or indirect readout mechanisms dominate in certain protein-DNA complexes, and, if identified, can enable reliable DNA binding site predictions [37•]. A similar approach characterized the role of intermolecular interactions and the elastic properties of DNA in protein-DNA specificity [38]. Deformation energies, derived from either energetic parameters associated with different base pair steps [39•,40] or from all-atom conformational energies [41], have also been shown to increase the accuracy of affinity predictions. Overall, structure-based predictions of binding specificity seem to work quite well if a template with an appropriate docking geometry is available, at least in the case of zinc-finger proteins [42,43•].
Studies of this type are of potentially great importance since they offer a fundamentally new, non-statistical approach for the prediction of transcription factor binding sites. Their success will depend on continued improvements in the ability to calculate the sequence dependence of DNA conformation and protein-DNA interaction energies. Complexities in these areas result in part from the absence of a force field specifically designed for protein-DNA interactions and from the fact that the protein-DNA interface is not always closely packed and may contain water molecules that need to be taken into account. Indeed there have been only limited reports of the prediction of protein side chain conformations in the protein-DNA interface. This problem constitutes an important challenge although there have been recent reports of promising advances [43•,44,45]. All-atom predictions can provide fundamental insights, but they also suffer from inaccuracies in the force field and, in addition, they are generally too slow for genome-wide analysis. This problem can be reduced by using knowledge-based constraints on protein-DNA contacts to predict transcription factor binding sites [46••].
Nucleosome positioning
There is much interest in understanding how and predicting where nucleosomes are positioned on the genome. The wrapping of nucleosomal DNA around the histone core requires deformations in the double-helix. Helical parameters [47] were employed to describe inter-base pair translations and rotations that can accommodate this distortion and to suggest a structural mechanism that leads to the formation of a superhelical pitch [48••]. This is an important development because bending in circular DNA was understood on a base-pair level but the superhelical pitch was not. Knowledge of the specific dinucleotide deformations that are necessary for nucleosome formation made it possible to predict experimentally identified binding sites with a three-base pair or higher accuracy [49].
The occupancy of nucleosomes at different positions along the yeast genome was experimentally analyzed in vivo [7•]. It was found, based on statistical analysis of the periodicity and phase relationship of certain dinucleotides in nucleosomal DNA, that nucleosome sites can be predicted with approximately 50% accuracy [7•,50]. However, a similar analysis of C. elegans nucleosomes singled out different positional preferences for dinucleotides at nucleosomal DNA [51] than found in the yeast study [7•]. Despite many advances, there is still much controversy in this area. Current approaches are based on the properties of dinucleotides, but it is likely that more molecular detail is required for solving the nucleosome positioning problem.
Conclusions
Protein-DNA recognition is the result of a complex interplay of physical interactions that act at varying levels of nucleotide sequence specificity. At the simplest level, there are non-specific electrostatic interactions between positive regions on a protein surface and the negatively charged DNA that bring a protein to the vicinity of DNA and may facilitate diffusion along its surface (see e.g. recent discussion in [52•,53,54]). At the next level, there can be family-specific recognition of particular nucleotide sequences; for example the binding of the recognition helix of many Hox proteins to the major groove of DNA. This recognition is specific to an entire family but is non-specific in the sense that major groove binding alone is not sufficient to distinguish one Hox protein from another. We have seen here that finer grained recognition occurs in the minor groove through the recognition of differences in electrostatic potential mediated by differences in minor groove shape. On the protein side, shape is recognized by the specific placement of basic amino acids in conformations that enable them to interact optimally with subtle changes in electrostatic potential [2••]. The binding mechanisms of Hox proteins appear then to be cooperative, with family-wide major groove binding providing much of the affinity while protein-specific minor groove binding provides much of the protein-level specificity.
Mechanistic details will likely vary among families but, as is true in so many other cases, biological systems seem to exploit whatever is available to achieve specificity. This reality complicates any attempt to come up with simple rules for protein-DNA recognition, and poses challenges to detailed simulations, since individual contributions are often relatively small. On the other hand, the identification of the range of possible mechanisms and their association with the strategies used by individual families has the potential to lead to the development of new computational approaches that can be applied to specific problems. In this article we have focused on one such mechanism involving the effect of DNA sequence on the detailed structure of the double-helix.
The insights gained from studies of the conformational properties of specific sequences, both in free DNA and in complexes, should provide invaluable insights in the development of fast algorithms that can predict specificity on a genome-wide scale. Such approaches should prove to become important complements to new high-throughput methods that probe protein-DNA binding specificity. More generally, the integration of structural information, simulation technologies, binding data, and bioinformatics approaches will play an increasingly important role in our understanding of the remarkable ability of the seemingly uniform double-helix to accomplish so many tasks that rely on subtle variations in its structure and composition.
Acknowledgments
We thank Professors Zippora Shakked and Richard Mann for many valuable discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and annotations
- 1.Sarai A, Kono H. Protein-DNA recognition patterns and predictions. Annu Rev Biophys Biomol Struct. 2005;34:379–398. doi: 10.1146/annurev.biophys.34.040204.144537. [DOI] [PubMed] [Google Scholar]
- 2••.Joshi R, Passner JM, Rohs R, Jain R, Sosinsky A, Crickmore MA, Jacob V, Aggarwal AK, Honig B, Mann RS. Functional specificity of a hox protein mediated by the recognition of minor groove structure. Cell. 2007;131:530–543. doi: 10.1016/j.cell.2007.09.024. This paper establishes sequence-dependent minor groove shape as the molecular basis of Hox specificity. The paper introduces local shape readout as a new mode of protein-DNA recognition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3•.Dixit SB, Beveridge DL, Case DA, Cheatham TE, 3rd, Giudice E, Lankas F, Lavery R, Maddocks JH, Osman R, Sklenar H, et al. Molecular dynamics simulations of the 136 unique tetranucleotide sequences of DNA oligonucleotides. II: sequence context effects on the dynamical structures of the 10 unique dinucleotide steps. Biophys J. 2005;89:3721–3740. doi: 10.1529/biophysj.105.067397. This work reports structural information on all 136 unique tetra-nucleotides based on MD simulations with the AMBER94 force field. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4•.Fujii S, Kono H, Takenaka S, Go N, Sarai A. Sequence-dependent DNA deformability studied using molecular dynamics simulations. Nucleic Acids Res. 2007;35:6063–6074. doi: 10.1093/nar/gkm627. This study reports structural information on all 136 unique tetra-nucleotides based on MD simulations with the AMBER99 force field. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5•.Rohs R, Sklenar H, Shakked Z. Structural and energetic origins of sequence-specific DNA bending: Monte Carlo simulations of papillomavirus E2-DNA binding sites. Structure. 2005;13:1499–1509. doi: 10.1016/j.str.2005.07.005. This paper provides insights into the molecular basis of indirect readout mechanisms and predicts sequence-dependent DNA bending based on MC simulations. [DOI] [PubMed] [Google Scholar]
- 6•.Czapla L, Swigon D, Olson WK. Effects of the nucleoid protein HU on the structure, flexibility, and ring-closure properties of DNA deduced from Monte Carlo simulations. J Mol Biol. 2008;382:353–370. doi: 10.1016/j.jmb.2008.05.088. This paper uses MC simulations based on a coarse-grained model to show that the non-specific binding of the histone-like HU protein to DNA induces structural distortions of the binding site. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7•.Segal E, Fondufe-Mittendorf Y, Chen L, Thastrom A, Field Y, Moore IK, Wang JP, Widom J. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. This study determines nucleosome sites in vivo and tries to predict these sites based on an approach that employs dinucleotide occupancy statistics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu Z, Delaglio F, Tjandra N, Zhurkin VB, Bax A. Overall structure and sugar dynamics of a DNA dodecamer from homo- and heteronuclear dipolar couplings and 31P chemical shift anisotropy. J Biomol NMR. 2003;26:297–315. doi: 10.1023/a:1024047103398. [DOI] [PubMed] [Google Scholar]
- 9.Nelson HC, Finch JT, Luisi BF, Klug A. The structure of an oligo(dA). oligo(dT) tract and its biological implications. Nature. 1987;330:221–226. doi: 10.1038/330221a0. [DOI] [PubMed] [Google Scholar]
- 10.Hizver J, Rozenberg H, Frolow F, Rabinovich D, Shakked Z. DNA bending by an adenine--thymine tract and its role in gene regulation. Proc Natl Acad Sci U S A. 2001;98:8490–8495. doi: 10.1073/pnas.151247298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crothers DM, Shakked Z. DNA bending by adenine-thymine tracts. In: Neidle S, editor. Oxford Handbook of Nucleic Acid Structures. Oxford University Press; 1999. pp. 455–470. [Google Scholar]
- 12.Gorin AA, Zhurkin VB, Olson WK. B-DNA twisting correlates with base-pair morphology. J Mol Biol. 1995;247:34–48. doi: 10.1006/jmbi.1994.0120. [DOI] [PubMed] [Google Scholar]
- 13.Olson WK, Gorin AA, Lu XJ, Hock LM, Zhurkin VB. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc Natl Acad Sci U S A. 1998;95:11163–11168. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burkhoff AM, Tullius TD. Structural details of an adenine tract that does not cause DNA to bend. Nature. 1988;331:455–457. doi: 10.1038/331455a0. [DOI] [PubMed] [Google Scholar]
- 15••.Greenbaum JA, Pang B, Tullius TD. Construction of a genome-scale structural map at single-nucleotide resolution. Genome Res. 2007;17:947–953. doi: 10.1101/gr.6073107. This study uses hydroxyl radical cleavage data to provide genome-wide structural information on the solvent-accessible surface area of DNA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheatham TE., 3rd Simulation and modeling of nucleic acid structure, dynamics and interactions. Curr Opin Struct Biol. 2004;14:360–367. doi: 10.1016/j.sbi.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Cheatham TE, 3rd, Kollman PA. Molecular dynamics simulation of nucleic acids. Annu Rev Phys Chem. 2000;51:435–471. doi: 10.1146/annurev.physchem.51.1.435. [DOI] [PubMed] [Google Scholar]
- 18•.Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE, 3rd, Laughton CA, Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. This work attempts to correct the AMBER force field imbalance regarding α/γ-backbone torsion angle flips. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lankas F, Lavery R, Maddocks JH. Kinking occurs during molecular dynamics simulations of small DNA minicircles. Structure. 2006;14:1527–1534. doi: 10.1016/j.str.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 20.Perez A, Luque FJ, Orozco M. Dynamics of B-DNA on the microsecond time scale. J Am Chem Soc. 2007;129:14739–14745. doi: 10.1021/ja0753546. [DOI] [PubMed] [Google Scholar]
- 21.Orozco M, Noy A, Perez A. Recent advances in the study of nucleic acid flexibility by molecular dynamics. Curr Opin Struct Biol. 2008;18:185–193. doi: 10.1016/j.sbi.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 22.Babin V, Baucom J, Darden TA, Sagui C. Molecular dynamics simulations of DNA with polarizable force fields: convergence of an ideal B-DNA structure to the crystallographic structure. J Phys Chem B. 2006;110:11571–11581. doi: 10.1021/jp061421r. [DOI] [PubMed] [Google Scholar]
- 23.Chocholousova J, Feig M. Implicit solvent simulations of DNA and DNA-protein complexes: agreement with explicit solvent vs experiment. J Phys Chem B. 2006;110:17240–17251. doi: 10.1021/jp0627675. [DOI] [PubMed] [Google Scholar]
- 24.Prabhu NV, Panda M, Yang Q, Sharp KA. Explicit ion, implicit water solvation for molecular dynamics of nucleic acids and highly charged molecules. J Comput Chem. 2008;29:1113–1130. doi: 10.1002/jcc.20874. [DOI] [PubMed] [Google Scholar]
- 25.Bomble YJ, Case DA. Multiscale modeling of nucleic acids: insights into DNA flexibility. Biopolymers. 2008;89:722–731. doi: 10.1002/bip.21000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sklenar H, Wustner D, Rohs R. Using internal and collective variables in Monte Carlo simulations of nucleic acid structures: chain breakage/closure algorithm and associated Jacobians. J Comput Chem. 2006;27:309–315. doi: 10.1002/jcc.20345. [DOI] [PubMed] [Google Scholar]
- 27•.Rohs R, Bloch I, Sklenar H, Shakked Z. Molecular flexibility in ab initio drug docking to DNA: binding-site and binding-mode transitions in all-atom Monte Carlo simulations. Nucleic Acids Res. 2005;33:7048–7057. doi: 10.1093/nar/gki1008. This paper presents the first ab-initio drug-DNA docking method with complete flexibility of both the ligand and receptor molecule throughout the docking process. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mackerell AD, Jr, Nilsson L. Molecular dynamics simulations of nucleic acid-protein complexes. Curr Opin Struct Biol. 2008;18:194–199. doi: 10.1016/j.sbi.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29•.Pan Y, Nussinov R. Structural basis for p53 binding-induced DNA bending. J Biol Chem. 2007;282:691–699. doi: 10.1074/jbc.M605908200. This paper used MD simulations to provide insights into the architecture of the complex formed by the p53 tetramer with its consensus DNA binding site. [DOI] [PubMed] [Google Scholar]
- 30.Pan Y, Nussinov R. p53-Induced DNA bending: the interplay between p53-DNA and p53-p53 interactions. J Phys Chem B. 2008;112:6716–6724. doi: 10.1021/jp800680w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma B, Pan Y, Gunasekaran K, Venkataraghavan RB, Levine AJ, Nussinov R. Comparison of the protein-protein interfaces in the p53-DNA crystal structures: towards elucidation of the biological interface. Proc Natl Acad Sci U S A. 2005;102:3988–3993. doi: 10.1073/pnas.0500215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32••.Kitayner M, Rozenberg H, Kessler N, Rabinovich D, Shaulov L, Haran TE, Shakked Z. Structural basis of DNA recognition by p53 tetramers. Mol Cell. 2006;22:741–753. doi: 10.1016/j.molcel.2006.05.015. This study presents the first crystal structures of p53 tetramer-DNA complexes. [DOI] [PubMed] [Google Scholar]
- 33.Swigon D, Coleman BD, Olson WK. Modeling the Lac repressor-operator assembly: the influence of DNA looping on Lac repressor conformation. Proc Natl Acad Sci U S A. 2006;103:9879–9884. doi: 10.1073/pnas.0603557103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arya G, Schlick T. Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model. Proc Natl Acad Sci U S A. 2006;103:16236–16241. doi: 10.1073/pnas.0604817103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rocchia W, Sridharan S, Nicholls A, Alexov E, Chiabrera A, Honig B. Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: applications to the molecular systems and geometric objects. J Comput Chem. 2002;23:128–137. doi: 10.1002/jcc.1161. [DOI] [PubMed] [Google Scholar]
- 36.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 37•.Paillard G, Lavery R. Analyzing protein-DNA recognition mechanisms. Structure. 2004;12:113–122. doi: 10.1016/j.str.2003.11.022. This work suggests that optimal base sequences of protein binding sites can be derived from the relative energetic contributions of direct and indirect readout. [DOI] [PubMed] [Google Scholar]
- 38.Gromiha MM, Siebers JG, Selvaraj S, Kono H, Sarai A. Role of inter and intramolecular interactions in protein-DNA recognition. Gene. 2005;364:108–113. doi: 10.1016/j.gene.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 39•.Morozov AV, Havranek JJ, Baker D, Siggia ED. Protein-DNA binding specificity predictions with structural models. Nucleic Acids Res. 2005;33:5781–5798. doi: 10.1093/nar/gki875. This approach includes inter- and intra-base pair deformation energy in the development of a knowledge-based potential to predict protein-DNA binding specificity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahmad S, Kono H, Arauzo-Bravo MJ, Sarai A. ReadOut: structure-based calculation of direct and indirect readout energies and specificities for protein-DNA recognition. Nucleic Acids Res. 2006;34:W124–127. doi: 10.1093/nar/gkl104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jamal Rahi S, Virnau P, Mirny LA, Kardar M. Predicting transcription factor specificity with all-atom models. Nucleic Acids Res. 2008;36:6209–6217. doi: 10.1093/nar/gkn589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siggers TW, Silkov A, Honig B. Structural alignment of protein--DNA interfaces: insights into the determinants of binding specificity. J Mol Biol. 2005;345:1027–1045. doi: 10.1016/j.jmb.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 43•.Siggers TW, Honig B. Structure-based prediction of C2H2 zinc-finger binding specificity: sensitivity to docking geometry. Nucleic Acids Res. 2007;35:1085–1097. doi: 10.1093/nar/gkl1155. This paper reports the use of protein-DNA structures to predict binding specificity and considers the possibility of predicting position weight matrices (PWM) for an entire protein family based on the structures of just a few family members. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Havranek JJ, Duarte CM, Baker D. A simple physical model for the prediction and design of protein-DNA interactions. J Mol Biol. 2004;344:59–70. doi: 10.1016/j.jmb.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 45.Ashworth J, Havranek JJ, Duarte CM, Sussman D, Monnat RJ, Jr, Stoddard BL, Baker D. Computational redesign of endonuclease DNA binding and cleavage specificity. Nature. 2006;441:656–659. doi: 10.1038/nature04818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46••.Morozov AV, Siggia ED. Connecting protein structure with predictions of regulatory sites. Proc Natl Acad Sci U S A. 2007;104:7068–7073. doi: 10.1073/pnas.0701356104. This work integrates homology modeling of transcription factors with a database of protein-DNA contact geometry in genome-wide transcription factor binding site prediction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu XJ, Olson WK. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat Protoc. 2008;3:1213–1227. doi: 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48••.Tolstorukov MY, Colasanti AV, McCandlish DM, Olson WK, Zhurkin VB. A novel roll-and-slide mechanism of DNA folding in chromatin: implications for nucleosome positioning. J Mol Biol. 2007;371:725–738. doi: 10.1016/j.jmb.2007.05.048. This paper reveals the detailed relationship between dinucleotide geometry and nucleosomal topology and superhelical pitch. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tolstorukov MY, Choudhary V, Olson WK, Zhurkin VB, Park PJ. nuScore: a web-interface for nucleosome positioning predictions. Bioinformatics. 2008;24:1456–1458. doi: 10.1093/bioinformatics/btn212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, Leproust EM, Hughes TR, Lieb JD, Widom J, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2008 doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gabdank I, Barash D, Trifonov EN. Nucleosome DNA Bendability Matrix (C. elegans) J Biomol Struct Dyn. 2009;26:403–412. doi: 10.1080/07391102.2009.10507255. [DOI] [PubMed] [Google Scholar]
- 52•.Iwahara J, Zweckstetter M, Clore GM. NMR structural and kinetic characterization of a homeodomain diffusing and hopping on nonspecific DNA. Proc Natl Acad Sci U S A. 2006;103:15062–15067. doi: 10.1073/pnas.0605868103. This NMR study shows that, while the HoxD9 homeodomain binds to DNA both specifically and non-specifically with a similar binding geometry, arginine side chains at the interface are more flexible when in contact with the non-specific sequence. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gorman J, Greene EC. Visualizing one-dimensional diffusion of proteins along DNA. Nat Struct Mol Biol. 2008;15:768–774. doi: 10.1038/nsmb.1441. [DOI] [PubMed] [Google Scholar]
- 54.Givaty O, Levy Y. Protein sliding along DNA: dynamics and structural characterization. J Mol Biol. 2009;385:1087–1097. doi: 10.1016/j.jmb.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 55.Lavery R, Sklenar H. Defining the structure of irregular nucleic acids: conventions and principles. J Biomol Struct Dyn. 1989;6:655–667. doi: 10.1080/07391102.1989.10507728. [DOI] [PubMed] [Google Scholar]
- 56.Petrey D, Honig B. GRASP2: visualization, surface properties, and electrostatics of macromolecular structures and sequences. Methods Enzymol. 2003;374:492–509. doi: 10.1016/S0076-6879(03)74021-X. [DOI] [PubMed] [Google Scholar]