

Abstract
MHC-class II genes determine susceptibility in human type-1 diabetes. In their context, presentation of target antigen(s) results in autoimmunity and β-cell destruction. An animal model, in which human β-cell autoantigen(s) are presented to effector-cells in the context of human MHC-class II diabetes susceptibility genes, would be desirable for studying molecular mechanisms of disease and developing antigen-specific immune-interventions. We report the development of antigen-specific insulitis in double-transgenic mice carrying the HLA-DQ8 diabetes susceptibility haplotype and expressing the human autoantigen GAD65 in pancreatic β-cells. Immunization with human GAD65 cDNA resulted in severe insulitis and low antibody levels in double-transgenic mice while control mice were mostly insulitis free. CFA/protein immunization resulted in high antibody levels and modest insulitis. Pancreatic lymphocytic infiltration progressed through stages (exocrine pancreas followed by peri and intra-insulitis). Adoptive transfer of splenocytes from DNA-immunized mice resulted in development of insulitis in recipient transgenics. Our results show that immunization with a clinically relevant, type-1 diabetes human autoantigen, in a humanized genetic setting, results in the development of an immune response that homes to islets of Langerhans. This animal model will facilitate studies of autoimmunity to GAD65 in the context of HLA-DQ8, and development of methods to induce tolerance and prevent insulitis.
Keywords: Mouse model, autoimmune diabetes, type 1, MHC class II, GAD, GAD65
Introduction
Genetic susceptibility and target specificity of an immune attack characterize most organ-specific autoimmune diseases. Type-1 diabetes (T1D) is an organ-specific autoimmune disease that results from an autoimmune destruction of pancreatic β-cells in a process that can span several years and results in glucose intolerance and disease when the majority of β-cells have been depleted. The destruction is marked by circulating antibodies to β-cell autoantigens in the blood and by a massive infiltration of mononuclear lymphocytes into the islets of Langerhans while β-cells still remain, and their retraction when β-cells are completely destroyed. For T1D, the genetic susceptibility is linked to major histocompatibility (MHC)-class II molecules [1]. The strongest association is with HLA DR3, DQ2 (DQB1*0201) and HLA DR4 (DRB1*0401), DQ8 (DQB1*0302) haplotypes in Caucasian populations [2]. HLA DQ8 is believed to be the dominant susceptibility tissue type in humans [3]. The most significant link lies in the presence or absence of an aspartic acid at position 57 of the HLA-DQ β-chain [of which DQ8 is the best studied example, 4]. In T1D, specific target autoantigens of the immune attack have been identified and extensively studied. Two major autoantigens in the human disease have been identified by immunoprecipitation of islet-cell proteins by T1D and prediabetes sera. They were first described jointly as a 64kD autoantigen immunoprecipitated by about 80% of T1D sera [5–9; 10 and 11]. One of the 64kD antigens was identified as the smaller isoform of the gamma-amino-butyric acid (GABA)-synthesizing enzyme, glutamic acid decarboxylase, GAD65 [9]. This protein is recognized by 70–80% of patients’ sera. A second component of the 64kD antigen recognized by human-diabetes sera was identified as a putative tyrosine phosphatase, and named IA-2 [11–16]. This antigen is recognized by 60–70% of patients’ sera. More than 90% of T1D patients have antibodies to one or both of these antigens in the period preceding the clinical onset of T1D. Autoantibodies to insulin are also found at a high incidence in young T1D patients [19 and 20]. A highly homologous isoform of GAD65, GAD67, is recognized by virtue of cross-reacting antibodies in 11–18% of patients, but is not an independent autoantigen in human diabetes [21 and 22]. Whereas mouse β-cells predominantly express GAD67, human β-cells only express the GAD65 isoform [23 and 24].
Multiple models are available that mimic immune-mediated diabetes to varying degrees. The spontaneous models, the Biobreeding (BB) rat [25] and the non-obese diabetic (NOD) mouse [26 for review] have been instructive for elucidating basic molecular mechanisms involved in autoimmune destruction of pancreatic β-cells. However, these models do not carry human MHC-class II molecules and the nature of the primary target antigen remains unclear. The NOD mouse has several features, which distinguish it from the human disease. For example, the induction of organ-specific autoimmunity in humans may be caused by human pathogens and/or toxins, autoimmunity seems to be the default mechanism in the NOD mouse. Thus mice in a clean, pathogen-free environment have a high incidence of disease, whereas a variety of regimens which stimulate the immune system, such as viral infections, prevent disease [27]. More than 125 methods for preventing or curing disease in the NOD mouse have been described; however, most are not applicable to humans [26]. In the case of the BB rat, spontaneous T-cell mediated diabetes is significantly distinct from the human disease in that it is accompanied by autoantibodies to lymphocytes and a severe lymphocytopenia which is essential for development of β-cell autoimmunity and diabetes in this model [25]. In an attempt to develop better models of diabetes, ‘humanized’ transgenic mice that express diabetes-susceptibility human MHC-class II molecules were developed [28 and 29 as examples]. Since these animals did not develop spontaneous diabetes, many were backcrossed into the NOD background. However, such backcrossing failed to induce diabetes [28 and 30] in most cases. Other animal models of T1D, some of them carrying human MHC-class II diabetes-susceptibility genes, were developed by inducing expression of ectopic antigens in pancreatic islets using the rat insulin promoter (RIP) [31 and 32 as examples]. While diabetes did not develop spontaneously in those models, autoimmunity and β-cell destruction was induced following immunization with an antigen [33] or infection with a virus expressing the antigen [34]. Overall, none of these models accurately mimic the human disease in which an immune response against a target human β-cell autoantigen(s) in the context of human MHC-class II antigens is mounted and insulitis/diabetes ensues.
We hypothesized that a combination of human, diabetes-susceptibility MHC-class II molecules and a human β-cell autoantigen like GAD65 fully expressed as a transgene in mouse β-cells would increase the susceptibility for diabetes in mice. A transgenic mouse line that expresses human GAD65 (hGAD65) has been developed [35]. These mice express hGAD65 from the rat insulin II promoter (RIP). Expression of GAD65 and GABA in the pancreatic β-cells of these mice results in a modest decrease in first-phase insulin secretion but autoimmunity and diabetes do not develop [35]. The expression of GAD65 is restricted to pancreatic β-cells and the level of expression is similar to endogenous expression of GAD65 in human β-cells [as opposed to negligible expression in mice, 36]. We have bred hGAD5 transgenics with mice in which endogenous mouse MHC-class II antigens have been replaced with the human HLA-DQ8 susceptibility locus for diabetes. DNA immunization of these double transgenics with cDNA encoding hGAD65 resulted in induction of autoimmunity and lymphocytic homing to islets of Langerhans. In contrast, numerous protocols, including DNA and protein immunization of autoantigens, carried out in mice and in non-human primates who did not express the unique combination of a human β-cell target autoantigen and a human MHC-class II diabetes-susceptibility gene failed to generate insulitis and/or diabetes [9].
METHODS
Mice
HLA-DQA1*0301/DQB1*0302 (DQ8) transgenic, murine MHC-class II molecule–deficient (mII-) C57BL/6 mice (kindly provided by Dr. Wen, Yale, New Haven, CT) [31] were crossed with RIP7-hGAD65 line 1 transgenic C57BL/6 mice [35] to generate DQ8+/mII−/RIP7-hGAD65+ double-transgenic mice. Mice were bred to homozygosity for DQ8 and RIP7-hGAD65 genes. PBMCs from 50 F2 transgenics were screened from the crossing of heterozygous (F1) DQ8+/−/mII+/−/RIP7-hGAD65+/− mice. DQ8 homozygosity was determined by screening simultaneously for DQ8 expression and absence of mII antigens using monoclonal anti-HLA-DQ antibody conjugated with fluorescein isothiocyanate (FITC) and monoclonal anti-murine MHC-class II (mII) antibody conjugated with phycoerythrin (PE) respectively (Pharmingen, Becton & Dickinson Biosciences, Mountain View, CA). Cell-associated fluorescence was measured by flow cytometry (FACS). RIP7-hGAD65 expression was analyzed using tail biopsy and PCR analysis of genomic DNA. RNA dot blot analysis with radiolabeled probe was used to establish homozygosity which was later confirmed by PCR of backcrossed littermates. All mice used in this study were >N6 crosses. All animal protocols were approved by the University of California San Francisco, the University of Wisconsin and the Veterans Affair animal research committees.
Protein and cDNA used for immunization
For DNA immunization, the cDNA fragment encoding full-length hGAD65 was inserted into the CpG-rich pCI vector (Promega, Madison, WI). Plasmids were grown in E. coli, followed by alkaline lysis of bacteria, and purification with maxi plasmid prep kit (Promega, Madison, WI). Recombinant-hGAD65 protein expressed in Saccharomyces cerevisiae strain c13ABYS86, was kindly provided by Drs. M. Powell, B. Rees-Smith, and J. Furmaniak, FIRS Laboratories, RSR Ltd, Cardiff, UK. The purified protein was conformationally stable and was recognized by a series of human monoclonal antibodies recognizing conformational epitopes in GAD65 (not shown).
A cDNA encoding mouse interleukin 12 [mIL12, kindly provided by Dr. Watanabe, NIH, NCI, Bethesda, MD, 37] was also sub-cloned in the pCI vector (Promega, Madison, WI). Polyinosinic-polycytidylic acid (poly I:C) was from Sigma (St. Louis, MO).
Histology and Immunohistochemistry
Pancreata were snap-frozen in Tissue Tek (Miles Laboratories, Elkhart, IN), and 5μm-thick sections stained with hematoxilin and eosin. A minimum of 80 islets per individual pancreas were assessed for insulitis and graded as follows: 0, no insulitis; 1, peri-insulitis; 2, insulitis in <50% of the islet; 3, insulitis in >50% of the islet [38]. Graded islets were expressed as a percentage of the total number of islets in each mouse group. For example 10 weeks post-inmunization, in the DNA-immunized double-transgenic group, we found 11 % of their total islets with no insulitis (Grade 0); 53% with peri-insulitis (Grade 1); 25% with Grade 2 insulitis and 11% with Grade 3. Results were reported by 3 independent observers blinded to the immunization protocol and genotype. In addition, pancreas cryostat sections were stained with biotinylated MAb directed against CD4, CD8 and CD68 (Serotec, Raleigh, NC) followed by streptavidin-FITC conjugate and TO-PRO 3 as a counter-stain (Molecular Probes, Eugene, OR) and examined by confocal microscopy. For in vivo bromodeoxyuridine (BrdU) labeling, histology, and immunostaining, mice were given two intraperitoneal injections of 200μl of a 4mg/ml solution of 5-bromo-2′-deoxyuridine (Sigma, St. Louis, MO) 4 hours apart. The pancreata and spleens were collected 12 hours later. Quantization of the percentage of islet-infiltrating cells and splenocytes that incorporated BrdU was done on frozen tissue using BrdU staining kit (Amersham Biosciences, Piscataway, NJ).
T-cell proliferation assays
Peripheral Blood Mononuclear Cells (PBMCs) and splenocytes were isolated from whole blood and spleen respectively. Mononuclear cells were separated by standard gradient centrifugation in Ficoll-Hypaque, washed in RPMI medium and rested overnight in AIM-V medium at 106/ml concentration. 100μl (~105 cells) were added per well to 96 well tissue culture treated plates in triplicates and incubated with 10μl of either culture media, anti-CD3 (Becton Dickinson Biosciences, Mountain View, CA) or hGAD65 protein (1 and 5 μg) for 48 hours at 37°C in a tissue culture incubator. Cells were pulsed ON with 25μl of 20μCi/ml 3H labeled thymidine (Amersham Biosciences). Cells were harvested and radioactivity measured in a plate scintillation counter (Perkin Elmer, USA).
Antibody measurement
Human GAD65 was expressed from the corresponding wild type cDNA in an in vitro transcription/translation system (TNT SP6 Coupled Reticulocyte Lysate System from Promega, Madison, WI) in the presence of 35S-methionine (1000Ci/mmol; Amersham Biosciences). Unincorporated radioactivity was removed by gel filtration of the transcription/translation mixture on a PD10 column, (Amersham Biosciences) in immunoprecipitation (IP) buffer (10mM Hepes, 10mM benzamidine/HCl, 150mM NaCl, 0.5mM methionine, 5mM EDTA, 0.1% w/v BSA, and 0.5% v/v Triton X-114). Aliquots containing 50,000 cpm of freshly 35S-labelled protein in 50μl of IP buffer were incubated overnight at 4°C with 5μl mouse serum or GAD6 mMAb (positive control). Immune complexes were isolated by adsorption to 25μl of Protein A Sepharose and Protein G Sepharose 50% v/v mixture (Protein A-G Sepharose; Amersham Biosciences) in IP buffer for 45 minutes at 4°C. The Sepharose beads were washed five times in IP buffer to remove unbound protein. Immune complexes bound to Protein A-G Sepharose were eluted by boiling in an SDS-sample buffer (0.1M Tris HCl, pH 6.8, 5% w/v sodium dodecyl sulphate, 0.005% w/v Bromophenol blue, 20% v/v glycerol, 0.5% v/v β-mercaptoethanol), followed by scintillation counting using Ecolume (ICN, Costa Mesa, CA). Normal mouse serum (5μl) was used as a negative control in all immunoprecipitation assays. Four standard deviations above that value were considered positive. A world standard control for measurement of mouse GAD65 antibodies [GAD6 mMAb, 39] was used as a positive control in all immunoprecipitation experiments. In order to determine the GAD6 mMAb dilution at which maximum immunoprecipitation of 35S-labelled GAD65 was reached, increasing concentrations of GAD6 mMAb were tested prior to the mouse serum experiments. The results were transformed to reactivity units (RU) by defining the maximum amount of GAD65 immunoprecipitated by GAD6 antigen as 100%. The experiments were carried out in duplicate and repeated at least three times.
Adoptive transfers
Recipient mice used for adoptive transfer experiments were irradiated (600 rads, CS source) 1 day before the transfer. Splenocytes (105 in 200μl of sterile PBS per recipient) from donor mice were intravenously injected (tail vein) into littermate recipient mice immediately after being isolated.
Glucose measurements
Blood sugar was monitored once a week for the first 4 weeks and every other week thereafter using a Ascencia Elite (Bayer, Mishawaka, IN) glucometer. 10–20μl of blood were drawn directly into an heparinized microtiter tube after nicking the tail vein.
Statistical analysis
One tailed probability of the chi-square distribution was used to compare results between different groups.
RESULTS
Induction of antigen specific insulitis with hGAD65 DNA in DQ8-RIP7hGAD65 mice
A colony of homozygous double-transgenic mice carrying RIP7-hGAD65 [35] and DQ8 [31] was established. Spontaneous diabetes was not observed in mice carrying one or both transgenes during a 12 month follow up. Initially, we tested the ability of two different immunization methods to generate insulitis in groups of double-transgenic (DQ8-RIP7hGAD65+/+), single-transgenic (either DQ8+/+ or RIP7hGAD65+/+) and non-transgenic C57BL/6 controls. Ten 6–8 week old animals in each group received 2 intradermal (i.d.) injections (1 week apart) of GAD65 encoded in a plasmid (50μg of plasmid in a total volume of 50μl of PBS) designed to induce strong T-cell helper 1 (Th1) responses [40]. This approach has been shown to induce cytotoxic T lymphocyte (CTL) responses that persist for several months after initial immunizations and can be boosted by additional DNA immunizations several weeks or months later while antibody levels remain low [41]. Another ten, 6–8 week old animals, were immunized intradermally with 10μg of protein in complete Freund’s adjuvant (CFA). Immunization was repeated at 1 week with 10μg of protein in incomplete Freund’s adjuvant (IFA). In the NOD mouse model, it has been shown that Th1 CD4+ cells are critical for development of destructive insulitis. To induce an innate immune response and enhance the development of a Th1 CD4+ cells, all animals received i.d, injections of a plasmid encoding mIL12 DNA [37] 50μg in 50μl PBS once every 4 days × 4 starting on the day of the first immunization. Mice also received 100μg of poly I:C, in PBS intraperitoneally for 10 days (two sets of 5 with 2 resting days in between) starting the day of the first GAD65 immunization [33]. Poly I:C (also known as synthetic RNA) induces innate immune responses similar to the ones observed post viral infections [viral mimicry, 33].
Animals were followed for 10 weeks post-immunization. Weekly non-fasting glucose measurements were observed to fluctuate in a broader range in immunized double-transgenics than in single-transgenics or controls over the 10 week follow up (135 ± 55 vs. 100 ± 20 vs. 106 ± 6 mg/dl respectively). At 8 weeks after immunization, DNA-immunized mice had significantly higher blood glucoses (170 + 8 mg/dl) than protein-immunized (145 + 6 mg/dl p<0,001) double-transgenics and controls.
All animals were euthanized at 10 weeks post-immunization. Pancreas was obtained for histology (Fig. 1). Hematoxilin/eosin staining of frozen pancreatic sections revealed lymphocytic infiltration of islets mostly in DNA-immunized double-transgenics. Insulitis scores rated as, Grade 0, no insulitis; Grade 1, peri-insulitis; Grade 2, insulitis in <50% of the islet; and Grade 3, insulitis in >50% of the islet [39] were assigned to all experimental groups (Fig. 2). All ten double-transgenic, DNA-immunized animals developed severe intra-insulitis (Grades 2 or 3). Animals in this group had between 25 and 50 % of their islets with Grades 2 and 3 infiltration. In contrast, only two of ten protein-immunized double-transgenic animals developed Grade 2 insulitis (12 % of their islets) and one of these mice had both Grade 2 and 3 (few islets) insulitis All other groups had sporadic (less than 1 %) or none Grade 2 or 3 insulitis (Fig. 1 and 2). Staining of pancreatic sections with antibodies to CD4 and CD8, showed presence of both CD4 and CD8 positive T cells in the peri and intra-islet infiltrates indicative of a targeted, active inflammatory process.
Figure 1. Pancreas histology of protein-immunized and DNA-immunized mice.
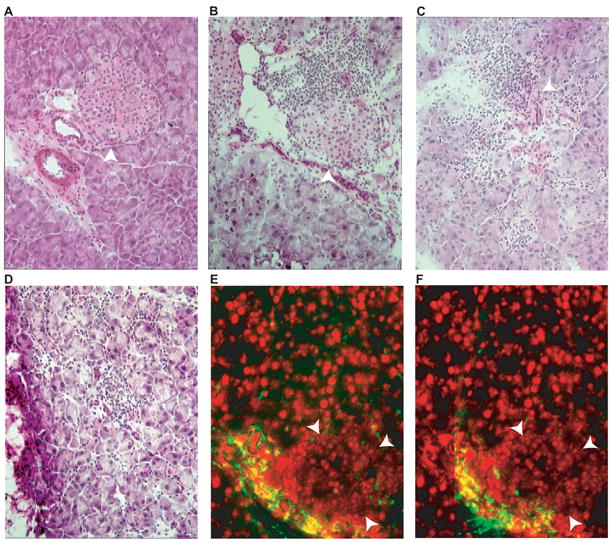
Frozen sections of pancreas were stained with hematoxylin and eosin (A-D) or immune-stained for CD4 (E) or CD8 (F). (A), DNA-immunized non-transgenic mouse pancreas, (B), protein-immunized DQ8-RIP7-hGAD65, double-transgenic mouse pancreas. (C-D), DNA-immunized DQ8-RIP7-GAD65 double-transgenic mouse pancreas. Note the more aggressive characteristic of the lymphocytic infiltration in C (some islet tissue preserved), and D (complete islet destruction) of the GAD65 DNA-immunized double-transgenic as compared with the peri-insular infiltration of protein-immunized double-transgenic mouse (B). Confluent nuclei in E and F (red) correspond to islet area (arrow delineated). Islets are indicated with arrowheads.
Figure 2. Summary of islet infiltrate scores following GAD65 DNA or protein immunization protocols in double-transgenic (DT), DQ8 and RIP7-GAD65, single- transgenic, ST), or non-transgenic control mice.

Insulitis scores were rated as, Grade 0, no insulitis (white bars); grade 1, peri-insulitis (dotted bars); grade 2, insulitis in <50% of the islet (stripped bars); grade 3, insulitis in >50% of the islet (black bars) [39]. The percentage of islets in each scoring category is shown. All 10 DT, DNA-immunized animals developed Grade 2 and 3 insulitis. Only two of ten protein-immunized DT animals developed Grade 2 insulitis and one these mice had both grade 2 and 3 insulitis. All other groups had sporadic or no Grade 2 or 3 insulitis. * Indicates statistically significant differences (p ≤ 0.005) within the same grade, as compared to controls and all other groups.
Blood samples were analyzed for GAD65 autoantibodies (Fig. 3) and PBMCs isolated. PBMCs and splenocytes were prepared for analyses of T cell responses (Fig. 4).
Figure 3. Humoral immunity in immunized mice.

GAD65 antibodies in DNA or protein immunized mice. Animals were named by a number and the first initial of their gender (f or m). A world standard control for measurement of GAD65 antibodies (GAD6 mMAb) was used as a positive control in all immunoprecipitation experiments. The results were transformed to reactivity units (RU) by defining the maximum amount of GAD65 radioactivity precipitated by GAD6 mMAb as 100%. The experiments were carried out in duplicate and repeated at least three times. The antibody levels of hGAD65 protein-immunized animals (right panel) were consistently higher than those of the hGAD65 DNA-immunized group (left panel). Not all animals developed detectable antibodies in either group (5 of 10 in DNA-immunized vs. 7 of 10 in protein-immunized groups).
Figure 4. Cellular immunity in immunized mice.

T-cell proliferation assays were performed using peripheral blood mononuclear cells (PBMCs, left panel) and splenocytes (right panel). DNA-immunized animals’ results shown. Radioactivity from radiolabeled thymidine incorporation to proliferating T cells is expressed in CPM × 103 (Y axis, left margin). DT indicates double transgenics (DQ8-RIP7-hGAD65) while ST indicates single transgenics (GAD65 ST shown). None = no antigen added (negative control), Anti-CD3 was used to stimulate proliferation (positive control). Mean and standard deviation of triplicate results from 5 animals in each group (double or single transgenics) is shown. All double-transgenic animals tested had significant central (splenocytes) antigen specific proliferative responses to hGAD65 (top right) when compared to single-transgenic mice (bottom right).
Antibodies to GAD65 were detected in all immunized groups, suggesting lack of active tolerance to the GAD65 protein in controls and transgenics (regardless of the presence of the GAD65 transgene). Within the groups of double-transgenic animals, 7 of 10 protein-immunized and 5 of 10 DNA-immunized mice developed detectable antibodies to GAD65. Furthermore, amongst the GAD65 antibody-positive animals, the levels for protein-immunized animals were significantly higher (115 +/− 52 RU in the protein-immunized vs. 65 +/− 30 RU in the DNA-immunized group) (Fig. 3). Among single-transgenics, 4 of 5 mice immunized with protein developed antibodies to GAD65 and the levels were 76 +/− 6 RU. None of 5 single-transgenics who received a DNA immunization developed antibodies. Finally, among non-transgenics, 4 of 5 mice receiving protein immunization developed antibodies while none of the mice receiving DNA-immunizations had detectable antibodies.
T-cell proliferation assays were performed using PBMCs and splenocytes from selected animals. Proliferative responses from DNA-immunized animals are shown in Fig. 4. While proliferative responses to hGAD65 in PBMCs were moderate, and similar in all groups (Fig. 4 and results not shown), splenocyte response to hGAD65 was significantly higher (p ≤ 0.05) in double-transgenic than in single (Fig. 4 right panels) and non-transgenic mice.
The presence of GAD65 autoantibodies and T cell proliferative responses in DNA-immunized and protein-immunized animals suggest that an immune response to GAD65 has been induced by both regimens. However, while protein immunization generated stronger antibody responses, DNA immunization induced stronger GAD65 specific T-cell proliferative responses. The detection of insulitis in double-transgenics, demonstrated that the immune response had homed to the pancreas and islets in all animals in the double-transgenic group immunized with DNA. In comparison, protein-immunized double-transgenics failed to show high grade insulitis. Furthermore in single and non-transgenics, only grade 0 or grade 1 (peri-insulitis) was observed. Thus, the combination of the DQ8 and RIP7-hGAD65 transgenes and the administration of GAD65 as DNA in a regimen that induces Th1 CD4+ cells and CTL responses in other mouse models [40 and 41] were most effective for development of insulitis.
DQ8-RIP7-hGAD65 double transgenics and progression of antigen specific insulitis
In order to study the development and progression of the insulitis in GAD65 DNA- immunized double-transgenics, 20 animals (equal number of male and female) were immunized at 6–8 weeks of age and two weeks later using the protocol described above. Two to three animals were euthanized at 11 days, 5 weeks, 9 weeks, 13 weeks, 17 weeks, 25 weeks and 37 weeks post-immunization and the pancreas frozen for histology. Frozen sections were stained with hematoxilin and eosin as described above. As early as 11 days post-immunization, generalized pancreatic infiltration was detected in both euthanized animals (Fig. 5, A). At 5 weeks post immunization peri-insular lymphocytic infiltration was first detected (Fig. 5, B). At 9 weeks, the infiltration was mainly localized to the peri-islet area but 10 % of the islets showed intra-insulitis (Fig. 5, C) similar to what we observed at week 10 in the first phase study (see Fig. 1). At 13 weeks, intra-islet infiltration was massive in approximately 25% of islets (Fig. 5, D & E). Animals euthanized at 17, 25, and 37 weeks showed progressively increased percentage of intra-islet infiltration (40% vs. 60% vs. 85% respectively). BrdU treatment of another 2 animals euthanized at week 17 revealed mitotic figures (Fig. 5, F) exclusively in the islets, indicative of active cell proliferation. The generalized pancreas infiltration observed in the early weeks after immunization, had almost completely disappeared at week 25 and was localized to the peri- or intra-islet areas. Immunostaining with anti-CD4 and anti-CD8 of section from animals euthanized at 25 weeks showed positive staining for both markers (Fig. 5 G and H respectively). Pancreata from animals euthanized at week 37, revealed similar characteristics as at week 25 but with a higher percentage of infiltrated islets.
Figure 5. Progression of insulitis in DNA-immunized double-transgenic mice.
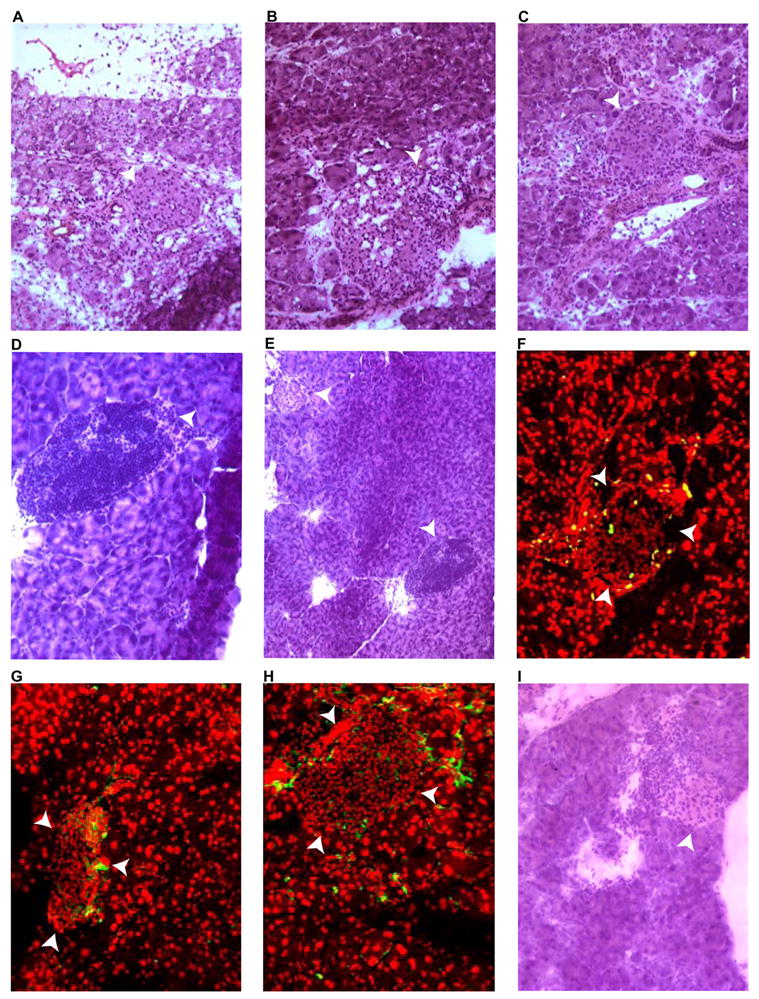
Frozen pancreatic sections, obtained at different time points following DNA immunization, were stained with hematoxilin and eosin (A-E and I), anti-BrdU (F), anti-CD4 (G), or anti-CD8 (H). White arrowheads indicate islets. A, eleven days post-immunization (infiltration present). B, week 5 post-immunization pancreas appeared massively infiltrated. C, week 9 post-immunization (more localized infiltration noted). D, week 13 post-immunization specimen shows a severely infiltrated islet. E: panoramic view of D showing the concomitant presence of normal islet (top left corner). F, post-BrdU treatment of an animal euthanized at week 17 (proliferating cells are exclusively detected in the islet). G and H, pancreas 25 weeks post-immunization (G=anti-CD4, H=anti-CD8). I, pancreas section of a recipient of adoptively transferred splenocytes 5 weeks post transfer.
Adoptive transfer of insulitis
We addressed the question of whether splenocytes from DNA-immunized double-transgenics, could home to islets and generate lymphocytic infiltration in non-immunized littermates. The spleen was removed from DNA-immunized double-transgenics 13 weeks after the first immunization (2 male donors) and 105 lymphocytes were transferred to non-immunized irradiated littermates (4 female recipients). Analyses of pancreatic sections of recipient mice 1 week after transfer revealed generalized and peri-islet infiltrates. At five weeks most (75%) of the islets were infiltrated (Fig. 5 I). Transfer of splenocytes from control non-immunized double-transgenic mice, did not generate insulitis (results not shown). These results demonstrate that splenocytes in DNA-immunized but not in non-immunized mice specifically induce insulitis in recipient mice.
DISCUSSION
MHC-class II genes are critical determinants of genetic susceptibility in human T1D. Presentation of primary target antigen(s) in the context of these genes is an essential component of the pathogenesis of the disease [reviewed in 42]. An animal model of T1D, in which primary human β-cell autoantigen(s) are presented to effector cells in the context of human MHC-class II susceptibility genes, would be useful for studies of molecular mechanisms of disease and for testing antigen specific immune-interventions applicable to humans.
We have generated mice that express high levels of hGAD65 in β-cells and at the same time have their endogenous mouse MHC-class II antigens replaced by the human HLA-DQ8 susceptibility locus for diabetes. After introducing genetic susceptibility for T1D with the human HLA genes and human quantitative expression of hGAD65 at the β-cell level, we reasoned we needed to mimic environmental triggers of the disease. Immunization with hGAD65 cDNA sub-cloned in a CpG-rich plasmid together with immunization with mIL12 and poly I:C (viral mimicry) produced a strong cellular immune response that homed to the islets of Langerhans. As in human T1D, no statistically significant gender differences were noted. Interestingly, hGAD65 protein/CFA immunization had a much milder degree of islet infiltration. Antibodies on the other hand, were present in high levels in protein/CFA-immunized animals (as opposed to the DNA-immunized animals in which antibody levels were present at much lower concentrations and not always detectable). This observation may imply a deviation to a Th2 type of helper response with protein immunization. Overall, the hGAD65 antibody response to either DNA or protein immunizations demonstrated a lack of tolerance to hGAD65, while being expressed as a transgene.
Although the DNA-immunized double-transgenics experienced episodic hyperglycemia, none of the animals developed diabetes during the 37 weeks of the study. The lymphocytic infiltration of the pancreas progressed from localizing mainly to the exocrine pancreas (11 days), moving into the endocrine pancreas (5–9 weeks), localizing to the peri-islet area (9–13 weeks), and becoming intra-insular (17–37 weeks). From week 9 onwards, islets containing different degrees of lymphocytic infiltration coexisted in the same pancreas. Quantification of the degree of insulitis over time (Fig. 5) showed that instead of retracting after the immunization stimulus finished, the insulitis progressed and became more pronounced over time. Furthermore, the presence of BrdU retained cells indicated active proliferation. Mitotic figures within islets confirmed active proliferation (Fig. 5 F). The positive BrdU staining within islets may represent lymphocytic proliferation although the possibility of islet cell regeneration cannot be excluded [43]. The characteristics of the infiltration with the simultaneous presence of CD4 and CD8 cells indicate the presence of an active immune attack [44]. At week 25, insulin staining (not shown) indicated the presence of remaining islet function. Thus, the immune attack either did not efficiently eliminate the majority of the β-cells or β-cell regeneration occurred at a sufficient rate to replenished the β-cell mass. Other possible explanation for the lack of a full-blown diabetes phenotype may be inherent to the mouse strain chosen. C57BL/6 may be best described, as a strain of low diabetes susceptibility. Despite the decrease insulin secretion seen in elder C57BL/6 mice, this line is relatively resistant to, for example, obesity-induced diabetes. Other strains like BTBR are more diabetes susceptible. The BTBR strain seems to have a relative deficiency of β cell neogenesis. When obese, these mice develop severe diabetes (fasting glucose >400 mg/dL). By contrast, C57BL/6 mice are able to compensate for the obesity-induced insulin resistance by increasing pancreatic insulin secretion and thus maintain only slightly elevated plasma glucose levels (<250 mg/dL)[45].
While the mechanisms involved in progression from insulitis to insulin dependent diabetes are still under investigation, our model is suitable for testing factors implicated in other models and in humans, including, but not limited to upregulation of MHC class I antigens [46], and/or interferons [47], and viral infections [48].
Finally, adoptive transfer of splenocytes from DNA-immunized mice resulted in insulitis of naïve-recipient double-transgenics. Progression of disease was observed when comparing early islet specimens (within a week) to specimens obtained 5 week post-adoptive transfer (Fig. 5 I).
Taken together, our results show that while protein immunization with a clinically relevant human autoantigen, in a regimen that induces strong antibody responses fails to induce significant insulitis, immunization with cDNA encoding GAD65 in a regimen that induces modest antibody responses, results in the development of progressive insulitis. The results demonstrate the requirement for both expression of hGAD65 in pancreatic β-cells and its presentation in the context of a human MHC-class II diabetes-susceptibility genes for development of severe insulitis.
Acknowledgments
To Drs. Haleh Bassiri, Virginie Menard Rose and Teresa Lopez for technical support.
To Dr. Matthias von Herrath for critical review. This work was funded by NIH-grants (SB, JCJ), a grant from Adolphus Andrews (SB), Mr. and Mrs. Stiehl (SB) and by VA grants (JCJ). This work was presented in part at The Endocrine Society Annual Meeting, Toronto, Canada 2007. (oral communication in “Bench to Bedside” session).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.She JX. Susceptibility to type I diabetes: HLA-DQ and DR revisited. Immunol Today. 1996;17:323–329. doi: 10.1016/0167-5699(96)10014-1. [DOI] [PubMed] [Google Scholar]
- 2.Aly TA, Ide A, Jahromi MM, Barker JM, Fernando MS, Babu SR, Yu L, Miao D, Erlich HA, Fain PR, Barriga KJ, Norris JM, Rewers MJ, Eisenbarth GS. Extreme genetic risk for type 1A diabetes. Proc Natl Acad Sci U S A. 2006;103:14074–14079. doi: 10.1073/pnas.0606349103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Todd JA, Bell JI, McDevitt HO. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature. 1987;329:599–604. doi: 10.1038/329599a0. [DOI] [PubMed] [Google Scholar]
- 4.Herman AE, Tisch RM, Patel SD, Parry SL, Olson J, Noble JA, Cope AP, Cox B, Congia M, McDevitt HO. Determination of glutamic acid decarboxylase 65 peptides presented by the type I diabetes-associated HLA-DQ8 class II molecule identifies an immunogenic peptide motif. J Immunol. 1996;163:6275–6282. [PubMed] [Google Scholar]
- 5.Baekkeskov S, Nielsen JH, Marner B, Bilde T, Ludvigsson J, Lernmark A. Autoantibodies in newly diagnosed diabetic children immunoprecipitate human pancreatic islet cell proteins. Nature. 1982;298:167–169. doi: 10.1038/298167a0. [DOI] [PubMed] [Google Scholar]
- 6.Baekkeskov S, Landin M, Kristensen JK, Srikanta S, Bruining GJ, Mandrup-Poulsen T, de Beaufort C, Soeldner JS, Eisenbarth G, Lindgren F, Sundquist G, Lernmark A. Antibodies to a 64,000 Mr human islet cell antigen precede the clinical onset of insulin-dependent diabetes. J Clin Invest. 1987;79:926–934. doi: 10.1172/JCI112903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baekkeskov S, Warnock G, Christie M, Rajotte RV, Larsen PM, Fey S. Revelation of specificity of 64K autoantibodies in IDDM serums by high-resolution 2-D gel electrophoresis. Unambiguous identification of 64K target antigen. Diabetes. 1989;38:1133–1141. doi: 10.2337/diab.38.9.1133. [DOI] [PubMed] [Google Scholar]
- 8.Baekkeskov S, Aanstoot HJ, Christgau S, Reetz A, Solimena M, Cascalho M, Folli F, Richter-Olesen H, DeCamilli P, Camilli PD. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature. 1990;347:151–156. doi: 10.1038/347151a0. [DOI] [PubMed] [Google Scholar]
- 9.Baekkeskov S, Kanaani J, Jaume JC, Kash S. Does GAD have a unique role in triggering IDDM? J Autoimmun. 2000;15:279–286. doi: 10.1006/jaut.2000.0443. [DOI] [PubMed] [Google Scholar]
- 10.Atkinson MA, Maclaren NK, Scharp DW, Lacy PE, Riley WJ. 64,000 Mr autoantibodies as predictors of insulin-dependent diabetes. Lancet. 1990;335:1357–1360. doi: 10.1016/0140-6736(90)91241-2. [DOI] [PubMed] [Google Scholar]
- 11.Christie MR, Vohra G, Champagne P, Daneman D, Delovitch TL. Distinct antibody specificities to a 64-kD islet cell antigen in type 1 diabetes as revealed by trypsin treatment. J Exp Med. 1990;172:789–794. doi: 10.1084/jem.172.3.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Payton MA, Hawkes CJ, Christie MR. Relationship of the 37,000- and 40,000-M(r) tryptic fragments of islet antigens in insulin-dependent diabetes to the protein tyrosine phosphatase-like molecule IA-2 (ICA512) J Clin Invest. 1995;96:1506–1511. doi: 10.1172/JCI118188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Passini N, Larigan JD, Genovese S, Appella E, Sinigaglia F, Rogge L. The 37/40-kilodalton autoantigen in insulin-dependent diabetes mellitus is the putative tyrosine phosphatase IA-2. Proc Natl Acad Sci U S A. 1995;92:9412–9416. doi: 10.1073/pnas.92.20.9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonifacio E, Genovese S, Braghi S, Bazzigaluppi E, Lampasona V, Bingley PJ, Rogge L, Pastore MR, Bognetti E, Bottazzo GF. Islet autoantibody markers in IDDM: risk assessment strategies yielding high sensitivity. Diabetologia. 1995;38:816–822. doi: 10.1007/s001250050358. [DOI] [PubMed] [Google Scholar]
- 15.Lan MS, Wasserfall C, Maclaren NK, Notkins AL. IA-2, a transmembrane protein of the protein tyrosine phosphatase family, is a major autoantigen in insulin-dependent diabetes mellitus. Proc Natl Acad Sci U S A. 1996;93:6367–6370. doi: 10.1073/pnas.93.13.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LaGasse J, Jelinek L, Sexson S, Lofton-Day C, Breininger J, Sheppard P, Kindsvogel W, Hagopian WA. An islet-cell protein tyrosine phosphatase is a likely precursor to the 37-kDa autoantigen in type 1 diabetes: human and macaque sequences, tissue distribution, unique and shared epitopes, and predictive autoantibodies. Mol Med. 1997;3:163–173. [PMC free article] [PubMed] [Google Scholar]
- 17.Rabin DU, Pleasic SM, Palmer-Crocker R, Shapiro JA. Cloning and expression of IDDM-specific human autoantigens. Diabetes. 1992;41:183–186. doi: 10.2337/diab.41.2.183. [DOI] [PubMed] [Google Scholar]
- 18.Rabin DU, Pleasic SM, Shapiro JA, Yoo-Warren H, Oles J, Hicks JM, Goldstein DE, Rae PM. Islet cell antigen 512 is a diabetes-specific islet autoantigen related to protein tyrosine phosphatases. J Immunol. 1994;152:3183–3188. [PubMed] [Google Scholar]
- 19.Karjalainen J, Knip M, Mustonen A, Akerblom HK. Insulin autoantibodies at the clinical manifestation of type 1 (insulin-dependent) diabetes-a poor predictor of clinical course and antibody response to exogenous insulin. Diabetologia. 1988;31:129–133. doi: 10.1007/BF00276844. [DOI] [PubMed] [Google Scholar]
- 20.Castano L, Eisenbarth GS. Type-I diabetes: a chronic autoimmune disease of human, mouse, and rat. Annu Rev Immunol. 1990;8:647–679. doi: 10.1146/annurev.iy.08.040190.003243. [DOI] [PubMed] [Google Scholar]
- 21.Hagopian WA, Michelsen B, Karlsen AE, Larsen F, Moody A, Grubin CE, Rowe R, Petersen J, McEvoy R, Lernmark A. Autoantibodies in IDDM primarily recognize the 65,000-M(r) rather than the 67,000-M(r) isoform of glutamic acid decarboxylase. Diabetes. 1993;42:631–636. doi: 10.2337/diab.42.4.631. [DOI] [PubMed] [Google Scholar]
- 22.Velloso LA, Kampe O, Hallberg A, Christmanson L, Betsholtz C, Karlsson FA. Demonstration of GAD-65 as the main immunogenic isoform of glutamate decarboxylase in type 1 diabetes and determination of autoantibodies using a radioligand produced by eukaryotic expression. J Clin Invest. 1993;91:2084–2090. doi: 10.1172/JCI116431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, Richter W, Aanstoot HJ, Shi Y, Fu Q, Rajotte R, Warnock G, Baekkeskov S. Differential expression of GAD65 and GAD67 in human, rat, and mouse pancreatic islets. Diabetes. 1993;42:1799–1808. doi: 10.2337/diab.42.12.1799. [DOI] [PubMed] [Google Scholar]
- 24.Petersen JS, Hejnaes KR, Moody A, Karlsen AE, Marshall MO, Hoier-Madsen M, Boel E, Michelsen BK, Dyrberg T. Detection of GAD65 antibodies in diabetes and other autoimmune diseases using a simple radioligand assay. Diabetes. 1994;43:459–467. doi: 10.2337/diab.43.3.459. [DOI] [PubMed] [Google Scholar]
- 25.Malaisse WJ, Courtois P, Scott FW. Insulin-dependent diabetes and gut dysfunction: the BB rat model. Horm Metab Res. 2004;36:585–594. doi: 10.1055/s-2004-825920. [DOI] [PubMed] [Google Scholar]
- 26.Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med. 1999;5:601–604. doi: 10.1038/9442. [DOI] [PubMed] [Google Scholar]
- 27.Tisch R, McDevitt HO. Antigen-specific immunotherapy: is it a real possibility to combat T-cell-mediated autoimmunity? Proc Natl Acad Sci U S A. 1994;91:437–438. doi: 10.1073/pnas.91.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raju R, Munn SR, David CS. T cell recognition of human pre-proinsulin peptides depends on the polymorphism at HLA DQ locus: a study using HLA DQ8 and DQ6 transgenic mice. Hum Immunol. 1997;58:21–29. doi: 10.1016/s0198-8859(97)00212-7. [DOI] [PubMed] [Google Scholar]
- 29.Patel SD, Cope AP, Congia M, Chen TT, Kim E, Fugger L, Wherrett D, Sonderstrup-McDevitt G. Identification of immunodominant T cell epitopes of human glutamic acid decarboxylase 65 by using HLA-DR (alpha1*0101,beta1*0401) transgenic mice. Proc Natl Acad Sci USA. 1997;94:8082–8087. doi: 10.1073/pnas.94.15.8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abraham RS, Wilson SB, de Souza NF, Jr, Strominger JL, Munn SR, David CS. NOD background genes influence T cell responses to GAD 65 in HLA-DQ8 transgenic mice. Hum Immunol. 1999;60:583–590. doi: 10.1016/s0198-8859(99)00057-9. [DOI] [PubMed] [Google Scholar]
- 31.Wen L, Wong FS, Burkly L, Altieri M, Mamalaki C, Kioussis D, Flavell RA, Sherwin RS. Induction of insulitis by glutamic acid decarboxylase peptide–specific and HLA-DQ8–restricted CD4+ T cells from human DQ transgenic mice. J Clin Invest. 1998;102:947–957. doi: 10.1172/JCI2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajagopalan G, Kudva YC, Chen L, Wen L, David CS. Autoimmune diabetes in HLA-DR3/DQ8 transgenic mice expressing the co-stimulatory molecule B7-1 in the beta cells of islets of Langerhans. Int Immunol. 2003;15:1035–1044. doi: 10.1093/intimm/dxg103. [DOI] [PubMed] [Google Scholar]
- 33.Moriyama H, Wen L, Abiru N, Liu E, Yu L, Miao D, Gianani R, Wong FS, Eisenbarth GS. Induction and acceleration of insulitis/diabetes in mice with a viral mimic (polyinosinic-polycytidylic acid) and an insulin self-peptide. Proc Natl Acad Sci USA. 2002;99:5539–5544. doi: 10.1073/pnas.082120099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oldstone MB, von Herrath M, Evans CF, Horwitz MS. Virus-induced autoimmune disease: transgenic approach to mimic insulin-dependent diabetes mellitus and multiple sclerosis. Curr Top Microbiol Immunol. 1996;206:67–83. doi: 10.1007/978-3-642-85208-4_5. [DOI] [PubMed] [Google Scholar]
- 35.Shi Y, Kanaani J, Menard-Rose V, Ma YH, Chang PY, Hanahan D, Tobin A, Grodsky G, Baekkeskov S. Increased expression of GAD65 and GABA in pancreatic beta-cells impairs first-phase insulin secretion. Am J Physiol Endocrinol Metab. 2000;279:684–694. doi: 10.1152/ajpendo.2000.279.3.E684. [DOI] [PubMed] [Google Scholar]
- 36.Kash SF, Condie BG, Baekkeskov S. Glutamate decarboxylase and GABA in pancreatic islets: lessons from knock-out mice. Horm Metab Res. 1999;31:340–344. doi: 10.1055/s-2007-978750. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe M, Fenton RG, Wigginton JM, McCormick KL, Volker KM, Fogler WE, Roessler PG, Wiltrout RH. Intradermal delivery of IL-12 naked DNA induces systemic NK cell activation and Th1 response in vivo that is independent of endogenous IL-12 production. J Immunol. 1999;163:1943–1950. [PubMed] [Google Scholar]
- 38.Morimoto J, Hiroyuki Yoneyama H, Shimada A, Shigihara T, Yamada S, Oikawa Y, Matsushima K, Saruta T, Narumi S. CXC Chemokine Ligand 10 Neutralization Suppresses the Occurrence of Diabetes in Nonobese Diabetic Mice through Enhanced Cell Proliferation without Affecting Insulitis. J Immunol. 2004;173:7017–7024. doi: 10.4049/jimmunol.173.11.7017. [DOI] [PubMed] [Google Scholar]
- 39.Chang YC, Gottlieb DI. Characterization of the proteins purified with monoclonal antibodies to glutamic acid decarboxylase. J Neurosci. 1988;8:2123–2130. doi: 10.1523/JNEUROSCI.08-06-02123.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lekutis C, Shiver JW, Liu MA, Letvin NL. HIV-1 env DNA vaccine administered to rhesus monkeys elicits MHC-class II-restricted CD4+ T helper cells that secrete IFN-gamma and TNF-alpha. J Immunol. 1997;158:4471–4477. [PubMed] [Google Scholar]
- 41.Shiver JW, Davies ME, Yasutomi Y, Perry HC, Freed DC, Letvin NL, Liu MA. Anti-HIV env immunities elicited by nucleic acid vaccines. Vaccine. 1997;15:884–887. doi: 10.1016/s0264-410x(96)00251-4. [DOI] [PubMed] [Google Scholar]
- 42.Jaume JC. Endocrine autoimmunity. In: Gardner DG, Shoback DM, editors. Greenspan’s Basic & Clinical Endocrinology. McGraw-Hill Medical; New York: 2007. pp. 59–79. [Google Scholar]
- 43.Beith JL, Alejandro EU, Johnson JD. Insulin stimulates primary beta-cell proliferation via Raf-1 kinase. Endocrinol. 2008;149:2251–2260. doi: 10.1210/en.2007-1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes. 1993;42:44–55. doi: 10.2337/diab.42.1.44. [DOI] [PubMed] [Google Scholar]
- 45.Clee SM, Nadler ST, Attie AD. Genetic and genomic studies of the BTBR ob/ob mouse model of type 2 diabetes. Am J Ther. 2005;12:491–498. doi: 10.1097/01.mjt.0000178781.89789.25. [DOI] [PubMed] [Google Scholar]
- 46.Nejentsev S, Howson JM, Walker NM, Szeszko J, Field SF, Stevens HE, Reynolds P, Hardy M, King E, Masters J, Hulme J, Maier LM, Smyth D, Bailey R, Cooper JD, Ribas G, Campbell RD, Clayton DG, Todd JA. Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature. 2007;450:887–892. doi: 10.1038/nature06406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hultcrantz M, Hühn MH, Wolf M, Olsson A, Jacobson S, Williams BR, Korsgren O, Flodström-Tullberg M. Interferons induce an antiviral state in human pancreatic islet cells. Virology. 2007;367:92–101. doi: 10.1016/j.virol.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 48.Filippi C, von Herrath M. How viral infections affect the autoimmune process leading to type 1 diabetes. Cell Immunol. 2005;233:125–132. doi: 10.1016/j.cellimm.2005.04.009. [DOI] [PubMed] [Google Scholar]