

Abstract
Estimates of people suffering from overweight (one billion) and obesity (300 million) are increasing. The accumulation of triglycerides in the liver, in the absence of excess alcohol intake, has been described in the early sixties. It was not until 1980, however, that Ludwig et al named this condition nonalcoholic steatohepatitis (NASH). Subsequently, nonalcoholic fatty liver disease (NAFLD) has been used as a general name for conditions ranging from simple steatosis through steatohepatitis to end-stage liver disease (cirrhosis). Many studies have demonstrated the significant correlation with obesity and insulin resistance. Other studies have revealed a significant correlation between hepatic steatosis, cardiovascular disease and increased intima-media thickness. WHO estimated that at least two million patients will develop cirrhosis due to hepatic steatosis in the years to come. Longitudinal cohort studies have demonstrated that those patients with cirrhosis have a similar risk to develop hepatocellular carcinoma as those with other causes of cirrhosis. Taken all together, NAFLD has become the third most important indication for liver transplantation. Therefore, training programmes in internal medicine, gastroenterology and hepatology should stress the importance of diagnosing this entity and treat properly those at risk for developing complications of portal hypertension and concomittant cardiovascular disease. This review will focus on the clinical characteristics, pathophysiology, imaging techniques and the readily available therapeutic options.
Keywords: Non-alcoholic fatty liver disease, Non-alcoholic steatohepatitis, Insulin resistance, Liver, Obesity, Steatosis
INTRODUCTION
When living creatures have a surplus of food at their disposition, they start to hoard energy for future scarce times. The breakthrough in agriculture and founding of villages around 9500 BC in several independent parts of the world not only laid the pavement for modern society, but also gave people the opportunity to cultivate and store more food than needed for their support and their families. This excess of energy has led to the current situation nowadays where the caloric availability exceeds the caloric needs by far. The human body still possesses the quality to store excess energy in adipocytes, a quality that now works in its disadvantage. The extreme storage of excess energy and a reduction in physical activity have led to a worldwide epidemic of obesity. The World Health Organization (WHO) estimated that the number of overweight (BMI > 25) individuals is over one billion, of whom 300 million are obese (BMI > 30)[1]. In future prospects, this number will increase further[2,3]. Although the biggest concern of most obese or overweight persons is about their appearance, obesity has been recognised as a pathogenic factor for a vast array of other pathologiesc, such as Diabetes mellitus, Insulin resistance, Hypertension, Dyslipidemia, Endocrine changes, Kidney stones, Cancer (overall and specific), Coronary disease, Heart failure, Myocardial steatosis, Atrial fibrillation/flutter, Stroke, Venous thrombosis, Hepatobiliary disease, GERD/esophageal cancer, Osteoarthritis, Skin changes, Gout, Dementia, Psychosocial etc. A subset of diseases often occurring in the same patient have been clustered, now known as the metabolic syndrome[4–6]. Vehement research has unravelled major parts of the pathophysiological mechanisms underlying obesity and metabolic syndrome, although many issues have not been explained yet[7,8]. It has been shown that non-alcoholic fatty liver disease (NAFLD) has a strong relation with metabolic syndrome[9–16]. NAFLD mainly with accumulation of triglycerides in the liver, in the absence of excess alcohol intake, has been described in the early sixties[17,18]. It was not until 1980, however, that Ludwig et al named this condition non-alcoholic steatohepatitis (NASH)[19]. Subsequently, NAFLD has been used as a general name for conditions ranging from simple steatosis through steatohepatitis to end-stage liver disease (cirrhosis)[20]. The first is rather benign[21,22], the second is of significant clinical importance[23] and the last one has an increased risk of hepatocellular carcinoma[24,25]. An almost universal association with hepatic and adipose tissue insulin resistance (IR) has been established in a number of studies[26–34]. Browning et al[35] used 1H-NMR-spectroscopy to measure the hepatic triglyceride content in a multi-ethnic urban US population-based study. Hepatic steatosis was found in 31% of their population and increased up to 67% in obese subjects. Other studies[36–38] have confirmed this positive correlation between BMI, waist-to-hip-ratio and hepatic steatosis. A few studies focusing on the natural history of NAFLD showed that only 1%-5% of patients with simple steatosis eventually develop actual cirrhosis[39,40]. WHO estimated that at least 2 million people will develop cirrhosis due to hepatic steatosis. Taken all together, NAFLD has become the third most important indication for liver transplantation and will become the leading indication in the next decades. In this respect, the finding of NASH in young obese children is very alarming[41]. Training in paediatrics, internal medicine, gastroenterology and hepatology should emphasize the awareness of this entity to avoid complications of portal hypertension, minimize the need for liver transplantation and prevent the associated cardiovascular disease.
This review will focus on the clinical characteristics, pathophysiology, imaging techniques and therapeutic opportunities of this disease.
CLINICAL CHARACTERISTICS
Most patients with NAFLD are asymptomatic and the symptoms are usually non specific when they occur. Frequent complaints are fatigue and vague right upper quadrant abdominal discomfort. Because of the latter, steatosis is often found at ultrasound examinations made for suspicion of biliary disease. Ultrasound abnormalities and elevated alanine transaminase (ALT) levels are often found at routine check-up or when patients present themselves with physical complaints due to other diseases. Given that NAFLD is widely accepted as a part of metabolic syndrome, or at least being related with it, most patients present with other pathologies linked to this syndrome. Once other pathologies (Table 1) are ruled out as a cause of steatosis, NAFLD can be allocated as the most common cause for elevated ALT levels and/or steatosis. Mildly raised levels of ALT have been found in hospitalized NASH patients, but not higher than four times the upper limit of normal (ULN)[42–46]. These levels fluctuate but never return to normal values. Abnormal AST levels have also been found, especially in cirrhotic patients. Gamma-glutamyl transpeptidase (γGT) and alkaline phosphatase levels can increase although in unknown frequency. In conclusion, these mild laboratory abnormalities would not be very helpful in diagnosing this disease due to their low sensitivity and specificity.
Table 1.
Possible causes for steatosis hepatis
| Causes | |
| Metabolic | |
| Abetalipoproteinemia | |
| Glycogen storage diseases | |
| Weber-Christian disease | |
| Wolmans disease | |
| Acute fatty liver of pregnancy | |
| Lipodystrophy | |
| Iron overload syndromes | |
| α-1-antitripsin deficiency | |
| Nutritional | |
| Malnutrition | |
| Total parenteral nutrition | |
| Severe weight loss | |
| Refeeding syndrome | |
| Jejuno-ileal bypass | |
| Gastric bypass | |
| Jejunal diverticulosis with bacterial overgrowth | |
| Inflammatory | |
| HIV1 | |
| Chronic Hepatitis C infection | |
| Drugs | |
| Methotrexate | |
| Diltiazem | |
| HAART2 | |
| Amiodarone | |
| Glucocorticoids | |
| Toxins | |
| Alcohol | |
| Environmental hepatotoxins (e.g. toxic mushroom) | |
| Wilsons disease | |
| Autoimmune | |
| Autoimmune hepatitis | |
| Celiac disease |
HIV: Human immunodeficiency virus;
HAART: Highly active anti-retroviral therapy.
At more advanced disease stages, liver stigmata like jaundice, spider naevi and erythema palmare may develop. In these patients, laboratory abnormalities are consistent with progressed liver disease.
PATHOPHYSIOLOGY
Healthy subjects
Within the body’s system, the liver plays a crucial role in controlling fatty-acid and triglyceride (TG) metabolism by synthesizing, storing, secreting and oxidizing free fatty acids (FFA). The liver responds to and manages fatty acids that originate from ingested foods, adipose stores and its own de novo production. Oxidation of FFA is considered the main energy source for gluconeogenesis in a fasting state. TG is incorporated into very low dense lipoprotein (VLDL) particles while being transported out of the liver to peripheral tissues. Fatty acids are mainly stored in adipose tissues of human beings. In healthy individuals, fasting lipolysis causes release of TG into the plasma nonesterified fatty acid (NEFA) pool, while adipocytes will take up fatty acids. Postprandial pancreas-released insulin increases lipogenesis and decreases lipolysis and fatty acid oxidation in mitochondria. The second source of fatty acids contributing to the total liver supply is hepatic de novo lipogenesis (DNL). In healthy individuals, this source is a minor contributor while fasting and insulin levels are low. The third source of fatty acids is the absorption of dietary fats.
In 1998, Day et al[47] launched the “two-hit-theory”, stating that two succeeding wallops have to be delivered to the liver to cause NASH. The first hit, development of hepatic steatosis, is the accumulation of TG consisting of 3 fatty acids and a glycerol backbone, in the hepatocytes. The development of hepatic steatosis is a form of ectopic lipid accumulation, resulting from a disturbance in the balance between supply, formation, consumption and hepatic oxidation or disposal of TG. Consumption includes mitochondrial β-oxidation, production of ketone bodies, and secretion of TG in VLDL particles. Many animal and human studies have shown that there is an inextricable relation between obesity and insulin resistance (IR)[48–52]. IR is a key pathogenic factor for the development of hepatic steatosis[26–34].
Insulin resistance
Insulin resistance (IR) is the disruption of signalling pathways in cells, leading to a diminished ability to execute normal cellular responses to insulin. For details of the insulin pathway, the reader is referred to excellent reviews by Herman et al[53] and Taniguchi et al[54]. In summary, the insulin receptor is tyrosine-phosphorylated upon binding to insulin, which in turn causes tyrosine phosphorylation of the insulin receptor substrate (IRS) proteins. There are two important IRS: IRS-1 and IRS-2. IRS-1 is the initiator in the pathway of glucose metabolism. Upon phosphorylation, IRS-1 induces stimulation of the phosphatidylinositol 3-kinase (PI3K)-AKT/protein kinase B (PKB) pathway, resulting in recruitment of glucose transporters (GLUT). IRS-2 cranks up lipid metabolism in cells and is a main regulator in DNL via sterol regulatory element binding protein 1c (SREBP-1c). SREBP-1c is a member of the SREBP family, a group of transcription factors that play a fundamental role in cellular lipid metabolism[55]. Three different SREB proteins have been identified. These SREB proteins activate the complete program of cholesterol and fatty acid synthesis in the liver[56]. SREBP-1c is the isoform that plays a role in synthesis of fatty acids and TG in the liver, by stimulating the formation of enzymes, most important acetyl CoA carboxylase (AAC) and fatty acid synthase (FAS)[57]. Most SREBP-1c- stimulated enzymes are also regulated by carbohydrate response element binding protein (ChREBP)[58]. For a long time, it was assumed that a defect in muscle tissue is the first step in the origination of insulin resistant states. In the last decade, however, this doubtful honour shifted towards the adipocyte. Besides the lump storage of fat, in the mid 90’s, the exocrine functions of fat were recognised, and it became clear that fat is the choirmaster in the aetiology of IR[59]. It has been found that mice lacking GLUT4 in adipocytes develop IR in muscle and liver tissue[60], suggesting that fat cells secrete a substance that can induce IR in other tissues.
Adipocytes excrete a number of bioactive peptides that are collectively called “adipokines”. Leptin (Greek for ‘thin’), discovered by Zhang et al in 1994[61] is the prototype, but since then other adipokines, such as adiponectin[62], ghrelin[63], resistin[64] and recently retinol binding protein 4 (RBP4)[65], have been identified and characterized. RBP4 works partly by blocking the action of insulin in muscle and liver[65,66]. Depending on the amount of lipids, a stored adipocyte releases adipokines when its maximum storage is reached (leptin, RBP4) or more capacity available (adiponectin). Each adipocyte secretes a small amount of these peptides into its direct surroundings. In obese states, adipocytes also excrete inflammatory cytokines[52]. It has been found that TNF-α is over expressed in obese people[67]. Since this discovery, more inflammatory mediators have been recognised and investigated[68]. Excretion of inflammatory cytokines attracts macrophages, probably as a natural response to the clearance of the extreme swollen body and malfunctioning of fat cells. Macrophages themselves also release inflammatory substances. In the copious blood flow in adipose tissue, these peptides readily manoeuvre into the blood, which enables them to exert a number of endocrine and autocrine functions. Together, all adipocytes make up the largest endocrine organ resulting in a considerable influence of adipokines on body function[69–71]. Especially because adipocytes grow and proliferate in an overfed situation, this will lead to more excreted adipokines navigating lipids to certain specific areas of the body. Muscle and liver tissue are the main sites for ectopic fat accumulation[72]. In myocytes and hepatocytes, FFA cause IR in genetically susceptible subjects through defects in the insulin signalling pathway. Although the search for specific defects in the pathogenesis is complicated by the complexity of insulin signalling cascades, one of the major problems is a disturbance in the IRS1/PI3-kinase/Akt/GLUT pathway. IRS-1 tyrosine phosphorylation leads to serine phosphorylation, thereby interrupting the pathway for the transport of glucose via the GLUT transporters to the membrane. A number of inflammatory kinases have been found to induce this inhibitory serine phosphorylation, such as IKB-kinase-β (IKK-β), jun-kinase-1 (JNK-1) and suppressor-of-cytokine-signalling-3 (SOCS3). Interestingly, SOCS3 receptors have also been found in the hypothalamus, where it may be involved in leptin signalling. JNK-1 is found to be an important mediator in the development of inflammation in obese tissue[73]. Özcan et al[74] found that the protein is triggered by endoplasmatic reticulum (ER) stress. In obese states, the metabolic demand on the ER, is maximal and sometimes even more. As a response to the continuously high workload, the ER initiates a complex response system, referred to as the unfolding protein response (UPR). This UPR leads to the activation of JNK-1, IKK-β and TNF-α. More evidence comes available that the ER in this way translates the metabolic stress into an inflammatory signal.
Interestingly, it has been found that in hepatic IR states, the IRS-2 signalling is relatively intact, insulin down-regulates the IRS-2 receptor, resulting in over-expression of SREBP-1c and up-regulation of DNL[75].
Supply
Plasma FFA: The plasma nonesterified fatty acid (NEFA) pool contributes to the majority of fatty acids that flow to the liver in the fasting state, thus providing the bulk of FFA secreted by the liver in VLDL particles[76]. The storage of TG and FFA in adipose tissue is mediated by insulin, especially in visceral fat. In healthy individuals, consumption of a meal induces an increase in plasma insulin concentration and subsequent suppression of adipocyte lipolysis, thereby reducing the plasma NEFA pool. Adipose tissues, especially visceral adipocytes, function as a depot for energy that can be released in times of need. IR develops after long-term excess energy intake, thus decreasing the inhibitory effects of insulin on peripheral lipolysis and increasing the availability of FFA. FFA is released into the blood stream by flow of visceral adipocytes to the liver without any circumbendibus. Paradoxically, the contribution of FFA derived from the plasma pool flowing into the liver is relatively smaller in NAFLD patients compared to healthy subjects. This is due to the increased contribution of other mechanisms in these patients.
Dietary fat intake: Dietary fats are supplied to the liver by two different routes. Chronic intake of energy-enriched food challenges the processing capacity of adipocytes, with an overflow of NEFA into the plasma as a result[77]. Lipid accumulation in non-adipose tissue, mainly muscle and the liver, is a characteristic of obesity, but is also seen in lipodystrophy. Where in obesity the adipocytes overflow, in lipodystrophy there are no or insufficient adipocytes to store lipids, both being a factor for the increased ectopic lipid accumulation. A second route is via remnant chylomicrons. FFA and monoglycerides are absorbed separately and packaged into TG in intestinal epithelial cells. They are then secreted in chylomicrons (lipoproteins with a very high lipid content), which release FFA to adipose and muscle cells, mediated by lipoprotein lipase. Chylomicrons depleted of most lipids (known as chylomicron remnants) are absorbed by the liver. Studies have shown that in the remnant delivered to the liver, up to 50% of the FFA can still present, which then have to be processed by the liver. Dietary fat intake is responsible for approximately 15% of the FFA supply to the liver[78].
De novo lipogenesis (DNL): The term lipogenesis refers to the biosynthesis of lipids. DNL indicates that synthesis of fatty acids occurs in various non-fat precursors. Most important precursors are glucose, aminoacids and ethanol which produce acetyl-CoA during their catabolism and are therefore susceptible to conversion to fatty acids in the intermediary metabolism. SREBP-1c plays a key role in the regulation of DNL and is activated by insulin, endocannabinoid receptor CB-1[79], liver X receptor (LXR)-α[80], oxysterol binding protein[81] and suppressor of cytokine signalling (SOCS)-3[82]. Leptin and glucagon have antagonising effects. The suppressing effect of leptin on SREBP-1c seems paradoxal, as obese persons often exhibit high levels of leptin and high expression of SREBP-1c. This is the consequence of, on the one hand, an increase in leptin production by the expanding mass of fat cells, and on the other hand, a decrease in leptin sensitivity[83].
LXR-α is an oxysterol-activated nuclear receptor. Activation of LXR-α induces SREBP-1c transcription through the co-activation of retinoid X receptor (RXR)-α. Grefhorst et al[84] showed that exogenous administration of LXR-α ligands results in extensive hepatic steatosis.
The observation of an increase in appetite in association with the use of cannabis has lead to the hypothesis that the endocannabinoid pathway might play a role in energy intake and fat metabolism. The most prominent receptors are the cannabinoid receptor 1 (CB-1) and cannabinoid receptor 2 (CB-2). CB-2 is mostly expressed in the immune system, while CB-1 is found to be involved in the SREBP-1c pathway in liver and brain. In these pathways, the effect of CB-1 is twofold that of CB-2. Regulation of the hypothalamic-driven feeding behaviour[85,86], has direct effects on energy intake. A second effect is on hepatic fatty acid synthesis, hepatic TG quantity and activation of the released fatty acids from adipose tissue[79,87].
SOCS3 is an adipocyte-excreted cytokine that up-regulates hepatic SREBP-1c. Although the mechanisms have not been fully elucidated, TNF-α, interleukin (IL)-6 and leptin seem to augment excretion of SOCS3, whereas adiponectin is found to have inhibitory effects[82]. In healthy individuals, DNL is a minor supplier of fatty acids to hepatocytes in the fasting state, when insulin levels are low. Less than 5% of the total supply of fatty acids originates from DNL. In the postprandial state, insulin stimulates DNL which then accounts for over 26% of the FFA supplies. This more or less diurnal rhythm is not seen in NAFLD patients where the contribution of DNL is continuously 26%[78].
Oxidation
In normal conditions, mitochondria take up FFA as a substrate for β-oxidation while fasting fatty acid oxidation is the main substrate for the production of energy used in gluconeogenesis. In IR states, the amount of FFA available for oxidation exceeds the mitochondrial capacity. A bulk of acetyl CoA enters the citric acid cycle, resulting in the delivery of electrons to the respiratory chain, where they generate reactive oxygen species (ROS).
Outflow
In physiological conditions, transport of TG from hepatocytes occurs through formation of VLDL by the ER in two steps. The first step is the lipidation of apolipoprotein B (ApoB), which creates a so-called pre-VLDL. This lipidation of ApoB is catalyzed by the microsomal triglyceride transfer protein (MTTP). The pre-VLDL is transported to the smooth ER and further lipidated before its migration to the cell membrane again. MTTP is the catalysor in this process. The second step is progression of ApoB to pre-VLDL, which is dependent on the amount of TG available. If insufficient lipids are available, the ApoB protein will degrade. Insulin is a strong promotor of ApoB degradation via the PI3K pathway and can thus influence the number of VLDL particles synthesized. SREBP-1c inhibits the formation of MTTP, thereby reducing the amount of VLDL particles produced. In IR states, the PI3K pathway is eliminated to a certain extent but the up-regulated SREBP-1c leads to a decrease in VLDL synthesis. The size of the particle is dependent on the amount of TG stored in cells. It has been shown that VLDL particles in fatty livers are sufficiently larger, most likely as a result of the decreased production[84]. It is likely that export of TG is impaired in IR states.
From hepatic steatosis to NASH
Steatohepatitis is characterized microscopically by hepatic fat accumulation, mixed lobular inflammation, ballooning degeneration of hepatocytes, Mallory bodies, glycogenated hepatocyte nuclei, and pericellular fibrosis. The characteristic “chicken wire” pattern of pericellular fibrosis affects portal areas only at later stages. Accumulation of FFA and TG in hepatocytes by mechanisms described above spreads over the bed for the second hit in Day’s widely accepted theory[42]. By far, not all fatty livers progress to steatohepatitis[39,88]. Although a considerable amount of evidence is available for environmental influences, there is inevitably a genetic compound that contributes to the origination and progression of the disease. Factors responsible for the progression from simple fatty liver to NASH are extensively researched. TNF-α expression, lipid peroxidation and mitochondrial dysfunction are likely to be involved. The mechanism is triggered and starts a sequence that leads to inflammatory response and release of inflammatory cytokines, eventually resulting in the development of fibrosis and cirrhosis. Since the lipid-laden liver of steatotic patients does not increase in size, the entire lipid content must come in place of existing structures and compress them somewhat. To resolve this disadvantageous condition, hepatocytes need to clear FFA and TG, for which they can use two mechanisms. The first is the above described excretion of TG through formation of VLDL particles. It is postulated that this mechanism is impaired in hepatic steatosis. The other mechanism is the metabolism of FFA by mitochondrial β-oxidation[89]. In summary, these metabolic changes in steatotic livers result in the formation of reactive oxygen species (ROS) by mitochondria as a result of increased mitochondrial β-oxidation, hepatic microsomal cytochrome P450 2E1(CYP2E1) up-regulation and formation of Kupffer cell ROS.
The mechanism by which mitochondrial β-oxidation is up-regulated in steatotic livers still remains unclear, but it is thought that especially DNL-derived FFA and peroxisome proliferator-activated receptor (PPAR)-α are important stimulators of carnitine palmitatoyltranferase-1 (CPT-1) responsible for the entry of FFA into mitochondria. The massive influx of FFA from peripheral tissue and mostly the increased DNL within hepatocytes exceeds the metabolic capabilities of mitochondria, with the formation of ROS and an inflammatory response as a result.
CYP2E1 is predominantly found in the ER, but significant amounts are present in the cytosol and mitochondria where it stimulates microsomal fatty acid oxidation. Increased CYP2E1 activity and expression are found in NASH patients[90], but the mechanisms behind this remain unclear. Recently Rahman et al found that CCAAT/Enhancer binding protein (C/EBPbeta) expression may be an important factor in the upregulation of CYP2E1[91]. Other authors have postulated the influence of IR[92,93] and increased ketogenesis[94].
In various models, steatosis endotoxin receptors on Kupffer cells are increased, which may trigger the assemblage of NAD(P)H oxidase on the plasma membrane of hepatocytes and thereby causing ROS formation[95].
The surplus of FFA within the liver causes the formation of excess amounts of ROS. Mitochondria in non-fat-laden hepatocytes also produce rather large amounts of ROS, but enzymatic processes can change ROS into “safe water”. Excessive ROS formation can lead to an overburden of this escape mechanism and ROS can leave the mitochondria. In the cytosol, ROS enhances lipid peroxidation products and mitochondrial DNA (mtDNA). Increased mitochondrial ROS formation could also directly oxidize mitochondrial DNA, proteins and lipids, and trigger hepatic TNF-α formation by activating nuclear factor (NF)-κB and deplete antioxidants, thus further increasing mitochondrial ROS formation. Induction of the inflammatory cascade can also be the result of the reduced availability of anti-inflammatory products as adiponectin[96].
Another worsening and possible triggering factor might be the tumour necrosis factor (TNF)-α[97,98]. In obese and IR patients, serum levels of TNF-α are proportionally increased and one of the striking differences between patients with NASH and those with simple steatosis is the serum level of TNF-α[99]. Not hepatic, but mainly adipose tissue is the main supplier of TNF-α. In normal conditions, hepatocytes that frequently come into contact with a variety of endo- and exotoxins are not very sensitive to TNF-α, but in NASH patients it might be possible that the increased levels cause leakage of the mitochondrial outer membrane and thereby increasing ROS formation[89]. TNF-α also has direct effects on IRS phosphorylation and stimulates SOCS3 formation via interleukin 6 (IL-6) and inhibitory κB-kinase (IKK), thereby worsening IR and steatosis[100,101] (Figure 1).
Figure 1.

Pathogenesis of nonalcoholic steatohepatitis during insulin resistance. FFA is supplied to the liver through dietary intake, and lipolysis in adipocytes via chylomicron remnants. Transcription of SREBP-1c is chronically up-regulated resulting in DNL. Simultaneous inhibition of VLDL synthesis results in disruption of triglycerides export. The surplus of fatty acids is stored in triglycerides or metabolized via peroxisomal and mitochondrial oxidation. The excessive oxidation will lead to production of ROS and oxidative stress. This will trigger the inflammatory response and apoptosis as well activation of stellate cells.
DIAGNOSIS
General
As noted before, NAFLD patients are mostly asymptomatic because slightly elevated liver enzymes are accidentally found. There is some debate about the question when further investigations are performed, especially in liver biopsy. Generally, it is reasonable to undertake action when the ALT level is > 2 × ULN measured at two different occasions[102]. When the diagnosis of NAFLD is considered, it is important to exclude other pathological conditions that are associated with elevated ALT levels and/or steatosis (Table 1). It is especially difficult to find the difference between alcoholic and non-alcoholic liver diseases, as not all patients are honest about their alcohol intake and there is no adequate diagnostic difference between the two diseases. An abdominal ultrasound is performed to exclude hepatobiliary obstructions or tumours.
Imaging techniques
Ultrasonography, computerized tomography (CT) scan, and magnetic resonance imaging (MRI) can all be used to diagnose hepatic steatosis. Ultrasonography, the most commonly used and least expensive method, can be used to diagnose moderate to severe steatosis. Studies in the 1980’s found that its sensitivity and specificity vary from 60% to 94% and 88% to 95%, respectively. Palmentieri et al[103] investigated a subgroup of patients with hepatic steatosis > 30%, showing that the sensitivity and specificity of US increase up to 90% and 97% , respectively. Although the diagnostic capacity of ultrasonography increases with higher degrees of steatosis[103–105], accurate quantification of hepatic fat and comparison between simple hepatic steatosis and steatohepatitis are impossible. Un-enhanced CT imaging can accurately detect and quantify the amount of steatosis in patients[106,107]. Grey scales (representing the amount of radiation absorbed) of the liver and spleen are measured and expressed in Houndsfield units (HU). For quantification of the amount of fat-infiltrated hepatocytes, the difference in grey scale between the liver and spleen can be measured in HU. This measurement correlates well with the percentage of hepatocytes with fatty infiltration[107], where the hepatic attenuation decreases with the increased fat. For steatosis > 33%, the sensitivity and specificity are 82%-93% and 100%, respectively[106]. There is no difference in diagnostic value between a non-contrast CT scan and a contrast-enhanced one, with specifically more attenuation in the blood vessels than in the liver parenchyma. Contrast-enhanced CT scan does not provide more information. In fact, its sensitivity and specificity for steatosis are lower than those of un-enhanced CT imaging. Another drawback of contrast-enhanced CT scan of steatosis is the greater difficulty in its measurement due to the more complicated protocol.
The most accurate available technique for detection and quantification of hepatic steatosis is NMR[108–110]. T1-weighted dual-echo chemical shift gradient- echo NMR is commonly used to obtain images. The main advantage of this technique is the possibility of acquiring in-phase (water) and opposed-phase (fat) images in one breath hold, thereby reducing the influence of breathing movements and contrast absorption. Besides, this technique is useful for follow-up due to the lack of the use of fluoroscopy. Qualitative measurement or detection of steatosis is assessed on opposed-phase images. On T1- weighted images, a shorter relaxation time represents the higher signal intensity (SI). In healthy individuals, the SI of the liver is higher than that of the spleen. Steatosis causes a drop in SI on opposed-phase images. When SI of the liver equals to that of the spleen, a diagnosis of mild steatosis is made. Moderate or severe steatosis is diagnosed when SI of the liver is less than that of the spleen. Quantification of steatosis is possible with MRI by calculating the mean SI of the liver on in-phase and opposed-phase images. A number of regions of interest (ROI) are drawn in sections of the liver, from which the mean can be calculated. Using the same method, the mean SI of the spleen or fat issue surrounding the liver can be calculated as a reference. The difference in SI on opposed-phase and in-phase images can be calculated and expressed as a percentage of fatty infiltration of the liver. MRI shows a good correlation with histological examination, the sensitivity and specificity of MRI are 100% and 92.3%, respectively[110].
Another relatively new NMR technique under development, widely used to quantify hepatic triglyceride content, is magnetic resonance spectroscopy (MRS)[111,112]. MRS of in vivo biological tissues was first reported in 1973 and used in the field of chemistry before NMR was introduced in hospitals. The principle of MRS is based on the differences in resonance frequencies of protons. The electron cloud surrounding molecules shields protons to varying degrees depending on the specific molecule structure and the specific position of protons in the molecule. This shielding causes protons in different molecules or even in different places of the same molecule to have a slightly different resonance frequency. Instead of using resonance frequencies for creating anatomical images, the differences in spectroscopy frequency are used to identify different chemical compounds. By using these differences, protons in water molecules can be differentiated from protons in lipids. Quantifying the amount of a certain biochemical component is possible by calculating the area under the “fat resonance peak” and comparing it to the “water resonance peak” (Figure 2).
Figure 2.
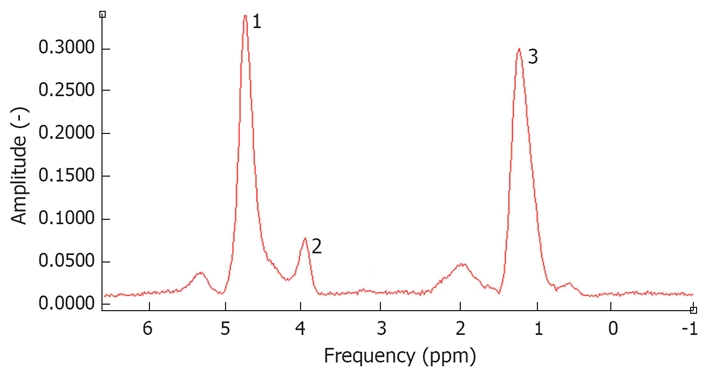
Spectrum of a fatty liver measured by 1H-magnetic resonance spectroscopy. The water peak is at 4.3 ppm. 1: Residual water partially suppressed; 2: Glycerol/phospholipids; 3: (-CH2-)n of saturated fat.
Technically, coronal, axial and sagital images of the liver are acquired and a volume of interest is defined, avoiding major blood vessels and bile ducts.
Unfortunately, to date, no conventional diagnostic imaging method that can accurately distinguish NASH from simple steatosis is available[111,113]. Transient elastography (fibroscan) is a technique under development. Studies in chronic hepatitis C (CHC) patients showed that cirrhosis (severe fibrosis) can adequately be distinguished from mild fibrosis, but this accuracy is much less in distinguishing various degrees of fibrosis[114]. Another drawback of the fibroscan method is that it is difficult to use and inaccurate in obese patients. A combination of serologic markers is under investigation to assess the severity of fibrosis[115]. Although results seem promising, further study is wanted.
THERAPEUTIC OPTIONS
Life style modifications and weight reduction
Since the majority of patients suffer from obesity, insulin resistance and concomitant cardiovascular disease, weight reduction of approximately 10% has been advised by the American Gastroenterological Association[116]. An analysis by Wang et al[117] of all published articles and meeting abstracts have revealed no randomized controlled trials. Besides, the use of variable primary endpoints and control groups worsened the analysis of this comprehensive review. Although on a theoretical basis, reduced caloric intake, exercise and weight loss would eventually improve hepatic steatosis, very scarce evidence is available to support this hypothesis. The limited data are due to the small number and lack of histological evidence. Recently, Huang et al[118] analyzed the effect of a 12-mo standardized nutritional counseling in 16 of 23 patients and found that the mean weight reduction is 2.9 kg with histological improvement in 9 patients.
No data are available on the long-term effect of weight loss on liver-related diseases such as cirrhosis or its complications.
Pharmacological interventions
Drug-induced weight reduction: The only two registered drugs for pharmacological weight reduction, Orlistat (n = 4) and Sibutramine (n = 1), have been investigated in a few small non-randomized studies[119–123]. Orlistat, a gastric and pancreatic lipase inhibitor resulting in fat malabsorption (approximately 30%), has been studied in one case series[119], three pilot studies[120–123] and one RCT[124]. Overall, patients can achieve impressive weight loss (10-15 kg) with improvements in liver enzymes but variable results in histology.
Sibutramine, a serotonin and norepinephrin reuptake inhibitor, acts on enhancing satiety via central mechanisms. There is only one published study on NAFLD in 25 patients demonstrating weight loss, improvement in liver enzymes and hepatic steatosis on ultrasound. Unfortunately, repeated liver biopsy was not performed.
Antioxidants: Since the pathogenesis of NAFLD is thought to be in a two-hit fashion, it is believed that oxidative stress causes a second hit leading to inflammation. In vitro and in vivo animal and human studies[125,126] have been performed on the effects of vitamins E and C as antioxidants. The promising results in one study were counteracted by another. Harisson et al[126] reported that a between group analysis cannot show any beneficial effect of the combination of vitamins E and C after 6 mo, compared to placebo. Recently, Lirussi et al[127] identified 6 trials that were analyzed according to the intention-to-treat principle and found that despite the significant improvements in liver enzymes and minor adverse events, radiological and histological data are too limited to support or repudiate the use of antioxidants in patients with NAFLD.
Ursodeoxycholic acid (UDCA): UDCA, approved as a drug of choice in treatment of patients with PBC, exerts its effect by reducing the portion of hydrophobic bile acids contributing to oxidative stress. Four clinical trials[128–131], of which only one assessed histology[129] and had a low-bias risk, have been conducted. No significant differences in the degree of steatosis, inflammation or fibrosis could be found between the treated and placebo groups. Unfortunately, these studies had no heterogeneity with respect to inclusion criteria, sample size, duration of treatment and methods of outcome assessment. Therefore, the Cochrane analysis by Orlando et al[132] concludes that the data are insufficient to use UDCA in treatment of patients with NAFLD.
Metformin: Metformin, a biguanide, has been shown to be an effective drug for the treatment of patients with type 2 diabetes mellitus[133]. Its administration improves hepatic steatosis and hepatomegaly. In addition to this observation, human pilot studies performed with variable results[134,135] could not demonstrate a beneficial effect of metformin compared to a calorie-restricted diet. Similar to the previous mentioned therapies, histological data are limited to support an association between improvements in liver enzymes and histological findings.
Thiazolidinediones (TZD): This class of agents acts as agonists of the peroxisome proliferator-activated receptor gamma on ameliorating insulin resistance and glucose and lipid metabolism. The first generation of TZD, e.g. troglitazone, appears to be effective on ALT levels, although it has been withdrawn from the market due to its severe hepatotoxicity[136,137]. In addition, the second generation of TZD (pioglitazone and rosiglitazone) is safer. A Cochrane analysis by Angelico et al[138], (excluding trials treating patients with type 2 diabetes mellitus) extracted only one RCT by Sanyal et al[139], showing that combination therapy with vitamin E and pioglitazone is significantly superior to vitamin E alone in terms of the degree of hepatic steatosis, but not other histological variables.
An open label trial of rosiglitazone in 26 biopsy-proven NASH[140] and two pilot studies with pioglitazone[141,142] (n = 73) during 48 wk demonstrated improvements both in liver chemistry and in histological features like steatosis, necroinflammation and fibrosis. Weight gain seems to be the most important adverse event. Recently, the use of rosiglitazone has been associated with the slightly increased bone loss in postmenopausal women and elderly diabetics[143–145] and an increased risk of cardiovascular events, i.e. myocardial infarction[143,146,147]. Further research focusing on larger randomized controlled trials with this class of drugs will be valuable.
Lipid lowering drugs: Use of statins or fibrates has not been investigated in large randomized trials. Primary endpoints of these studies are liver enzymes but not histology. No definite conclusions can be made from these limited studies[121,148].
Adipokines: As mentioned before, adipocytes act as hormonal active tissue producing cytokines, like leptin, resistin, adiponectin and tumor necrosis factor-α (TNF-α). It was reported that levels of TNF-α are increased in patients with NASH[99]. TNF-α, as a potential proinflammatory cytokine, promotes insulin resistance and thereby hepatic steatosis[101]. Some animal and even fewer human studies focusing on the effect of blocking this adipocytokine showed that patients treated with pentoxifyllin for 6 or 12 mo have a significant improvement in liver enzymes[149,150]
Administration of synthetic adiponectin, exposing opposite effects to TNF-α, in two animal models can ameliorate hepatomegaly, steatosis and elevated ALT-levels[151].
Leptin, a 16-kDa protein hormone, has been shown to play a pivotal role in energy homeostasis by activating and inhibiting certain neurons[152]. It has been shown that administration of leptin infusions in patients with generalized lipodystrophy significantly ameliorates insulin resistance, glucose and triglyceride levels as well as hepatic steatosis[153,154]. No such studies have been performed yet in NAFLD patients.
Resistin, another adipocytokine, is a subject of contro-versy regarding to its causal role in obesity and type 2 diabetes mellitus. Due to this controversy, no published data are available about its beneficial effects on inhibition of resistin.
FUTURE DIRECTIONS
Up until now, the exact treatment strategy for the treatment of patients with NAFLD has not been well established in RCT. Research topics in this field are challenging. In the United States and Europe, some research groups are focusing on comparing different treatment options and identifying those patients most in need for treatment. Evolving new imaging techniques like proton magnetic resonance spectroscopy might differentiate between those patients having type 1 NAFLD (defined as simple steatosis without features of inflammation) and type 2 NAFLD (containing patients with variable grades of inflammation and fibrosis eventually resulting in cirrhosis).
Several registered drugs aiming at improving metabolic syndrome should be further investigated on their exact anti-steatotic effects. The most promising drugs are insulin sensitizers (especially Thiazolidinediones), which should be further investigated in a larger RCT aiming at establishing the optimal dosage, time of treatment and adverse effects.
Since the discovery of two cannabinoid receptor antagonist receptors (CB1 and CB2) in the late 1980’s and the beginning of the 1990’s in the brain[155,156] and gastrointestinal tract[157], it has gained more and more interests from both researchers and clinicians. CB1 has been extensively found in the central nervous system, affecting many neurological and psychological phenomena, like appetite, mood and spatial coordination of muscle tone[158]. In contrast, CB2 detected in peripheral cells of the immune system (lymphocytes, monocytes and neutrophils) exerts additional effects on the gut (inhibition of motility) and vasodilation[159,160]. Research in animal models revealed that activation of the endocannabinoid system leads (partially) to the development of portal hypertension and arterial hypotension through macrophages and platelets activated by bacterial lipopolysacharides[161,162]. Secondly, anandamide, an endogenous ligand for CB-receptors, appears to be up-regulated in patients with endotoxic shock[163] and subsequently hepatocellular apoptosis has been linked to anandamide and its lipid-lipid plasma membrane interaction, resulting in enhanced susceptibility to oxidative stress[164]. Thirdly, another hypothesis is that an activated CB-system may have influence on hepatic encephalopathy[165]. In humans, endocannabinoids have been used in the treatment of three patients with cholestatic-related intractable pruritus[166]. SR141716A (Rimonabant, Sanofi-Aventis, Paris, France) under investigation may be used in the treatment of patients with obesity and metabolic syndrome[167]. In obese Zucker rats, administration of this drug could ameliorate markers of hepatic damage (defined as increased liver enzymes, focal hepatic TNF-α and decreased adiponectin), and decrease hepatomegaly[167]. Preliminary results in humans are promising since the used drug is safe, effective in achieving weight reduction and amelioration of the lipid profile and metabolic syndrome[85,168,169].
In conclusion, the development of drugs acting both on the cannabinoid system influencing the central nervous system through inhibition of appetite and on the peripheral tissue ameliorating hepatic hemodynamics, inflammation and weight reduction accompanying improvement in metabolic syndrome, will lead to new research aims in the field of hepatology. In particular, the development and conductance of RCT with synthetic, non-psychotropic cannabinoids might result in optimizing treatment strategies for patients with NASH.
CONCLUSION
Nonalcoholic fatty liver disease, especially nonalcoholic steatohepatitis, forms a definite threat to human health. With the increase in obesity, an increase in NAFLD patients can be expected, eventually leading to an increased number of liver transplantations. Pathophysiological mechanism is the subject of research all around the world, leading to a continuous current of new evidence and more knowledge about the complex mechanisms behind the disease. At the same time, diagnostic methods for detecting steatosis and steatohepatitis are under development. 1H-magnetic resonance spectroscopy is accurate in the detection and quantification of fat in the liver, but the eagerly wanted non-invasive tool for the detection of fibrosis or inflammation in steatotic livers is not available. Therapeutic options do increase. Weight loss by dietary and lifestyle intervention remains the cornerstone of treatment, but motivated patients can be supported by medications. Most promising results are found with thiazolidinediones, but recent upheaval around rosiglitazone means a setback of this drug.
Peer reviewer: Deborah L Diamond, Dr, Department of Microbio-logy, University of Washington, Box 358070, Seattle, WA 98195-8070, United States
S- Editor Sun YL L- Editor Wang XL E- Editor Ma WH
References
- 1.World Health Organization. Obesity and Overweight. 2003 Available from: URL: http://www.who.int/dietphysicalactivity/media/en/gsfs_obesity.pdf. [Google Scholar]
- 2.James PT, Leach R, Kalamara E, Shayeghi M. The worldwide obesity epidemic. Obes Res. 2001;9 Suppl 4:228S–233S. doi: 10.1038/oby.2001.123. [DOI] [PubMed] [Google Scholar]
- 3.Ogden CL, Yanovski SZ, Carroll MD, Flegal KM. The epidemiology of obesity. Gastroenterology. 2007;132:2087–2102. doi: 10.1053/j.gastro.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 4.Alberti KG, Zimmet P, Shaw J. Metabolic syndrome--a new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet Med. 2006;23:469–480. doi: 10.1111/j.1464-5491.2006.01858.x. [DOI] [PubMed] [Google Scholar]
- 5.Day C. Metabolic syndrome, or What you will: definitions and epidemiology. Diab Vasc Dis Res. 2007;4:32–38. doi: 10.3132/dvdr.2007.003. [DOI] [PubMed] [Google Scholar]
- 6.Johnson LW, Weinstock RS. The metabolic syndrome: concepts and controversy. Mayo Clin Proc. 2006;81:1615–1620. doi: 10.4065/81.12.1615. [DOI] [PubMed] [Google Scholar]
- 7.Bergman RN, Kim SP, Catalano KJ, Hsu IR, Chiu JD, Kabir M, Hucking K, Ader M. Why visceral fat is bad: mechanisms of the metabolic syndrome. Obesity (Silver Spring) 2006;14 Suppl 1:16S–19S. doi: 10.1038/oby.2006.277. [DOI] [PubMed] [Google Scholar]
- 8.Laclaustra M, Corella D, Ordovas JM. Metabolic syndrome pathophysiology: the role of adipose tissue. Nutr Metab Cardiovasc Dis. 2007;17:125–139. doi: 10.1016/j.numecd.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Chavez-Tapia NC, Mendez-Sanchez N, Uribe M. The metabolic syndrome as a predictor of nonalcoholic fatty liver disease. Ann Intern Med. 2006;144:379; author reply 380. doi: 10.7326/0003-4819-144-5-200603070-00021. [DOI] [PubMed] [Google Scholar]
- 10.Hamaguchi M, Kojima T, Takeda N, Nakagawa T, Taniguchi H, Fujii K, Omatsu T, Nakajima T, Sarui H, Shimazaki M, et al. The metabolic syndrome as a predictor of nonalcoholic fatty liver disease. Ann Intern Med. 2005;143:722–728. doi: 10.7326/0003-4819-143-10-200511150-00009. [DOI] [PubMed] [Google Scholar]
- 11.Kida Y, Sato T. The metabolic syndrome as a predictor of nonalcoholic fatty liver disease. Ann Intern Med. 2006;144:379–380; author reply 380. doi: 10.7326/0003-4819-144-5-200603070-00022. [DOI] [PubMed] [Google Scholar]
- 12.Marchesini G, Babini M. Nonalcoholic fatty liver disease and the metabolic syndrome. Minerva Cardioangiol. 2006;54:229–239. [PubMed] [Google Scholar]
- 13.Moseley RH. Progress in understanding the pathogenesis of nonalcoholic fatty liver disease. Hepatology. 2005;41:204–206. doi: 10.1002/hep.20558. [DOI] [PubMed] [Google Scholar]
- 14.Neuschwander-Tetri BA. Fatty liver and the metabolic syndrome. Curr Opin Gastroenterol. 2007;23:193–198. doi: 10.1097/MOG.0b013e32801421a9. [DOI] [PubMed] [Google Scholar]
- 15.Targher G. Non-alcoholic fatty liver disease, the metabolic syndrome and the risk of cardiovascular disease: the plot thickens. Diabet Med. 2007;24:1–6. doi: 10.1111/j.1464-5491.2007.02025.x. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe T, Murata C, Watanabe Y. Metabolic syndrome from the view point of public health: with special reference to nonalcoholic fatty liver disease. Nippon Koshu Eisei Zasshi. 2005;52:934–942. [PubMed] [Google Scholar]
- 17.Thaler H. Fatty liver, its causes and concomitant diseases. Dtsch Med Wochenschr. 1962;87:1049–1055. doi: 10.1055/s-0028-1111865. [DOI] [PubMed] [Google Scholar]
- 18.Thaler H. The fatty liver and its pathogenetic relation to liver cirrhosis. Virchows Arch Pathol Anat Physiol Klin Med. 1962;335:180–210. [PubMed] [Google Scholar]
- 19.Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55:434–438. [PubMed] [Google Scholar]
- 20.Saadeh S, Younossi ZM. The spectrum of nonalcoholic fatty liver disease: from steatosis to nonalcoholic steatohepatitis. Cleve Clin J Med. 2000;67:96–97, 101-104. doi: 10.3949/ccjm.67.2.96. [DOI] [PubMed] [Google Scholar]
- 21.Day CP. Natural history of NAFLD: remarkably benign in the absence of cirrhosis. Gastroenterology. 2005;129:375–378. doi: 10.1053/j.gastro.2005.05.041. [DOI] [PubMed] [Google Scholar]
- 22.Ioannou GN. The natural history of NAFLD: impressively unimpressive. Gastroenterology. 2005;129:1805. doi: 10.1053/j.gastro.2005.09.041. [DOI] [PubMed] [Google Scholar]
- 23.Ratziu V, Poynard T. Assessing the outcome of nonalcoholic steatohepatitis? It’s time to get serious. Hepatology. 2006;44:802–805. doi: 10.1002/hep.21391. [DOI] [PubMed] [Google Scholar]
- 24.Bugianesi E. Non-alcoholic steatohepatitis and cancer. Clin Liver Dis. 2007;11:191–207, x-xi. doi: 10.1016/j.cld.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Mori S, Yamasaki T, Sakaida I, Takami T, Sakaguchi E, Kimura T, Kurokawa F, Maeyama S, Okita K. Hepatocellular carcinoma with nonalcoholic steatohepatitis. J Gastroenterol. 2004;39:391–396. doi: 10.1007/s00535-003-1308-3. [DOI] [PubMed] [Google Scholar]
- 26.Angelico F, Del Ben M, Conti R, Francioso S, Feole K, Fiorello S, Cavallo MG, Zalunardo B, Lirussi F, Alessandri C, et al. Insulin resistance, the metabolic syndrome, and nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 2005;90:1578–1582. doi: 10.1210/jc.2004-1024. [DOI] [PubMed] [Google Scholar]
- 27.Bugianesi E, McCullough AJ, Marchesini G. Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology. 2005;42:987–1000. doi: 10.1002/hep.20920. [DOI] [PubMed] [Google Scholar]
- 28.Camma C, Bruno S, Di Marco V, Di Bona D, Rumi M, Vinci M, Rebucci C, Cividini A, Pizzolanti G, Minola E, et al. Insulin resistance is associated with steatosis in nondiabetic patients with genotype 1 chronic hepatitis C. Hepatology. 2006;43:64–71. doi: 10.1002/hep.20983. [DOI] [PubMed] [Google Scholar]
- 29.Choudhury J, Sanyal AJ. Insulin resistance and the pathogenesis of nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8:575–594, ix. doi: 10.1016/j.cld.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 30.Choudhury J, Sanyal AJ. Insulin resistance in NASH. Front Biosci. 2005;10:1520–1533. doi: 10.2741/1636. [DOI] [PubMed] [Google Scholar]
- 31.Diehl AM, Clarke J, Brancati F. Insulin resistance syndrome and nonalcoholic fatty liver disease. Endocr Pract. 2003;9 Suppl 2:93–96. doi: 10.4158/EP.9.S2.93. [DOI] [PubMed] [Google Scholar]
- 32.Eguchi Y, Eguchi T, Mizuta T, Ide Y, Yasutake T, Iwakiri R, Hisatomi A, Ozaki I, Yamamoto K, Kitajima Y, et al. Visceral fat accumulation and insulin resistance are important factors in nonalcoholic fatty liver disease. J Gastroenterol. 2006;41:462–469. doi: 10.1007/s00535-006-1790-5. [DOI] [PubMed] [Google Scholar]
- 33.Tilg H, Hotamisligil GS. Nonalcoholic fatty liver disease: Cytokine-adipokine interplay and regulation of insulin resistance. Gastroenterology. 2006;131:934–945. doi: 10.1053/j.gastro.2006.05.054. [DOI] [PubMed] [Google Scholar]
- 34.Utzschneider KM, Kahn SE. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 2006;91:4753–4761. doi: 10.1210/jc.2006-0587. [DOI] [PubMed] [Google Scholar]
- 35.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 36.Rinella ME, Alonso E, Rao S, Whitington P, Fryer J, Abecassis M, Superina R, Flamm SL, Blei AT. Body mass index as a predictor of hepatic steatosis in living liver donors. Liver Transpl. 2001;7:409–414. doi: 10.1053/jlts.2001.23787. [DOI] [PubMed] [Google Scholar]
- 37.Sabir N, Sermez Y, Kazil S, Zencir M. Correlation of abdominal fat accumulation and liver steatosis: importance of ultrasonographic and anthropometric measurements. Eur J Ultrasound. 2001;14:121–128. doi: 10.1016/s0929-8266(01)00153-7. [DOI] [PubMed] [Google Scholar]
- 38.Stranges S, Dorn JM, Muti P, Freudenheim JL, Farinaro E, Russell M, Nochajski TH, Trevisan M. Body fat distribution, relative weight, and liver enzyme levels: a population-based study. Hepatology. 2004;39:754–763. doi: 10.1002/hep.20149. [DOI] [PubMed] [Google Scholar]
- 39.Adams LA, Lymp JF, St SJ, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 40.Liou I, Kowdley KV. Natural history of nonalcoholic steatohepatitis. J Clin Gastroenterol. 2006;40:S11–S16. doi: 10.1097/01.mcg.0000168644.23697.31. [DOI] [PubMed] [Google Scholar]
- 41.Wieckowska A, Feldstein AE. Nonalcoholic fatty liver disease in the pediatric population: a review. Curr Opin Pediatr. 2005;17:636–641. doi: 10.1097/01.mop.0000172816.79637.c5. [DOI] [PubMed] [Google Scholar]
- 42.Adams LA, Talwalkar JA. Diagnostic evaluation of nonalcoholic fatty liver disease. J Clin Gastroenterol. 2006;40:S34–S38. doi: 10.1097/01.mcg.0000168642.38945.f1. [DOI] [PubMed] [Google Scholar]
- 43.Chang Y, Ryu S, Sung E, Jang Y. Higher concentrations of alanine aminotransferase within the reference interval predict nonalcoholic fatty liver disease. Clin Chem. 2007;53:686–692. doi: 10.1373/clinchem.2006.081257. [DOI] [PubMed] [Google Scholar]
- 44.Kunde SS, Lazenby AJ, Clements RH, Abrams GA. Spectrum of NAFLD and diagnostic implications of the proposed new normal range for serum ALT in obese women. Hepatology. 2005;42:650–656. doi: 10.1002/hep.20818. [DOI] [PubMed] [Google Scholar]
- 45.Oh SY, Cho YK, Kang MS, Yoo TW, Park JH, Kim HJ, Park DI, Sohn CI, Jeon WK, Kim BI, et al. The association between increased alanine aminotransferase activity and metabolic factors in nonalcoholic fatty liver disease. Metabolism. 2006;55:1604–1609. doi: 10.1016/j.metabol.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 46.Ratziu V, Imbert-Bismut F, Messous D, Poynard T. The elusiveness of “normal” ALT in fatty liver. Hepatology. 2004;39:1172; author reply 1173. doi: 10.1002/hep.20187. [DOI] [PubMed] [Google Scholar]
- 47.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 48.Boden G. Free fatty acids-the link between obesity and insulin resistance. Endocr Pract. 2001;7:44–51. doi: 10.4158/EP.7.1.44. [DOI] [PubMed] [Google Scholar]
- 49.Seidell JC. Obesity, insulin resistance and diabetes--a worldwide epidemic. Br J Nutr. 2000;83 Suppl 1:S5–S8. doi: 10.1017/s000711450000088x. [DOI] [PubMed] [Google Scholar]
- 50.Sesti G. Pathophysiology of insulin resistance. Best Pract Res Clin Endocrinol Metab. 2006;20:665–679. doi: 10.1016/j.beem.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 51.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 52.Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology. 2007;132:2169–2180. doi: 10.1053/j.gastro.2007.03.059. [DOI] [PubMed] [Google Scholar]
- 53.Herman MA, Kahn BB. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. J Clin Invest. 2006;116:1767–1775. doi: 10.1172/JCI29027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 55.Ferre P, Foufelle F. SREBP-1c transcription factor and lipid homeostasis: clinical perspective. Horm Res. 2007;68:72–82. doi: 10.1159/000100426. [DOI] [PubMed] [Google Scholar]
- 56.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dentin R, Pegorier JP, Benhamed F, Foufelle F, Ferre P, Fauveau V, Magnuson MA, Girard J, Postic C. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem. 2004;279:20314–20326. doi: 10.1074/jbc.M312475200. [DOI] [PubMed] [Google Scholar]
- 58.Postic C, Dentin R, Denechaud PD, Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu Rev Nutr. 2007;27:179–192. doi: 10.1146/annurev.nutr.27.061406.093618. [DOI] [PubMed] [Google Scholar]
- 59.Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006;444:847–853. doi: 10.1038/nature05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, Minnemann T, Shulman GI, Kahn BB. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–733. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 62.Maeda K, Okubo K, Shimomura I, Funahashi T, Matsuzawa Y, Matsubara K. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (AdiPose Most abundant Gene transcript 1) Biochem Biophys Res Commun. 1996;221:286–289. doi: 10.1006/bbrc.1996.0587. [DOI] [PubMed] [Google Scholar]
- 63.Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 64.Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 65.Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, Kotani K, Quadro L, Kahn BB. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. 2005;436:356–362. doi: 10.1038/nature03711. [DOI] [PubMed] [Google Scholar]
- 66.Graham TE, Yang Q, Bluher M, Hammarstedt A, Ciaraldi TP, Henry RR, Wason CJ, Oberbach A, Jansson PA, Smith U, et al. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N Engl J Med. 2006;354:2552–2563. doi: 10.1056/NEJMoa054862. [DOI] [PubMed] [Google Scholar]
- 67.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vettor R, Milan G, Rossato M, Federspil G. Review article: adipocytokines and insulin resistance. Aliment Pharmacol Ther. 2005;22 Suppl 2:3–10. doi: 10.1111/j.1365-2036.2005.02587.x. [DOI] [PubMed] [Google Scholar]
- 69.Ronti T, Lupattelli G, Mannarino E. The endocrine function of adipose tissue: an update. Clin Endocrinol (Oxf) 2006;64:355–365. doi: 10.1111/j.1365-2265.2006.02474.x. [DOI] [PubMed] [Google Scholar]
- 70.Hutley L, Prins JB. Fat as an endocrine organ: relationship to the metabolic syndrome. Am J Med Sci. 2005;330:280–289. doi: 10.1097/00000441-200512000-00005. [DOI] [PubMed] [Google Scholar]
- 71.Scherer PE. Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes. 2006;55:1537–1545. doi: 10.2337/db06-0263. [DOI] [PubMed] [Google Scholar]
- 72.Yki-Jarvinen H. Ectopic fat accumulation: an important cause of insulin resistance in humans. J R Soc Med. 2002;95 Suppl 42:39–45. [PMC free article] [PubMed] [Google Scholar]
- 73.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 74.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 75.Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000;6:77–86. [PubMed] [Google Scholar]
- 76.Barrows BR, Timlin MT, Parks EJ. Spillover of dietary fatty acids and use of serum nonesterified fatty acids for the synthesis of VLDL-triacylglycerol under two different feeding regimens. Diabetes. 2005;54:2668–2673. doi: 10.2337/diabetes.54.9.2668. [DOI] [PubMed] [Google Scholar]
- 77.Unger RH. Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends Endocrinol Metab. 2003;14:398–403. doi: 10.1016/j.tem.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 78.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Osei-Hyiaman D, DePetrillo M, Pacher P, Liu J, Radaeva S, Batkai S, Harvey-White J, Mackie K, Offertaler L, Wang L, et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J Clin Invest. 2005;115:1298–1305. doi: 10.1172/JCI23057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yan D, Lehto M, Rasilainen L, Metso J, Ehnholm C, Yla-Herttuala S, Jauhiainen M, Olkkonen VM. Oxysterol binding protein induces upregulation of SREBP-1c and enhances hepatic lipogenesis. Arterioscler Thromb Vasc Biol. 2007;27:1108–1114. doi: 10.1161/ATVBAHA.106.138545. [DOI] [PubMed] [Google Scholar]
- 82.Ueki K, Kadowaki T, Kahn CR. Role of suppressors of cytokine signaling SOCS-1 and SOCS-3 in hepatic steatosis and the metabolic syndrome. Hepatol Res. 2005;33:185–192. doi: 10.1016/j.hepres.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 83.Unger RH. Longevity, lipotoxicity and leptin: the adipocyte defense against feasting and famine. Biochimie. 2005;87:57–64. doi: 10.1016/j.biochi.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 84.Grefhorst A, Elzinga BM, Voshol PJ, Plosch T, Kok T, Bloks VW, van der Sluijs FH, Havekes LM, Romijn JA, Verkade HJ, et al. Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles. J Biol Chem. 2002;277:34182–34190. doi: 10.1074/jbc.M204887200. [DOI] [PubMed] [Google Scholar]
- 85.Van Gaal LF, Rissanen AM, Scheen AJ, Ziegler O, Rossner S. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet. 2005;365:1389–1397. doi: 10.1016/S0140-6736(05)66374-X. [DOI] [PubMed] [Google Scholar]
- 86.Wierzbicki AS. Rimonabant: endocannabinoid inhibition for the metabolic syndrome. Int J Clin Pract. 2006;60:1697–1706. doi: 10.1111/j.1742-1241.2006.01210.x. [DOI] [PubMed] [Google Scholar]
- 87.Woods SC. The endocannabinoid system: mechanisms behind metabolic homeostasis and imbalance. Am J Med. 2007;120:S9–S17; discussion S29-S32. doi: 10.1016/j.amjmed.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 88.Ekstedt M, Franzen LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, Kechagias S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44:865–873. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 89.Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 90.Weltman MD, Farrell GC, Hall P, Ingelman-Sundberg M, Liddle C. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology. 1998;27:128–133. doi: 10.1002/hep.510270121. [DOI] [PubMed] [Google Scholar]
- 91.Rahman SM, Schroeder-Gloeckler JM, Janssen RC, Jiang H, Qadri I, Maclean KN, Friedman JE. CCAAT/enhancing binding protein beta deletion in mice attenuates inflammation, endoplasmic reticulum stress, and lipid accumulation in diet-induced nonalcoholic steatohepatitis. Hepatology. 2007;45:1108–1117. doi: 10.1002/hep.21614. [DOI] [PubMed] [Google Scholar]
- 92.Moncion A, Truong NT, Garrone A, Beaune P, Barouki R, De Waziers I. Identification of a 16-nucleotide sequence that mediates post-transcriptional regulation of rat CYP2E1 by insulin. J Biol Chem. 2002;277:45904–45910. doi: 10.1074/jbc.M207841200. [DOI] [PubMed] [Google Scholar]
- 93.Chalasani N, Gorski JC, Asghar MS, Asghar A, Foresman B, Hall SD, Crabb DW. Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology. 2003;37:544–550. doi: 10.1053/jhep.2003.50095. [DOI] [PubMed] [Google Scholar]
- 94.Gonzalez FJ. Role of cytochromes P450 in chemical toxicity and oxidative stress: studies with CYP2E1. Mutat Res. 2005;569:101–110. doi: 10.1016/j.mrfmmm.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 95.Pessayre D, Fromenty B, Mansouri A. Mitochondrial injury in steatohepatitis. Eur J Gastroenterol Hepatol. 2004;16:1095–1105. doi: 10.1097/00042737-200411000-00003. [DOI] [PubMed] [Google Scholar]
- 96.Nishida M, Funahashi T, Shimomura I. Pathophysiological significance of adiponectin. Med Mol Morphol. 2007;40:55–67. doi: 10.1007/s00795-007-0366-7. [DOI] [PubMed] [Google Scholar]
- 97.Miele L, Forgione A, Gasbarrini G, Grieco A. Noninvasive assessment of fibrosis in non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) Transl Res. 2007;149:114–125. doi: 10.1016/j.trsl.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 98.Diehl AM. Tumor necrosis factor and its potential role in insulin resistance and nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8:619–638, x. doi: 10.1016/j.cld.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 99.Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2001;48:206–211. doi: 10.1136/gut.48.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Day CP. From fat to inflammation. Gastroenterology. 2006;130:207–210. doi: 10.1053/j.gastro.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 101.Crespo J, Cayon A, Fernandez-Gil P, Hernandez-Guerra M, Mayorga M, Dominguez-Diez A, Fernandez-Escalante JC, Pons-Romero F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology. 2001;34:1158–1163. doi: 10.1053/jhep.2001.29628. [DOI] [PubMed] [Google Scholar]
- 102.Sanyal AJ. AGA technical review on nonalcoholic fatty liver disease. Gastroenterology. 2002;123:1705–1725. doi: 10.1053/gast.2002.36572. [DOI] [PubMed] [Google Scholar]
- 103.Palmentieri B, de Sio I, La Mura V, Masarone M, Vecchione R, Bruno S, Torella R, Persico M. The role of bright liver echo pattern on ultrasound B-mode examination in the diagnosis of liver steatosis. Dig Liver Dis. 2006;38:485–489. doi: 10.1016/j.dld.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 104.Joy D, Thava VR, Scott BB. Diagnosis of fatty liver disease: is biopsy necessary? Eur J Gastroenterol Hepatol. 2003;15:539–543. doi: 10.1097/01.meg.0000059112.41030.2e. [DOI] [PubMed] [Google Scholar]
- 105.Valls C, Iannacconne R, Alba E, Murakami T, Hori M, Passariello R, Vilgrain V. Fat in the liver: diagnosis and characterization. Eur Radiol. 2006;16:2292–2308. doi: 10.1007/s00330-006-0146-0. [DOI] [PubMed] [Google Scholar]
- 106.Karcaaltincaba M, Akhan O. Imaging of hepatic steatosis and fatty sparing. Eur J Radiol. 2007;61:33–43. doi: 10.1016/j.ejrad.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 107.Duman DG, Celikel C, Tuney D, Imeryuz N, Avsar E, Tozun N. Computed tomography in nonalcoholic fatty liver disease: a useful tool for hepatosteatosis assessment? Dig Dis Sci. 2006;51:346–351. doi: 10.1007/s10620-006-3136-9. [DOI] [PubMed] [Google Scholar]
- 108.Fishbein M, Castro F, Cheruku S, Jain S, Webb B, Gleason T, Stevens WR. Hepatic MRI for fat quantitation: its relationship to fat morphology, diagnosis, and ultrasound. J Clin Gastroenterol. 2005;39:619–625. doi: 10.1097/00004836-200508000-00012. [DOI] [PubMed] [Google Scholar]
- 109.Hussain HK, Chenevert TL, Londy FJ, Gulani V, Swanson SD, McKenna BJ, Appelman HD, Adusumilli S, Greenson JK, Conjeevaram HS. Hepatic fat fraction: MR imaging for quantitative measurement and display--early experience. Radiology. 2005;237:1048–1055. doi: 10.1148/radiol.2373041639. [DOI] [PubMed] [Google Scholar]
- 110.Kim SH, Lee JM, Han JK, Lee JY, Lee KH, Han CJ, Jo JY, Yi NJ, Suh KS, Shin KS, et al. Hepatic macrosteatosis: predicting appropriateness of liver donation by using MR imaging--correlation with histopathologic findings. Radiology. 2006;240:116–129. doi: 10.1148/radiol.2393042218. [DOI] [PubMed] [Google Scholar]
- 111.Machann J, Thamer C, Schnoedt B, Stefan N, Haring HU, Claussen CD, Fritsche A, Schick F. Hepatic lipid accumulation in healthy subjects: a comparative study using spectral fat-selective MRI and volume-localized 1H-MR spectroscopy. Magn Reson Med. 2006;55:913–917. doi: 10.1002/mrm.20825. [DOI] [PubMed] [Google Scholar]
- 112.Szczepaniak LS, Babcock EE, Schick F, Dobbins RL, Garg A, Burns DK, McGarry JD, Stein DT. Measurement of intracellular triglyceride stores by H spectroscopy: validation in vivo. Am J Physiol. 1999;276:E977–E989. doi: 10.1152/ajpendo.1999.276.5.E977. [DOI] [PubMed] [Google Scholar]
- 113.Saadeh S, Younossi ZM, Remer EM, Gramlich T, Ong JP, Hurley M, Mullen KD, Cooper JN, Sheridan MJ. The utility of radiological imaging in nonalcoholic fatty liver disease. Gastroenterology. 2002;123:745–750. doi: 10.1053/gast.2002.35354. [DOI] [PubMed] [Google Scholar]
- 114.Foucher J, Castera L, Bernard PH, Adhoute X, Laharie D, Bertet J, Couzigou P, de Ledinghen V. Prevalence and factors associated with failure of liver stiffness measurement using FibroScan in a prospective study of 2114 examinations. Eur J Gastroenterol Hepatol. 2006;18:411–412. doi: 10.1097/00042737-200604000-00015. [DOI] [PubMed] [Google Scholar]
- 115.Poynard T, Ratziu V, Charlotte F, Messous D, Munteanu M, Imbert-Bismut F, Massard J, Bonyhay L, Tahiri M, Thabut D, et al. Diagnostic value of biochemical markers (NashTest) for the prediction of non alcoholo steato hepatitis in patients with non-alcoholic fatty liver disease. BMC Gastroenterol. 2006;6:34. doi: 10.1186/1471-230X-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.American Gastroenterological Association medical position statement: nonalcoholic fatty liver disease. Gastroenterology. 2002;123:1702–1704. doi: 10.1053/gast.2002.36569. [DOI] [PubMed] [Google Scholar]
- 117.Wang RT, Koretz RL, Yee HF Jr Is weight reduction an effective therapy for nonalcoholic fatty liver? A systematic review. Am J Med. 2003;115:554–559. doi: 10.1016/s0002-9343(03)00449-2. [DOI] [PubMed] [Google Scholar]
- 118.Huang MA, Greenson JK, Chao C, Anderson L, Peterman D, Jacobson J, Emick D, Lok AS, Conjeevaram HS. One-year intense nutritional counseling results in histological improvement in patients with non-alcoholic steatohepatitis: a pilot study. Am J Gastroenterol. 2005;100:1072–1081. doi: 10.1111/j.1572-0241.2005.41334.x. [DOI] [PubMed] [Google Scholar]
- 119.Harrison SA, Ramrakhiani S, Brunt EM, Anbari MA, Cortese C, Bacon BR. Orlistat in the treatment of NASH: a case series. Am J Gastroenterol. 2003;98:926–930. doi: 10.1111/j.1572-0241.2003.07375.x. [DOI] [PubMed] [Google Scholar]
- 120.Harrison SA, Fincke C, Helinski D, Torgerson S, Hayashi P. A pilot study of orlistat treatment in obese, non-alcoholic steatohepatitis patients. Aliment Pharmacol Ther. 2004;20:623–628. doi: 10.1111/j.1365-2036.2004.02153.x. [DOI] [PubMed] [Google Scholar]
- 121.Hatzitolios A, Savopoulos C, Lazaraki G, Sidiropoulos I, Haritanti P, Lefkopoulos A, Karagiannopoulou G, Tzioufa V, Dimitrios K. Efficacy of omega-3 fatty acids, atorvastatin and orlistat in non-alcoholic fatty liver disease with dyslipidemia. Indian J Gastroenterol. 2004;23:131–134. [PubMed] [Google Scholar]
- 122.Hussein O, Grosovski M, Schlesinger S, Szvalb S, Assy N. Orlistat reverse fatty infiltration and improves hepatic fibrosis in obese patients with nonalcoholic steatohepatitis (NASH) Dig Dis Sci. 2007;52:2512–2519. doi: 10.1007/s10620-006-9631-1. [DOI] [PubMed] [Google Scholar]
- 123.Sabuncu T, Nazligul Y, Karaoglanoglu M, Ucar E, Kilic FB. The effects of sibutramine and orlistat on the ultrasonographic findings, insulin resistance and liver enzyme levels in obese patients with non-alcoholic steatohepatitis. Rom J Gastroenterol. 2003;12:189–192. [PubMed] [Google Scholar]
- 124.Zelber-Sagi S, Kessler A, Brazowsky E, Webb M, Lurie Y, Santo M, Leshno M, Blendis L, Halpern Z, Oren R. A double-blind randomized placebo-controlled trial of orlistat for the treatment of nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2006;4:639–644. doi: 10.1016/j.cgh.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 125.Lavine JE. Vitamin E treatment of nonalcoholic steatohepatitis in children: a pilot study. J Pediatr. 2000;136:734–738. [PubMed] [Google Scholar]
- 126.Harrison SA, Torgerson S, Hayashi P, Ward J, Schenker S. Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2003;98:2485–2490. doi: 10.1111/j.1572-0241.2003.08699.x. [DOI] [PubMed] [Google Scholar]
- 127.Lirussi F, Mastropasqua E, Orando S, Orlando R. Probiotics for non-alcoholic fatty liver disease and/or steatohepatitis. Cochrane Database Syst Rev. 2007;98:CD005165. doi: 10.1002/14651858.CD005165.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ersoz G, Gunsar F, Karasu Z, Akay S, Batur Y, Akarca US. Management of fatty liver disease with vitamin E and C compared to ursodeoxycholic acid treatment. Turk J Gastroenterol. 2005;16:124–128. [PubMed] [Google Scholar]
- 129.Lindor KD, Kowdley KV, Heathcote EJ, Harrison ME, Jorgensen R, Angulo P, Lymp JF, Burgart L, Colin P. Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: results of a randomized trial. Hepatology. 2004;39:770–778. doi: 10.1002/hep.20092. [DOI] [PubMed] [Google Scholar]
- 130.Mendez-Sanchez N, Gonzalez V, Chavez-Tapia N, Ramos MH, Uribe M. Weight reduction and ursodeoxycholic acid in subjects with nonalcoholic fatty liver disease. A double-blind, placebo-controlled trial. Ann Hepatol. 2004;3:108–112. [PubMed] [Google Scholar]
- 131.Santos VN, Lanzoni VP, Szejnfeld J, Shigueoka D, Parise ER. A randomized double-blind study of the short-time treatment of obese patients with nonalcoholic fatty liver disease with ursodeoxycholic acid. Braz J Med Biol Res. 2003;36:723–729. doi: 10.1590/s0100-879x2003000600007. [DOI] [PubMed] [Google Scholar]
- 132.Orlando R, Azzalini L, Orando S, Lirussi F. Bile acids for non-alcoholic fatty liver disease and/or steatohepatitis. Cochrane Database Syst Rev. 2007;36:CD005160. doi: 10.1002/14651858.CD005160.pub2. [DOI] [PubMed] [Google Scholar]
- 133.Lin HZ, Yang SQ, Chuckaree C, Kuhajda F, Ronnet G, Diehl AM. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat Med. 2000;6:998–1003. doi: 10.1038/79697. [DOI] [PubMed] [Google Scholar]
- 134.Uygun A, Kadayifci A, Isik AT, Ozgurtas T, Deveci S, Tuzun A, Yesilova Z, Gulsen M, Dagalp K. Metformin in the treatment of patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2004;19:537–544. doi: 10.1111/j.1365-2036.2004.01888.x. [DOI] [PubMed] [Google Scholar]
- 135.Bugianesi E, Gentilcore E, Manini R, Natale S, Vanni E, Villanova N, David E, Rizzetto M, Marchesini G. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am J Gastroenterol. 2005;100:1082–1090. doi: 10.1111/j.1572-0241.2005.41583.x. [DOI] [PubMed] [Google Scholar]
- 136.Kohlroser J, Mathai J, Reichheld J, Banner BF, Bonkovsky HL. Hepatotoxicity due to troglitazone: report of two cases and review of adverse events reported to the United States Food and Drug Administration. Am J Gastroenterol. 2000;95:272–276. doi: 10.1111/j.1572-0241.2000.01707.x. [DOI] [PubMed] [Google Scholar]
- 137.Menon KVN, Angulo P, Lindor KD. Severe cholestatic hepatitis from troglitazone in a patient with nonalcoholic steatohepatitis and diabetes mellitus. Am J Gastroenterol. 2001;96:1631–1634. doi: 10.1111/j.1572-0241.2001.03809.x. [DOI] [PubMed] [Google Scholar]
- 138.Angelico F, Burattin M, Alessandri C, Del Ben M, Lirussi F. Drugs improving insulin resistance for non-alcoholic fatty liver disease and/or non-alcoholic steatohepatitis. Cochrane Database Syst Rev. 2007;96:CD005166. doi: 10.1002/14651858.CD005166.pub2. [DOI] [PubMed] [Google Scholar]
- 139.Sanyal AJ, Mofrad PS, Contos MJ, Sargeant C, Luketic VA, Sterling RK, Stravitz RT, Shiffman ML, Clore J, Mills AS. A pilot study of vitamin E versus vitamin E and pioglitazone for the treatment of nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. 2004;2:1107–1115. doi: 10.1016/s1542-3565(04)00457-4. [DOI] [PubMed] [Google Scholar]
- 140.Neuschwander-Tetri BA, Brunt EM, Wehmeier KR, Oliver D, Bacon BR. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology. 2003;38:1008–1017. doi: 10.1053/jhep.2003.50420. [DOI] [PubMed] [Google Scholar]
- 141.Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, Gastaldelli A, Tio F, Pulcini J, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–2307. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 142.Promrat K, Lutchman G, Uwaifo GI, Freedman RJ, Soza A, Heller T, Doo E, Ghany M, Premkumar A, Park Y, et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology. 2004;39:188–196. doi: 10.1002/hep.20012. [DOI] [PubMed] [Google Scholar]
- 143.Grey A, Bolland M, Gamble G, Wattie D, Horne A, Davidson J, Reid IR. The peroxisome proliferator-activated receptor-gamma agonist rosiglitazone decreases bone formation and bone mineral density in healthy postmenopausal women: a randomized, controlled trial. J Clin Endocrinol Metab. 2007;92:1305–1310. doi: 10.1210/jc.2006-2646. [DOI] [PubMed] [Google Scholar]
- 144.Schwartz AV, Sellmeyer DE, Vittinghoff E, Palermo L, Lecka-Czernik B, Feingold KR, Strotmeyer ES, Resnick HE, Carbone L, Beamer BA, et al. Thiazolidinedione use and bone loss in older diabetic adults. J Clin Endocrinol Metab. 2006;91:3349–3354. doi: 10.1210/jc.2005-2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schwartz AV, Sellmeyer DE. Thiazolidinediones: new evidence of bone loss. J Clin Endocrinol Metab. 2007;92:1232–1234. doi: 10.1210/jc.2007-0328. [DOI] [PubMed] [Google Scholar]
- 146.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 147.Home PD, Pocock SJ, Beck-Nielsen H, Gomis R, Hanefeld M, Jones NP, Komajda M, McMurray JJ. Rosiglitazone evaluated for cardiovascular outcomes--an interim analysis. N Engl J Med. 2007;357:28–38. doi: 10.1056/NEJMoa073394. [DOI] [PubMed] [Google Scholar]
- 148.Basaranoglu M, Acbay O, Sonsuz A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J Hepatol. 1999;31:384. doi: 10.1016/s0168-8278(99)80243-8. [DOI] [PubMed] [Google Scholar]
- 149.Satapathy SK, Garg S, Chauhan R, Sakhuja P, Malhotra V, Sharma BC, Sarin SK. Beneficial effects of tumor necrosis factor-alpha inhibition by pentoxifylline on clinical, biochemical, and metabolic parameters of patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2004;99:1946–1952. doi: 10.1111/j.1572-0241.2004.40220.x. [DOI] [PubMed] [Google Scholar]
- 150.Adams LA, Zein CO, Angulo P, Lindor KD. A pilot trial of pentoxifylline in nonalcoholic steatohepatitis. Am J Gastroenterol. 2004;99:2365–2368. doi: 10.1111/j.1572-0241.2004.40064.x. [DOI] [PubMed] [Google Scholar]
- 151.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jequier E. Leptin signaling, adiposity, and energy balance. Ann N Y Acad Sci. 2002;967:379–388. doi: 10.1111/j.1749-6632.2002.tb04293.x. [DOI] [PubMed] [Google Scholar]
- 153.Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, Wagner AJ, DePaoli AM, Reitman ML, Taylor SI, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346:570–578. doi: 10.1056/NEJMoa012437. [DOI] [PubMed] [Google Scholar]
- 154.Ebihara K, Kusakabe T, Hirata M, Masuzaki H, Miyanaga F, Kobayashi N, Tanaka T, Chusho H, Miyazawa T, Hayashi T, et al. Efficacy and safety of leptin-replacement therapy and possible mechanisms of leptin actions in patients with generalized lipodystrophy. J Clin Endocrinol Metab. 2007;92:532–541. doi: 10.1210/jc.2006-1546. [DOI] [PubMed] [Google Scholar]
- 155.Devane WA, Dysarz FA 3rd, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- 156.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 157.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 158.Goutopoulos A, Makriyannis A. From cannabis to cannabinergics: new therapeutic opportunities. Pharmacol Ther. 2002;95:103–117. doi: 10.1016/s0163-7258(02)00250-4. [DOI] [PubMed] [Google Scholar]
- 159.Galiegue S, Mary S, Marchand J, Dussossoy D, Carriere D, Carayon P, Bouaboula M, Shire D, Le Fur G, Casellas P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- 160.Gallily R, Breuer A, Mechoulam R. 2-Arachidonylglycerol, an endogenous cannabinoid, inhibits tumor necrosis factor-alpha production in murine macrophages, and in mice. Eur J Pharmacol. 2000;406:R5–R7. doi: 10.1016/s0014-2999(00)00653-1. [DOI] [PubMed] [Google Scholar]
- 161.Varga K, Wagner JA, Bridgen DT, Kunos G. Platelet- and macrophage-derived endogenous cannabinoids are involved in endotoxin-induced hypotension. FASEB J. 1998;12:1035–1044. doi: 10.1096/fasebj.12.11.1035. [DOI] [PubMed] [Google Scholar]
- 162.Batkai S, Jarai Z, Wagner JA, Goparaju SK, Varga K, Liu J, Wang L, Mirshahi F, Khanolkar AD, Makriyannis A, et al. Endocannabinoids acting at vascular CB1 receptors mediate the vasodilated state in advanced liver cirrhosis. Nat Med. 2001;7:827–832. doi: 10.1038/89953. [DOI] [PubMed] [Google Scholar]
- 163.Wang Y, Liu Y, Ito Y, Hashiguchi T, Kitajima I, Yamakuchi M, Shimizu H, Matsuo S, Imaizumi H, Maruyama I. Simultaneous measurement of anandamide and 2-arachidonoylglycerol by polymyxin B-selective adsorption and subsequent high-performance liquid chromatography analysis: increase in endogenous cannabinoids in the sera of patients with endotoxic shock. Anal Biochem. 2001;294:73–82. doi: 10.1006/abio.2001.5015. [DOI] [PubMed] [Google Scholar]
- 164.Biswas KK, Sarker KP, Abeyama K, Kawahara K, Iino S, Otsubo Y, Saigo K, Izumi H, Hashiguchi T, Yamakuchi M, et al. Membrane cholesterol but not putative receptors mediates anandamide-induced hepatocyte apoptosis. Hepatology. 2003;38:1167–1177. doi: 10.1053/jhep.2003.50459. [DOI] [PubMed] [Google Scholar]
- 165.Gabbay E, Avraham Y, Ilan Y, Israeli E, Berry EM. Endocannabinoids and liver disease--review. Liver Int. 2005;25:921–926. doi: 10.1111/j.1478-3231.2005.01180.x. [DOI] [PubMed] [Google Scholar]
- 166.Neff GW, O’Brien CB, Reddy KR, Bergasa NV, Regev A, Molina E, Amaro R, Rodriguez MJ, Chase V, Jeffers L, et al. Preliminary observation with dronabinol in patients with intractable pruritus secondary to cholestatic liver disease. Am J Gastroenterol. 2002;97:2117–2119. doi: 10.1111/j.1572-0241.2002.05852.x. [DOI] [PubMed] [Google Scholar]
- 167.Gary-Bobo M, Elachouri G, Gallas JF, Janiak P, Marini P, Ravinet-Trillou C, Chabbert M, Cruccioli N, Pfersdorff C, Roque C, et al. Rimonabant reduces obesity-associated hepatic steatosis and features of metabolic syndrome in obese Zucker fa/fa rats. Hepatology. 2007;46:122–129. doi: 10.1002/hep.21641. [DOI] [PubMed] [Google Scholar]
- 168.Cleland JG, Ghosh J, Freemantle N, Kaye GC, Nasir M, Clark AL, Coletta AP. Clinical trials update and cumulative meta-analyses from the American College of Cardiology: WATCH, SCD-HeFT, DINAMIT, CASINO, INSPIRE, STRATUS-US, RIO-Lipids and cardiac resynchronisation therapy in heart failure. Eur J Heart Fail. 2004;6:501–508. doi: 10.1016/j.ejheart.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 169.Black SC. Cannabinoid receptor antagonists and obesity. Curr Opin Investig Drugs. 2004;5:389–394. [PubMed] [Google Scholar]