
Figure 2.
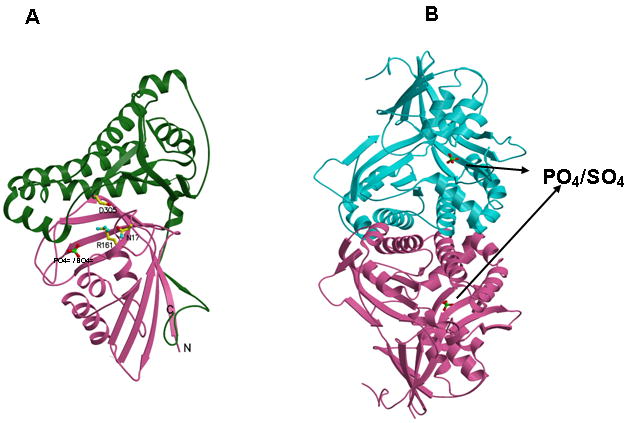
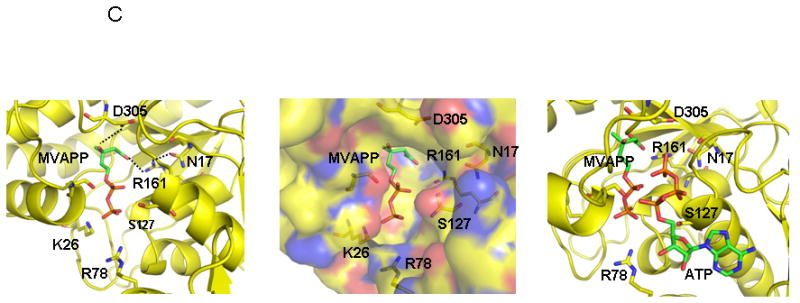
Human MDD structure and models of substrate complexes. A. The two domains of the monomer structure are shown with the N-terminal domain in pink and the C-terminal domain in green. R161, N17, D305, and the tightly bound phosphate/sulfate are shown in ball and stick models. N17 and R161 side chains are within hydrogen bonding distance (2.8 Å) of each other. B. The structure of a dimer of human MDD is shown in a view perpendicular to its 2-fold axis. Arrows indicate the position of bound phosphate/sulfate. The view of the pink monomer is rotated about 90° around the y-axis from the view shown in panel A. C. Structural models of human MDD-substrate complexes. Left panel depicts the active site of a MDD-MVAPP binary complex, with MVAPP docked into the protein using the Z-dock algorithm (http://zdock.bu.edu). Estimated distance between D305’s carboxyl and substrate’s C3 oxygen is ≤4.0 Å. For R161’s guanidinium and substrate’s C1 carboxyl oxygen, the estimate is ≤3.5 Å. Middle panel shows a surface representation of the active site region of the binary complex model, which suggests that the C1 end of MVAPP is situated in a pocket deep in the active site while the substrate phosphoryls may be more surface exposed. In the interior of the pocket are R161 and D305, which has been proposed to interact with MVAPP’s C3 hydroxyl [8]. Right panel represents a model of a ternary MDD-MVAPP-ATP complex. The position of ATP is based on an overlay of a structure of the mevalonate kinase-ATP complex [11] on the human MDD structure (Fig. 2A). The position of S127 agrees with the previous suggestion of its interaction with the phosphoryl chain of ATP [12]. The juxtaposition of ATP’s gamma phosphoryl group with respect to MVAPP’s C3 oxygen is in accord with production of a 3-phosphoMVAPP reaction intermediate [26] and supports the docking position of MVAPP in the binary complex model.