

Abstract
We investigated oxidative damage to the c-fos gene and to its transcription in the brain of Long—Evans rats using a transient focal cerebral ischemia and reperfusion (FCIR) model. We observed a significant (p < 0.001) increase in the immunoreactivity to 8-hydroxy-2′-guanine (oh8G) and its deoxy form (oh8dG) in the ischemic cortex at 0–30 min of reperfusion in all 27 animals treated with 15–90 min of ischemia. Treatment with a neuronal nitric oxide synthase (nNOS) inhibitor, 3-bromo-7-nitroindazole (60 mg/kg, i.p.), abolished the majority but not all of the oh8G/oh8dG immunoreactivity. Treatment with RNase A reduced the oh8G immunoreactivity, suggesting that RNA may be targeted. This observation was further supported by decreased levels of mRNA transcripts of the c-fos and actin genes in the ischemic core within 30 min of reperfusion using in situ hybridization. The reduction in mRNA transcription occurred at a time when nuclear gene damage, detected as sensitive sites to Escherichia coli Fpg protein in the transcribed strand of the c-fos gene, was increased 13-fold (p < 0.01). Our results suggest that inhibiting nNOS partially attenuates FCIR-induced oxidative damage and that nNOS or other mechanisms induce nuclear gene damage that interferes with gene transcription in the brain.
Keywords: Stroke, DNA repair, Oxidative stress, Gene expression, Neuroregeneration, Apoptosis
An elevated sensitivity to oxidative stress in certain populations of brain cells may be a key mechanism underlying several neurological disorders (Liu et al., 1989; Jenner et al., 1992; Neve, 1996; Parshad et al., 1996; Kisby et al., 1997). Numerous studies have correlated an increase in calcium influx, glutamate, and reactive oxygen species (ROS) with neuronal death. ROS levels are known to be elevated after brain injury of the ischemia—reperfusion type (Hall and Braughler, 1989; Rosenthal et al., 1992; Poiries, 1994; Yoshida et al., 1994; Zhang et al., 1994; O’Neill et al., 1996; Smith et al., 1996) and following administration of excitotoxic drugs (Schulz et al., 1995; Ayata et al., 1997; Back et al., 1998). ROS are known to damage mitochondrial DNA (Driggers et al., 1993; Mecocci et al., 1993; Yakes and Van Houten, 1997; Murakami et al., 1998), nuclear genes (Liu et al., 1996; Taffe et al., 1996; Chen et al., 1997), and RNA (Liu et al., 1996; Nunomura et al., 1999). Damage to nucleic acids may lead to abnormal gene expression and premature neuronal death (Wolozin et al., 1996; Citron et al., 1997; Lamb, 1997; Iadecola et al., 1999).
Activation of gene transcription in the ischemic brain following oxidative stress appears to signify a need for gene products to repair the injury. For example, transcription of one of the immediate early genes, the c-fos gene, is activated after head injury (An et al., 1993; Yang et al., 1994). The product of the c-fos transcript then forms activator protein-1, which in turn activates various cellular functions, including the production of growth factors (Cui et al., 1999) and DNA repair enzymes (Scanlon et al., 1991). Although ROS are by-products of normal cellular metabolism (Fraga et al., 1990; Park et al., 1992), excessive ROS formation in the brain under pathological conditions may alter gene expression and lead to abnormal production of proteins. Animal models of cerebral ischemia—reperfusion, which is one of the conditions that increase ROS formation, are useful not only in understanding the cellular response to ischemic injury, but also in understanding how ROS may contribute to other neurological disorders.
Nitric oxide (NO) transforms itself as the nitrosonium ion (NO+) or the free radical NO•, depending on the redox state in the brain (Lipton et al., 1993). The nitrosonium ion is neuroprotective (Lei et al., 1992), and the free radical NO• is neurotoxic (Beckman et al., 1990). NO can be generated from arginine, a reaction that is catalyzed by three isoforms of NO synthase (NOS). NOS from neurons (type I) and from endothelia (type III) are activated by calcium ions, levels of which can be elevated during ischemic injury. The expression of the inducible type II does not appear to be affected by the fluctuation in calcium ion levels. Neuronal NOS (nNOS) is specifically inhibited by 7-nitroindazole (Babbedge et al., 1993). NO produced by NOS can combine with superoxide ions to form peroxynitrite in the presence of hydrogen ions (Beckman et al., 1990; Lipton et al., 1993). The peroxynitrite can then generate hydroxyl radicals and NO2. Hydroxyl radicals are known to damage proteins and nucleic acids.
We have reported oxidative damage to DNA and RNA in mouse brain using the forebrain (global) ischemia—reperfusion model (Liu et al., 1996). Because the physiology of global cerebral ischemia may be different from that of focal ischemia and because the cellular response in the mouse may be different from the response that occurs in other rodents (Fujii et al., 1997), we have investigated oxidative injury to nucleic acids in focal cerebral ischemia (FCI) and reperfusion (FCIR) of the rat in the present study. In addition to examining the appearance of oxidative lesions in affected brain cells, we have studied DNA damage and repair in one representative nuclear gene, the c-fos gene, after oxidative stress induced by FCI.
MATERIALS AND METHODS
Brain injury model
A total of 95 male Long—Evans rats (Harlan, Indianapolis, IN, U.S.A.), weighing 225–250 g, were used. Anesthesia was induced with pentobarbital sodium (Nembutal; 80 mg/kg, i.p.). In this focal cerebral ischemia model, the right middle cerebral artery (MCA) and both common carotid arteries were occluded for 30–90 min (Chen et al., 1986; Liu et al., 1989, 1994). The occlusion was then released to allow reperfusion of the affected area. This model produces necrosis in the right cerebral cortex supplied by the MCA (Chen et al., 1986; Du et al., 1996). Animals that underwent the same surgical treatment but received no FCI were used as controls. Body temperature was monitored and maintained at 37 ± 0.5°C; all animals were kept in well-ventilated incubators at 24 ± 0.5°C during the reperfusion period. Postoperative animal care with free access to food and water was as described previously (Liu et al., 1989, 1994). Housing and anesthesia were in accordance with the U.S. Public Health Service Guide for the Care and Use of Laboratory Animals, U.S. Department of Agriculture regulations, and the American Veterinary Medicine Association Panel on Euthanasia guidelines.
To determine the effective dosage of 3-bromo-7-nitroindazole (3BR7NI; Alexis, San Diego, CA, U.S.A.) in the brain, 3BR7NI (30 mg/kg, i.p.) in soybean oil was injected into four animals. NOS activity in triplicates was determined in the crude extract of the cerebral cortex from each animal that was collected immediately (control; n = 2) or 60 min (n = 2) after injection (Yoshida et al., 1994). To study the effect of nNOS on generation of 8-hydroxy-2′-guanine (oh8G) and its deoxy form (oh8dG), we injected six animals with 3BR7NI and six animals with soybean oil alone (4 ml/kg, i.p.) at 5 and at 45 min after occlusion of the vessels.
For immunohistochemistry and in situ hybridization (ISH), the animals were anesthetized at the designated reperfusion end points and were perfused from the right ventricle with saline and 4% paraformaldehyde (200 ml), and the brains were removed and incubated in 20% sucrose overnight at 4°C before preparation of samples (An et al., 1993; Liu et al., 1994; Cui et al., 1999). Within 24 h of brain preparation, consecutive coronal sections (at 20 μm) of the brains were obtained 3.6–4.8 mm anterior to the bregma. Two brain samples, separated by 0.2 mm, were mounted on one poly-L-lysine-coated slide. The slides were stored at -20°C. All samples were analyzed within 5 weeks. For immunohistochemistry using the free-floating method, the brain sections (two to four tissue samples per brain, each separated by 0.2 mm) were placed in one of the 12-well plate for antibody incubation and staining.
For DNA isolation, the anesthetized animals were decapitated at the designated reperfusion end points. The brain was immediately washed in ice-cold saline and then was placed on ice for separation of the cerebral cortex, the hippocampus, the cerebellum, and the thalamus/hypothalamus from the brainstem. Each tissue was flash-frozen in liquid nitrogen and then transferred to a freezer for storage at -70°C. All experiments were repeated at least three times, and each end point contained a minimum of three animals (Liu et al., 1994; Cui et al., 1999).
Detection of oh8G/oh8dG immunoreactivity in situ
From a total of 27 animals, the presence of oh8G/oh8dG lesions was determined using the free-floating method. The brain tissue was treated with 2% H2O2 for 1 min at room temperature. The primary antibody was murine IgG against oh8G/oh8dG (1:100 dilution; QED Bioscience, San Diego, CA, U.S.A.) (Fraga et al., 1990; Park et al., 1992). The secondary antibody was a goat anti-mouse IgG-bodipy-FL conjugate (1:400 dilution; Molecular Probes, Eugene, OR, U.S.A.). The samples were counterstained with propidium iodide (PI; 0.5 μg/ml) in the presence of RNase A (20 μg/ml). The mounting medium was phenylenediamine (0.1%, vol/vol) in glycerol. Animals with and without FCIR were analyzed at the same time. For quality control in each analysis, two other separate samples from each brain were analyzed without the primary antibody, and the fluorescent signal intensity from these controls was the background intensity.
The fluorescent images were digitally captured using the Cooled Color Digital Camera (Diagnostic Instruments, Sterling Heights, MI, U.S.A.). The intensity of the green fluorescent signals was assessed a numerical pixel value under the green spectrum using Adobe PhotoShop. Because we used fluorescent staining and the signal came from the substrate, the signal appeared around the nuclei in the oh8G/oh8dG immunoreactivity staining: The green signal was generally stronger around the nucleus and became weaker toward cell surface. Cells with at least a threefold increase in signal value over the background (30 ± 18) were defined as oh8G/oh8dG-positive cells (Cui et al., 1999). Data were analyzed by eliminating a signal with <120 pixel values, and the remaining signal represented positive cells. We then counted the number of nuclei (outline void of green signal, but with the orange signal if counterstained using PI) that had green signal around it. The positive cells were counted from at least five different areas in the same cerebral cortex, and the data obtained were analyzed using GraphPad Prism (GraphPad Software, San Diego) for the mean ± SEM, t test, and ANOVA.
Animals that were oh8G/oh8dG-positive were defined as having brain specimens that showed a higher fluorescent signal in the ischemic cortex than in the contralateral cortex and in which the fluorescent signal could be abolished or significantly reduced by the primary antibody preadsorbed with antigen (oh8G—bovine serum albumin). The results were analyzed; a value of p < 0.05 was considered statistically significant.
Analysis of c-fos mRNA using ISH
The presence of c-fos or actin mRNA in the brain sections was detected in 23 animals using cRNA probes of the c-fos and actin cDNA clones, respectively, as previously described (An et al., 1993) except that the cRNA probe was labeled with [33P]UTP (107 cpm/ml). The actin gene was used a control. Previous work has shown that the actin gene is expressed at a relatively constant level following oxidative stress by FCIR (An et al., 1993; Cui et al., 1999). Probes in the antisense (cRNA) and sense (mRNA) orientations of both the c-fos and actin genes were hybridized to the brain tissue. After hybridization, the brain slices were digested using RNase A at 37°C for 30 min to remove nonspecific hybrids. The brain sections were placed in an autoradiography cassette and developed at the same time. The radiolabeled [33P]mRNA probe did not produce any signal, but the cRNA probe did.
Detection of DNA injury and repair in the c-fos gene
Damage to nuclear genes was determined using a fragment-shift assay (Bohr et al., 1985; Mellon et al., 1987; Driggers et al., 1993; Bhagwat and Gerlt, 1996; Liu et al., 1996; Taffe et al., 1996). Total DNA was isolated from the right (ipsilateral) cerebral cortex of 29 animals (nine control animals and four FCIR animals for each of five reperfusion times). A detailed description of our technique for DNA isolation has been previously described (Liu et al., 1996). The purified DNA was stored in TE buffer [10 mM Tris HCl (pH 8.0) and 1 mM EDTA] and was never exposed to phenol or chloroform before analysis. A total of 24 μg of DNA from each cortex was tested (Liu et al., 1996). We used a rat c-fos RNA probe transcribed from a cDNA clone in a pBC KS (-) vector, whose mRNA could be translated into Fos protein (Liu et al., 1994). The mRNA or cRNA probe was transcribed from the cDNA clone of rat c-fos gene in the presence of [α-32P]UTP (3,000 Ci/mmol; New England Nuclear, Wilmington, DE, U.S.A.) using T7 or T3 RNA polymerase (Promega Corp., Madison, WI, U.S.A.), respectively. Each RNA probe was purified using RNase-free DNase and a Sephadex G-50 column (An et al., 1993). The specific activities of the 32P-RNA probe were ∼109 cpm/μg. The presence of Escherichia coli formamidopyridine DNA N-glycolase (Fpg) protein-sensitive sites (FPGSS) in the nontranscribed strand of the c-fos gene was analyzed using the cRNA probe (1 × 108 cpm). To detect the presence of FPGSS in the transcribed strand of the c-fos gene, the same DNA blot was boiled in stripping buffer (15 mM NaCl, 1.5 mM sodium citrate, and 1% sodium dodecyl sulfate) for 10 min and then was hybridized to the mRNA probe (1 × 108 cpm) of the same gene. The autoradiogram was developed at -70°C for 16 h. The FPGSS frequency was calculated by ln(1/R), where R is the signal ratio of DNA strands (Mellon et al., 1987; Liu et al., 1996).
RESULTS
Presence of oh8G/oh8dG following FCIR with and without the nNOS inhibitor 3BR7NI
In the absence of RNase A treatment, the green fluorescent signal in the ischemic (right or ipsilateral) cortex of the FCIR animals was visible throughout the cytoplasm, with occasional nuclear staining observed at high magnifications. We then counterstained the samples with PI and a brief treatment with RNase A to locate the nuclei (Fig. 1). The fluorescent signal in the ischemic cortex, denoting oh8dG immunoreactivity, became sharper and was visible at the end of 15 (15/0) and 60 (60/0) min of FCI (Fig. 1A and B, respectively). The signal remained elevated at 15 min of reperfusion after 90 min of FCI (90/15 FCIR; Fig. 1C). An additional RNase A treatment reduced the cytoplasmic signal, but the green signal remained visible in some cells (Fig. 1D, arrow). The data suggested that RNA and DNA were targeted for the formation of oh8G and oh8dG.
FIG. 1.

oh8G/oh8dG immunoreactivity after RNase A or 3BR7NI. The fluorescent signal of oh8G/oh8dG in the right (ischemic or ipsilateral) cortices of six animals following FCIR (A—G) and one of the non-FCI (control) animals (H) is shown. Nuclei were counterstained using PI in the presence of RNase A (20 μg/ml, 5 min at room temperature) and appear as red fluorescent signals. The yellowish fluorescent signals indicate colocalization of the oh8dG immunoreactivity with the nuclear PI staining. Two typical cortical samples with 15/0 (A) and 60/0 (B) without reperfusion are shown, along with 90 min of FCI and 15 min of reperfusion (90/15 FCIR in C and D), except the brain samples in D were further treated with RNase A (5 mg/ml,1hat 37°C). A higher magnification is shown of the right cortical sample from one of three animals with 90/15 FCIR and oil (E) or 3BR7NI (F). Another FCIR sample (90/30) is also shown in G. Bar = 30 μm in A—D and 10 μm in E—H.
The average positive cell density (mean ± SEM) was 1,215 ± 100 cells/mm2 at 90/15 FCIR (Fig. 1E). At 90/30 FCIR (Fig. 1G), the positive cell density was decreased (400 ± 150 cells/mm2), and so was the staining intensity, but the green signal (arrow) was still readily observed when compared with non-FCI tissue (Fig. 1H). No green signal was observed in the ischemic cortex when the primary antibody was preadsorbed with oh8G—bovine serum albumin or when the primary antibody was not added. At 90/60 FCIR, no oh8G/oh8dG immunoreactivity was observed (data not shown). A very weak fluorescent signal was observed in the left (contralateral) cortex (Fig. 2A), whereas a strong fluorescent signal was readily observed in the ischemic cortex of the same animal (Fig. 2B).
FIG. 2.

Elevation of oh8G/oh8dG immunoreactivity in the cortex. The left (A; contralateral) and right (B; ischemic) cerebral cortices from one of six animals treated with 90/15 FCIR are shown in black and white. The oh8dG immunoreactivity appears as a white fluorescent signal. Bar = 120 μm.
To delineate the mechanism by which the oh8G/oh8dG lesions are formed, we investigated the effect of 3BR7NI. The NOS activity in the cytosolic extract of cerebral cortex was reduced by 92 ± 1% at 60 min after administration of 3BR7NI. Soybean oil, used as the vehicle for 3BR7NI and alone as a control, had no effect on the number or intensity of oh8G/oh8dG-positive cells at 90/15 FCIR (Table 1 and Fig. 1E). In the six animals treated with 90/15 or 90/30 FCIR (n = 3 each) and 3BR7NI, the nuclear oh8dG immunoreactivity was greatly reduced (Fig. 1F), but cytoplasmic immunoreactivity remained visible. The data suggest that NOS was not the only contributor of hydroxyl radicals. In the non-FCI controls and in the FCIR animals treated with 3BR7NI (Table 1), none of the 11 animals was positive for oh8dG immunoreactivity using the criteria specified in Materials and Methods, but 27 of the 28 FCIR animals without 3BR7NI were positive (p < 0.001).
TABLE 1.
Presence of oh8dG immunoreactivity in rat cortex
| oh8dG-positive |
||
|---|---|---|
| No | Yes | |
| No FCI (control) | 5 | 0 |
| Reperfusion of ≤30 min after FCI of |
||
| 15 min (n = 2) | 0 | 2 |
| 30 min (n = 8) | 1 | 7 |
| 60 min (n = 5) | 0 | 5 |
| 90 min (n = 7) | 0 | 7 |
| 90 min (n = 6, soybean oil) | 0 | 6 |
| Total | 1 | 27a |
| 90 min (n = 6, 3BR7NI) | 6 | 0 |
Animals with >200 positive cells/mm2 were defined as oh8dG-positive.
The results were analyzed using Fisher’s exact analysis:
p ≤ 0.001.
Expression of c-fos mRNA
Because oh8G in the RNA was observed in the ischemic cortex, we used ISH to investigate how oxidative stress after FCIR might affect mRNA transcription of the actin (constitutively expressed) and c-fos (inducible by stress) genes in the brains from 23 animals (three non-FCI controls and 20 FCIR animals). By using the [33P]cRNA probe for hybridization and RNase A (20 μg/ml) at the end of hybridization, we obtained a sharper signal than with the [32P]cRNA probe. This method avoids treatments with oxidative reagents that are commonly used in RNA isolation and detects intact mRNA defined by the length and sequence of the cRNA probe. The intensity of the signal using the radiolabeled cRNA probe reflects the amount of mRNA transcript present, with darker intensity indicating a greater amount of mRNA transcript (Fig. 3).
FIG. 3.

Expression of the c-fos and actin genes using ISH. The expression of the c-fos and actin genes in the cerebral cortex was determined for six treatment groups in a total of 23 animals. Radiograms of four coronal sections per end point (two examined for c-fos and two examined for actin, rostral-to-caudal view; sections in each box are 0.2 mm apart) from one representative animal in each group are shown. Each of the coronal sections used for the c-fos transcript detection was adjacent to the respective section used for the actin transcript detection.
The amount of the c-fos gene transcript varied strikingly in response to FCIR. In control (no ischemia) animals, c-fos mRNA was barely detectable (Figs. 3 and 4), indicating that the c-fos gene was not actively transcribed. Transcripts of the c-fos gene in the right ischemic cortex became significantly elevated (p < 0.001) at least 10-fold immediately after FCI (90/0 FCIR; n = 3; Fig. 4). The average amount of c-fos mRNA transcripts was decreased at 15 (n = 6) or 30 min (n = 3) of reperfusion from that observed at 90/0 (although still significantly elevated compared with the controls) and then increased over the next 45 min of reperfusion. At 90/60 FCIR (n = 2), the c-fos transcript had increased to a level not significant different (p > 0.05) from that observed immediately after occlusion (90/0 FCIR). At 120 min of reperfusion (n = 2), the signal intensity in the ischemic cortex had declined but remained detectable until 6 h of reperfusion (n = 4; data not shown). The c-fos gene transcripts in the hippocampal formation was detected at 90/15, and levels remained elevated at 90/60 FCIR but became no longer detectable at 90/120 FCIR (Fig. 3). Seventeen of 20 FCIR animals that were examined within 6 h of reperfusion after FCI expressed the c-fos mRNA transcript in the right ischemic hemisphere only, whereas the other three FCIR animals expressed c-fos mRNA bilaterally. Compared with the non-FCI animals, the average intensity of the c-fos mRNA signal in the ischemic cortex was significantly elevated in all animals with FCIR (Fig. 4, denoted by +). The reduction observed at 15 and 30 min of reperfusion was limited to the right MCA (Fig. 3). In fact, it appeared that the newly transcribed c-fos mRNA in the right MCA area was elevated immediately following FCI and then decreased to a level that could barely be seen at 15 min of reperfusion. At 30 min of reperfusion, the transcript in the MCA area became detectable but remained less intense than the surrounding penumbra. At ≥60 min of reperfusion, the intensity of the c-fos transcript in the ischemic core and the penumbra was approximately the same. The MCA area that demonstrated a marked reduction in c-fos mRNA expression was similar to the infarct area produced by this method (Chen et al., 1986; Garcia et al., 1993; Du et al., 1996).
FIG. 4.
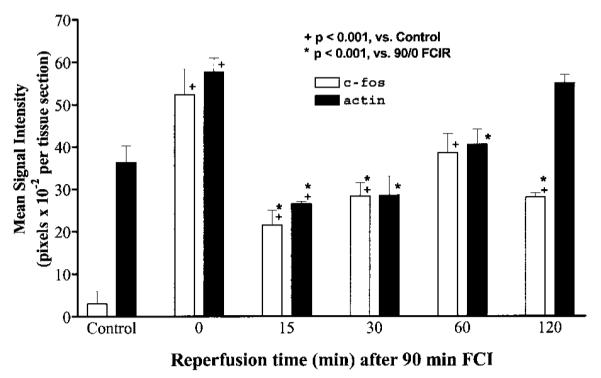
Quantification of c-fos and actin mRNA transcripts. The transcript intensity in the brain sections from the animals at each time point by ISH was measured using an image documentation system (AlphaImager; Alpha Innotech, San Leandro, CA, U.S.A.). At least two brain sections from each animal were examined in repeated ISH. Data are mean ± SEM (bars) values; the number of animals (n) was n = 3 each for control, 90/0, and 90/30 FCIR, n = 6 for 90/15 FCIR, and n = 2 for each for 90/60 and 90/120 FCIR. Figure 3 shows the typical expression of these two genes at each time point.
The intensity of the transcripts of the actin gene was uniform throughout cerebral cortex. The level of the transcript at 90/0 FCIR was ∼1.6-fold (p < 0.001) higher than that in the non-FCI controls (Fig. 4). The increase in actin mRNA at 90/0 FCIR could be seen in the striatum, the hippocampal formation, and the piri-form cortex (Fig. 3). At 90/15 FCIR, the average pixel value of the actin transcript was lower than the average value measured in the non-FCI controls or in the 90/0 FCIR animals (p < 0.001). The actin mRNA intensity then gradually increased over the remaining time points. A reduction of the actin gene expression could be seen in the ischemic core of the same MCA distribution area (Fig. 3, arrow), although the background signal, which was of a similar intensity to that in the non-FCI cortex, remained observable. When the intensities of the c-fos and actin gene transcripts were compared, there was no significant difference in the fluctuation from the end of FCI (90/0) through 60 min of reperfusion (Fig. 4; by two-way ANOVA).
DNA damage and repair in both strands of nuclear genes
Because the resolution of oh8G/oh8dG immunoreactivity is low, we also examined whether oxidative damage occurred in both strands of the c-fos gene in purified DNA from the right cerebral cortices of 20 FCIR and nine control animals. No significant difference was noted in the fragment shift of the c-fos gene from the control DNA with and without Fpg protein (Fig. 5), indicating that the isolation procedure and manipulation during analysis did not cause excess DNA breakage. We observed a significant fragment shift, as measured by a decrease in the intensity of the c-fos gene, in the ischemic cortical samples treated with Fpg protein (Fig. 5, Y samples), compared with DNA from the same animals not treated with Fpg protein (Fig. 5, N samples).
FIG. 5.

Presence of FPGSS in the c-fos gene. FPGSS were measured in genomic DNA from right cortices of nine sham-operated control animals and from right (ischemic) cortices of 20 rats that underwent 90 min of FCI and various durations of reperfusion. The Fpg protein-treated DNA samples (Y) were resolved in parallel with DNA samples with no Fpg protein treatment (N). The blots were hybridized to 32P-mRNA (for the transcribed strand) or 32P-cRNA (for the nontranscribed strand) of the c-fos gene.
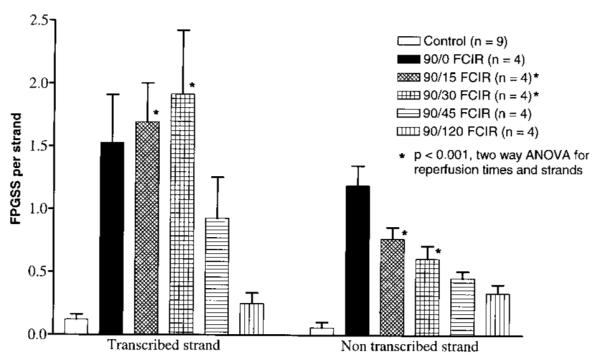
Figure 6 shows the frequency of genomic FPGSS on the c-fos gene as calculated from the data shown in Fig. 5. The frequency of genomic FPGSS (mean ± SEM) in the DNA from the nine non-FCI control brains was not statistically different in the transcribed (0.12 ± 0.04) and in the nontranscribed (0.06 ± 0.04) strands of the c-fos gene (Fig. 6). The FPGSS frequency at the 90/0 time point was increased at least 12-fold in the transcribed (1.53 ± 0.39) and nontranscribed (1.19 ± 0.16) strands (p < 0.001). The frequency in both strands at 90/0 was not significantly different. The FPGSS frequency in the transcribed strand at 90/15 (1.69 ± 0.31) and 90/30 (1.92 ± 0.51) remained significantly higher than that of the non-FCI control in the same strand. The genomic FPGSS in the transcribed strand at 45 min of reperfusion (0.93 ± 0.33) showed an apparent reduction (repair) by 50% from the 90/30 time point, although the frequency was still significantly higher than in the control. At 120 min of reperfusion, the FPGSS frequency (0.25 ± 0.09) in the transcribed strand was decreased to a level (0.25 ± 0.09) that was not significantly different from that measured in the non-FCI control. The data indicate that there was no net repair in the transcribed strand of the c-fos gene during the first 30 min of reperfusion and that ∼50% and 85% of the damage were removed by 45 and 120 min of reperfusion, respectively.
FIG. 6.
FPGSS frequencies in the c-fos gene. The signal intensity in the autoradiogram was measured using the AlphaImager. The pixel value of the autoradiogram background was subtracted. The genomic FPGSS frequency was calculated as described in Materials and Methods. The frequencies of genomic FPGSS in the c-fos gene at 15 and 30 min after 90 min of FCI were significantly different (F = 27) using two-way ANOVA: *p < 0.001.
The frequency of genomic FPGSS in the nontranscribed strand of the c-fos gene was reduced by an average of 50 ± 10% at 15 (0.77 ± 0.09), 30 (0.61 ± 0.10), and 45 min (0.45 ± 0.05) of reperfusion from that at the end of FCI (90/0). At 120 min of reperfusion (0.33 ± 0.07), ∼70% of FPGSS in the nontranscribed strand had been removed. The frequencies were statistically (p < 0.001 by t test) higher than that in the non-FCI controls (Fig. 6). The data suggest that 50% of the oxidative DNA injury in the nontranscribed strand was repaired within 30 min of reperfusion, followed by little or no repair over the next 90 min. The repair of FPGSS in the transcribed strand was slower than that in the nontranscribed strand during the initial 30 min immediately after cerebral ischemia (by two-way ANOVA). Moreover, the genomic FPGSS in both strands were not repaired completely within 120 min of reperfusion.
DISCUSSION
Our results indicate that oxidative damage (oh8G/oh8dG) to nucleic acids was present at the end of 15 min of FCI and that inhibition of nNOS activity reduced but did not abolish all of the oxidative damage. The oxidative injury induced in the nucleic acids, as detected by an elevated level of oh8G/oh8dG lesions, is most likely caused by reactive NO, superoxide anions, and/or derivatives of both that can generate hydroxyl radicals. Our results show that damage from ROS can be observed in nucleic acids as early as 15 min after vessel occlusion. Because calcium ion activates nNOS activity, our finding does not exclude the role of calcium influx in the formation of hydroxyl radicals. We also noted that a reduction in mRNA transcription of the c-fos gene, which was significantly increased immediately after FCI, can be observed at the same time when a maximal damage was observed in its nuclear gene, i.e., at the 15- and 30-min reperfusion time points. That transcription of mRNA resumed by the time of the majority of gene damage (as measured by oh8G/oh8dG immunoreactivity or FPGSS) had been removed/repaired at 60 min of reperfusion suggests that the expression of a gene during oxidative stress can be affected by damage to its gene.
Our findings suggest that FCIR-induced oh8G lesions in the RNA of the rat brain are similar to those injuries observed in human brains with Alzheimer’s disease (Nunomura et al., 1999), in a forebrain ischemia—reperfusion (global) model in the mouse (Liu et al., 1996), and in rat brains subjected to drug-induced oxidative stress (Schulz et al., 1995). We have previously reported that genomic DNA and RNA damage slowly increases after global ischemia, peaking at 10 min of reperfusion after 30 min of global ischemia (Liu et al., 1996). The earlier peak in nucleic acid damage that we observed in the rat model probably reflects differences in vascular perfusion. For example, in the rat, oxygen could be supplemented from blood circulation via the posterior vasculature (circle of Willis) during FCI to the area adjacent to the MCA territory. The C57BL6 mouse used in the forebrain ischemia—reperfusion model does not have effective collateral circulation through the circle of Willis (Fujii et al., 1997) and therefore may have less oxygen and electrons available during the ischemic period. The rapid increase in oxidative nucleic acid damage observed in the rat immediately after the occlusion is released suggests that perfusion via the circle of Willis during the ischemia period may increase the formation of ROS during this period. Because humans generally have a functional circle of Willis, the rat FCIR model may more closely reflect the molecular pathophysiology of stroke and other ischemic injuries than does the mouse model.
Our results using 3BR7NI, a specific inhibitor of nNOS, support what has been reported in other models of cerebral oxidative stress (Beckman et al., 1990; Yoshida et al., 1994; Schulz et al., 1995; Kamii et al., 1996; Ayata et al., 1997), i.e., that nNOS mediates oxidative damage to cerebral cortical cells after experimental FCIR. However, other factors, most likely superoxide anions or NO from calcium-independent type II NOS, are likely to be involved in oxidative damage (Murakami et al., 1998; Iadecola et al., 1999). We found that although 3BR7NI effectively blocked the generation of nuclear oh8dG injury, it did not completely abolish the cytosolic oh8G/oh8dG immunoreactivity. Our findings after FCI, as measured by oh8G/oh8dG immunoreactivity with and without nNOS inhibition (Fig. 1), agree with our experiments using a DNA fragment-shift assay that identifies FPGSS (Fig. 6). Although we have observed increased oh8G/oh8dG immunoreactivity after as little as 15 min of FCI, it is unclear whether 15 min of FCI in the rat generates an elevated efflux of L-glutamate and GABA in the MCA area, as has been suggested as the precipitating mechanism for ischemic injury by others (Shimizu-Sasamata et al., 1998), or if some other mechanism is involved. Nevertheless, the data suggest that elevated levels of oh8G/oh8dG and FPGSS, and calcium influx, are the first measurable signs of cell injury after FCIR.
The DNA lesions detected using Fpg protein include base modifications in the c-fos gene not repaired by the mammalian equivalents of the E. coli Fpg protein (Croteau et al., 1997; Roldan-Arjona et al., 1997; Rosenquist et al., 1997; Alamo et al., 1998). FPGSS is known to measure several manifestations of cerebral oxidative DNA injury, including lesions of 8-hydroxyadenine, 2,6-diamino-4-hydroxy-5-formamidopyrimidine, and apurinic/apyrimidinic (AP) sites, in addition to oh8dG lesions in DNA (Bhagwat and Gerlt, 1996; Liu et al., 1996). Because the detection of FPGSS uses a radiolabeled probe, it is very sensitive, but the lesions detected are limited to the probe that is used. At the same time, because the antibody recognizes both oh8G in RNA and oh8dG in DNA, the resolution on boundary is limited, especially using color imaging. Therefore, the FPGSS assay is specific at the gene level, whereas the oh8G/dG assay can show the location of oxidative stress in the tissue. The FPGSS assay therefore can supplement the result of oh8G/dG immunoreactivity, but the two assays cannot be directly compared because the FPGSS assay does not detect damage in RNA.
We estimate that there was at least one hit per gene strand or two FPGSS per c-fos gene by the end of 90 min of FCI. Our data indicate that the repair of oxidative DNA damage is rapid: 40% of the oxidative DNA injury in both strands of the c-fos gene was repaired within 45 min of reperfusion, and 70% was removed within 120 min of reperfusion. Our inability to detect oh8dG immunoreactivity at 60 min of reperfusion, therefore, may be a result of this repair activity. Alternatively, the lack of immunoreactivity at 60 min of reperfusion may reflect the low sensitivity of the oh8G/oh8dG assay. Indeed, ∼30% of the base damage that could be detected in the FPGSS assay was not observed using the immunohistochemical assay for oh8G/oh8dG. Nevertheless, the nature of the damage observed in our study is in agreement with studies that addressed oxidative DNA damage, including damage to mitochondrial DNA (Driggers et al., 1993; Taffe et al., 1996), to nuclear genes in cell culture (Nose and Nikaido, 1984; Hanawalt, 1994; Taffe et al., 1996), and to the γ-actin and DNA polymerase-β genes in the mouse brain (Liu et al., 1996). The recent discovery of rodent oh8dG glycosylase AP endonuclease in rat liver mitochondria suggests that mitochondrial FPGSS may be removed by a mechanism similar to the mechanism implicated in removal of nuclear FPGSS (Croteau and Bohr, 1997; Croteau et al., 1997; Roldan-Arjona et al., 1997; Rosenquist et al., 1997; Alamo et al., 1998). The reduction of cytosolic oh8dG we observed at 30 min of reperfusion could be a result of rodent oh8dG glycosylase AP endonuclease repair of mitochondrial DNA lesions.
Several physiological responses occur at the start of FCI, including influx of calcium, depolarization of the cell membrane, and formation of free radicals of oxygen. These responses activate gene transcription (Shimizu-Sasamata et al., 1998; Sharp et al., 1999). The increase in levels of transcripts of the c-fos and actin genes that we observed immediately following FCI (90/0) supports the possibility of prereperfusion activation of transcription in FCIR injury. The mechanism(s) that causes the decreased transcription of c-fos mRNA at ∼15 min after the initiation of reperfusion is not yet clear. Several hypotheses can be considered: (a) gradients in blood flow and energy metabolism, (b) the mRNA transcript being targeted by hydroxyl radicals, (c) the possible presence of altered (mutated) sequences in the mRNA transcript, and (d) concurrent inhibition of both transcription and repair on the damaged gene. It is, however, unlikely that a gradient in blood flow and energy metabolism would affect the transcribed and nontranscribed strands of the same gene differently. The hypothesis of a mutual exclusion of base excision repair and gene transcription, i.e., binding of base excision repair enzymes could prevent, inhibit, or exclude the binding of gene transcription factors, could explain the kinetics of DNA repair and gene transcription on the transcribed strand of the c-fos gene that were observed in this study. More research is needed to examine this hypothesis.
Lastly, we observed that the rate of repair in the transcribed strand was slower than that in the nontranscribed strand of the same gene at the first 30 min of reperfusion. We cannot exclude the possibility that there is a different rate of DNA repair in different types of brain cells (Gobbel et al., 1998) or that a mixed pathway of repair in the brain exists, one that includes components of both the base excision repair and nucleotide excision repair pathways (Frosina et al., 1996; Klungland and Lindahl, 1997; Wilson and Thompson, 1997). This pathway might be similar to the one that repairs UV-induced pyrimidine dimers in xeroderma pigmentosum patients who have a deficient nucleotide excision repair pathway (Robbins et al., 1983; Satoh et al., 1993) or one that fails to perform base excision repair on the oh8dG present in DNA (Readon et al., 1997). The importance of gene repair within a reasonable time after cerebral ischemia can be understood by the fact that a mutant transcript could be translated into mutant peptides, which in turn could initiate several molecular and cellular responses. Some of these may produce secondary injury, and others may be crucial to neuronal repair and regeneration after ischemic injury (Fornance et al., 1992; An et al., 1993; Sharp, 1994; Yang et al., 1994; Cui et al., 1999; Sharp et al., 1999). The activation of c-fos transcription, as well as the transcription of other immediate early genes and genes that are involved in DNA repair, e.g., DNA polymerase-β, may participate in a key role in determining these late responses for neuroregeneration.
In conclusion, we have presented evidence that two assays, oh8G/oh8dG immunoreactivity and genomic FPGSS assays, can be used as an early indicator of oxidative damage to nucleic acids after FCIR and other acute and chronic neurological insults. A better understanding of oxidative DNA and RNA injury and of its role in initiating or controlling subsequent pathophysiological response to ischemia could be pivotal to development of therapies to reduce the initial oxidative stress and injury, to optimize DNA repair, and to modulate subsequent cellular events.
Acknowledgment
We thank Mr. James Wolff and Mr. Jack Lin for excellent technical assistance, Dr. R. Grossman for suggestions, Dr. Charles Contant for consultation on statistical analysis, and Dr. Winifred Hamilton for editorial assistance. This work was supported in part by grants NS34810 (to P.K.L.) and CA67163 (to E.H.H.) from the National Institutes of Health and by funding from CytoChem (Seattle, WA, U.S.A.) (to E.H.H.). P.K.L. is an Established Investigator of the American Heart Association (award 9640202N).
Abbreviations used
- 3BR7NI
3-bromo-7-nitroindazole
- FCI
focal cerebral ischemia
- FCIR
focal cerebral ischemia and reperfusion
- Fpg
formamidopyridine DNA N-glycolase
- FPGSS
formamidopyridine DNA N-glycolase protein-sensitive sites
- ISH
in situ hybridization
- MCA
middle cerebral artery
- nNOS
neuronal nitric oxide synthase
- NO
nitric oxide
- NOS
nitric oxide synthase
- oh8G and oh8dG
8-hydroxy-2′-guanosine and its deoxy form respectively
- PI
propidium iodide
- ROS
reactive oxygen species
REFERENCES
- Alamo MJP, Jurado J, Francastel E, Laval F. Rat 7,8-dihydro-8-oxoguanine DNA glycosylase: substrate specificity, kinetics and cleavage mechanism at an apurinic site. Nucleic Acids Res. 1998;22:5199–5202. doi: 10.1093/nar/26.22.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An G, Lin T, Liu JS, Xue JJ, He YY, Hsu CY. Expression of c-fos and c-jun family genes after focal cerebral ischemia. Ann. Neurol. 1993;33:457–464. doi: 10.1002/ana.410330508. [DOI] [PubMed] [Google Scholar]
- Ayata C, Ayata G, Hara H, Matthews RT, Beal MF, Ferrante RJ, Endres M, Kim A, Christie RH, Waeber C, Huang PL, Hyman BT, Moskowitz MA. Mechanisms of reduced striatal NMDA excitotoxicity in type I nitric oxide synthase knock-out mice. J. Neurosci. 1997;17:6908–6917. doi: 10.1523/JNEUROSCI.17-18-06908.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbedge RC, Bland-Ward PA, Hart SL, Moore PK. Inhibition of rat cerebellar nitric oxide synthase by 7-nitro indazole and related substituted indazoles. Br. J. Pharmacol. 1993;110:225–228. doi: 10.1111/j.1476-5381.1993.tb13796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J. Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagwat M, Gerlt JA. 3′- and 5′-strand cleavage reactions catalyzed by the Fpg protein from Escherichia coli occur via successive β- and δ-elimination mechanism, respectively. Biochemistry. 1996;35:659–665. doi: 10.1021/bi9522662. [DOI] [PubMed] [Google Scholar]
- Bohr VA, Smith CA, Okumoto DS, Hanawalt PC. DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell. 1985;40:359–369. doi: 10.1016/0092-8674(85)90150-3. [DOI] [PubMed] [Google Scholar]
- Chen J, Jin K, Chen M, Pei W, Kawaguchi K, Greenberg DA, Simon RP. Early detection of DNA strand breaks in the brain after transient focal ischemia: implications for the role of DNA damage in apoptosis and neuronal cell death. J. Neurochem. 1997;69:232–245. doi: 10.1046/j.1471-4159.1997.69010232.x. [DOI] [PubMed] [Google Scholar]
- Chen ST, Hsu CY, Hogan EL, Maricq H, Balentine JD. A model of focal ischemic stroke in the rat: reproducible extensive cortical infarction. Stroke. 1986;17:738–743. doi: 10.1161/01.str.17.4.738. [DOI] [PubMed] [Google Scholar]
- Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Soyeon K, Schenk D, Fraser P, Hyslop PSG, Selkoe DJ. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat. Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- Croteau DL, Bohr VA. Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells. J. Biol. Chem. 1997;272:25409–25412. doi: 10.1074/jbc.272.41.25409. [DOI] [PubMed] [Google Scholar]
- Croteau DL, Rhys CMJ, Hudson EK, Dianov GL, Hansford RG, Bohr VA. An oxidative damage-specific endonuclease from rat liver mitochondria. J. Biol. Chem. 1997;272:27338–27344. doi: 10.1074/jbc.272.43.27338. [DOI] [PubMed] [Google Scholar]
- Cui J, Hsu CY, Liu PK. Suppression of postischemic hippocampal NGF expression by a c-fos antisense oligodeoxynucleotide. J. Neurosci. 1999;19:1335–1344. doi: 10.1523/JNEUROSCI.19-04-01335.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driggers WJ, LeDoux SP, Wilson GL. Repair of oxidative damage within the mitochondrial DNA of RINr 38 cells. J. Biol. Chem. 1993;268:22042–22045. [PubMed] [Google Scholar]
- Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after middle focal cerebral ischemia: a role for apoptosis? J. Cereb. Blood Flow Metab. 1996;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- Fornance AJ, Jackman J, Hollander MC, Hoffman-Liebermann B, Liebermann DA. Genotoxic-stress-response genes and growth-arrest genes. Ann. NY Acad. Sci. 1992;663:139–159. doi: 10.1111/j.1749-6632.1992.tb38657.x. [DOI] [PubMed] [Google Scholar]
- Fraga CG, Shigenaga MK, Park J-W, Degan P, Ames BN. Oxidative damage to DNA during aging: 8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proc. Natl. Acad. Sci. USA. 1990;87:4533–4537. doi: 10.1073/pnas.87.12.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandolo A, Dogliotti E. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 1996;271:9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- Fujii M, Hara H, Meng W, Vonsattel JP, Huang Z, Moskowitz MA. Strain-related differences in susceptibility to transient forebrain ischemia in SV-129 and C57black/6 mice. Stroke. 1997;28:1805–1810. doi: 10.1161/01.str.28.9.1805. [DOI] [PubMed] [Google Scholar]
- Garcia JH, Yoshida Y, Chen H, Li Y, Zhang ZG, Lian J, Chen S, Chopp M. Progression from ischemic injury to infarct following middle cerebral artery occlusion in the rat. Am. J. Pathol. 1993;142:623–635. [PMC free article] [PubMed] [Google Scholar]
- Gobbel GT, Bellinzona M, Vogt AR, Gupta N, Fike JR, Chan P. Response of postmitotic neurons to x-irradiation: implications for the role of DNA damage in neuronal apoptosis. J. Neurosci. 1998;18:147–155. doi: 10.1523/JNEUROSCI.18-01-00147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Braughler JM. Central nervous system trauma and stroke. II. Physiological and pharmacological evidence for involvement of oxygen radicals and lipid peroxidation. Free Radic. Med. Biol. 1989;6:303–313. doi: 10.1016/0891-5849(89)90057-9. [DOI] [PubMed] [Google Scholar]
- Hanawalt PC. Transcription-coupled repair and human disease. Science. 1994;266:1957–1958. doi: 10.1126/science.7801121. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Niwa K, Eckmam C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, Carlson GA. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat. Neurosci. 1999;2:157–161. doi: 10.1038/5715. [DOI] [PubMed] [Google Scholar]
- Jenner P, Dexter DT, Sian J, Schapira AH, Marsden CD. Oxidative stress as a cause of nigral cell death in Parkinson’s disease and incidental Lewy body disease. Ann. Neurol. 1992;32(Suppl):S82–S87. doi: 10.1002/ana.410320714. [DOI] [PubMed] [Google Scholar]
- Kamii H, Mikawa S, Murakami K, Kinouchi H, Yoshimoto T, Reola L, Carlson E, Epstein CJ, Chan PH. Effects of nitric oxide synthase inhibition on brain infarction in SOD-1-transgenic mice following transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1996;16:1153–1157. doi: 10.1097/00004647-199611000-00009. [DOI] [PubMed] [Google Scholar]
- Kisby GE, Milne J, Sweatt C. Evidence of reduced DNA repair in amyotrophic lateral sclerosis brain tissue. Neuroreport. 1997;8:1337–1340. doi: 10.1097/00001756-199704140-00004. [DOI] [PubMed] [Google Scholar]
- Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) EMBO J. 1997;16:3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb BT. Presenilins, amyloid-β and Alzheimer’s disease. Nat. Med. 1997;3:28–29. doi: 10.1038/nm0197-28. [DOI] [PubMed] [Google Scholar]
- Lei SZ, Pan Z, Aggarwal SK, Chen H, Hartman J, Sucher NJ, Lipton SA. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor channel complex. Neuron. 1992;8:1087–1099. doi: 10.1016/0896-6273(92)90130-6. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of NO and related nitroso compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- Liu PK, Salminen A, He Y, Jiang MH, Xue JJ, Liu JS, Hsu CY. Suppression of ischemia-induced Fos expression and AP-1 activity by an antisense oligodeoxynucleotide to c-fos mRNA. Ann. Neurol. 1994;36:566–576. doi: 10.1002/ana.410360405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PK, Hsu CY, Dizdaroglu M, Floyd RA, Kow YW, Karakaya A, Rabow LE, Cui JK. Damage, repair and mutagenesis in nuclear genes after mouse forebrain ischemia—reperfusion. J. Neurosci. 1996;16:6795–6806. doi: 10.1523/JNEUROSCI.16-21-06795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TH, Beckman JS, Freeman BA, Hogan EL, Hsu CY. Polyethylene glycol-conjugated superoxide dismutase and catalase reduce ischemic brain injury. Am. J. Physiol. 1989;256:H589–H593. doi: 10.1152/ajpheart.1989.256.2.H589. [DOI] [PubMed] [Google Scholar]
- Mecocci P, MacGarvey U, Kaufman AE, Beal MF. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann. Neurol. 1993;34:609–616. doi: 10.1002/ana.410340416. [DOI] [PubMed] [Google Scholar]
- Mellon I, Spivak G, Hanawalt PC. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell. 1987;51:241–249. doi: 10.1016/0092-8674(87)90151-6. [DOI] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J. Neurosci. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RL. Mixed signals in Alzheimer’s disease. Trends Neurosci. 1996;19:371–372. doi: 10.1016/S0166-2236(96)30011-8. [DOI] [PubMed] [Google Scholar]
- Nose K, Nikaido O. Transcriptionally active and inactive genes are similarly modified by chemical carcinogens or X-ray in normal human fibroblasts. Biochim. Biophys. Acta. 1984;781:273–278. doi: 10.1016/0167-4781(84)90093-9. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J. Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill MJ, Hicks C, Ward M. Neuroprotective effects of 7-nitroindazole in the gerbil model of global cerebral ischemia. Eur. J. Pharmacol. 1996;310:115–122. doi: 10.1016/0014-2999(96)00387-1. [DOI] [PubMed] [Google Scholar]
- Park E-M, Shigenaga MK, Degan P, Korn TS, Kitzler JW, Wehr CM, Kolachana P, Ames BN. Assay of excised oxidative DNA lesions: isolation of 8-oxoguanine and its nucleoside derivatives from biological fluids with a monoclonal antibody column. Proc. Natl. Acad. Sci. USA. 1992;89:3375–3379. doi: 10.1073/pnas.89.8.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parshad R, Sanford KK, Price FM. Fluorescence light-induced chromatid breaks distinguish Alzheimer disease cells from normal cells in tissue culture. Proc. Natl. Acad. Sci. USA. 1996;93:5146–5150. doi: 10.1073/pnas.93.10.5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poiries J. Apolipoprotein E in animal models of CNS injury and in Alzheimer’s disease. Trends Neurosci. 1994;17:525–530. doi: 10.1016/0166-2236(94)90156-2. [DOI] [PubMed] [Google Scholar]
- Readon JT, Bessho T, Kung HC, Bolton PH, Sancar A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: possible explanation for neurode-generation in xeroderma pigmentosum patients. Proc. Natl. Acad. Sci. USA. 1997;94:9463–9468. doi: 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins JH, Polinsky RJ, Moshell AN. Evidence that lack of deoxyribonucleic acid repair causes death of neurons in xeroderma pigmentosum. Ann. Neurol. 1983;13:682–684. doi: 10.1002/ana.410130621. [DOI] [PubMed] [Google Scholar]
- Roldan-Arjona T, Wei YF, Carter KC, Klungland A, Anselmino C, Wang R-P, Augustus A, Lindahl T. Molecular cloning and functional expression of a human cDNA encoding the antimutator enzyme 8-hydroxyguanine-DNA glycosylase. Proc. Natl. Acad. Sci. USA. 1997;94:8016–8020. doi: 10.1073/pnas.94.15.8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenquist TA, Zharkov DO, Grollman AP. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl. Acad. Sci. USA. 1997;94:7429–7434. doi: 10.1073/pnas.94.14.7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal RE, Chanderbahn R, Marshall G, Fiskum G. Prevention of post-ischemic brain lipid conjugated diene production and neurological injury by hydroxyethylstarch-conjugated deferoxamine. Free Radic. Biol. Med. 1992;12:29–33. doi: 10.1016/0891-5849(92)90055-l. [DOI] [PubMed] [Google Scholar]
- Satoh MS, Jones CJ, Wood RD, Lindahl T. DNA excision-repair defect of xeroderma pigmentosum prevents removal of a class of oxygen free radical-induced base lesions. Proc. Natl. Acad. Sci. USA. 1993;90:6335–6339. doi: 10.1073/pnas.90.13.6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlon KJ, Jiao L, Funato T, Wang W, Tone T, Rossi JJ, Kashani-Sabet M. Ribozyme-mediated cleavage of c-fos mRNA reduces gene expression of DNA synthesis enzymes and metallothionein. Proc. Natl. Acad. Sci. USA. 1991;88:10591–10595. doi: 10.1073/pnas.88.23.10591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Mathews RT, Jenkins BG, Ferrante RJ, Siwek D, Henshaw DR, Cipolloni PB, Mecocci P, Kowall NW, Rosen BR, Beal MF. Blockade of neuronal nitric oxide synthase protects against excitotoxicity in vivo. J. Neurosci. 1995;15:8419–8429. doi: 10.1523/JNEUROSCI.15-12-08419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp FR. The sense of antisense fos oligonucleotides. Ann. Neurol. 1994;36:555–556. doi: 10.1002/ana.410360403. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Massa SM, Swanson RA. Heat-shock protein protection. Trends Neurosci. 1999;22:97–99. doi: 10.1016/s0166-2236(98)01392-7. [DOI] [PubMed] [Google Scholar]
- Shimizu-Sasamata M, Bosque-Hamilton P, Huang PL, Moskowitz MA, Lo EH. Attenuated neurotransmitter release and spreading depression-like depolarizations after focal ischemia in mutant mice with disrupted type I nitric oxide synthase gene. J. Neurosci. 1998;18:9564–9571. doi: 10.1523/JNEUROSCI.18-22-09564.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Perry G, Richey PL. Oxidative damage in Alzheimer disease. Nature. 1996;382:121–122. doi: 10.1038/382120b0. [DOI] [PubMed] [Google Scholar]
- Taffe BG, Larminat F, Laval J, Croteau DJ, Anson RM, Bohr VA. Gene-specific nuclear and mitochondrial repair of formamidopyrimidine DNA glycosylase-sensitive sites in Chinese hamster ovary cells. Mutat. Res. 1996;364:183–192. doi: 10.1016/s0921-8777(96)00031-6. [DOI] [PubMed] [Google Scholar]
- Wilson DM, Thompson LH. Life without DNA repair. Proc. Natl. Acad. Sci. USA. 1997;94:12754–12757. doi: 10.1073/pnas.94.24.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B, Iwasaki R, Vito P, Ganjei JK, Lacana E, Sunderland T, Zhao B, Kusiak JW, Wasco W, Adamio L. Participation of presenilin 2 in apoptosis: enhanced basal activity conferred by Alzheimer mutation. Science. 1996;274:1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Mu S, Xue JJ, Whitson J, Salminen A, Dixon CE, Liu PK, Hayes RL. Increased expression of c-fos mRNA and AP-1 transcription factor after cortical impact injury in rats. Brain Res. 1994;664:141–147. doi: 10.1016/0006-8993(94)91964-x. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Limmroth V, Irikura K, Moskowitz MA. The NOS inhibitor, 7-nitroindazole, decreases focal infarct volume but not the response to topical acetylcholine in pial vessels. J. Cereb. Blood Flow Metab. 1994;14:924–929. doi: 10.1038/jcbfm.1994.123. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Chopp M, Gautam S, Zaloga C, Zhang RL, Schmidt HHHW, Pollock JS, Forstermann U. Upregulation of neuronal nitric oxide synthase and mRNA, and selective sparing of nitric oxide synthase-containing neurons after focal cerebral ischemia in rat. Brain Res. 1994;654:85–95. doi: 10.1016/0006-8993(94)91574-1. [DOI] [PubMed] [Google Scholar]