

Abstract
Ampullosporin A and alamethicin are two members of the peptaibol family of antimicrobial peptides. These compounds are produced by fungi and are characterized by a high content of hydrophobic amino acids, and in particular the α-tetrasubstituted amino acid residue α-aminoisobutyric acid. Here ampullosporin A and alamethicin were uniformly labeled with 15N, purified and reconstituted into oriented phophatidylcholine lipid bilayers and investigated by proton-decoupled 15N and 31P solid-state NMR spectroscopy. Whereas alamethicin (20 amino acid residues) adopts transmembrane alignments in 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) or 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) membranes the much shorter ampullosporin A (15 residues) exhibits comparable configurations only in thin membranes. In contrast the latter compound is oriented parallel to the membrane surface in 1,2-dimyristoleoyl-sn-glycero-3-phosphocholine and POPC bilayers indicating that hydrophobic mismatch has a decisive effect on the membrane topology of these peptides. Two-dimensional 15N chemical shift – 1H-15N dipolar coupling solid-state NMR correlation spectroscopy suggests that in their transmembrane configuration both peptides adopt mixed α-/310-helical structures which can be explained by the restraints imposed by the membranes and the bulky α-aminoisobutyric acid residues. The 15N solid-state NMR spectra also provide detailed information on the helical tilt angles. The results are discussed with regard to the antimicrobial activities of the peptides.
Introduction
Peptaibols, including alamethicin (Alm) and ampullosporin, are small hydrophobic peptides of fungal origin that are rich in α-aminoisobutyric acid (Aib) and many of which exhibit antimicrobial activities (reviewed in (1–4)). The closely related isoforms of alamethicin encompass 20 residues, nine of which are Aib residues in the case of the F50/7 variant investigated in this work. These peptides have been thoroughly studied in the past as they are considered a paradigm for channel formation in biological membranes. Structural analysis of alamethicin by x-ray crystallography (5), as well as NMR-spectroscopy in methanolic solution (6), or in the presence of sodium dodecyl sulfate micelles (7,8) all indicate that the conformations of alamethicin are predominantly helical with a flexible hinge region at the Gly-11 position (9) that results in a bend in the x-ray structures (5). In the center of the alamethicin sequence the G-X-X-P motif results in a break of the helix conformation and a less stable C-terminal structure which undergoes considerable conformational averaging (6,7,10). These findings have been confirmed by molecular dynamics simulations (11,12). In addition, Fourier transform infrared spectroscopy, Raman and circular dichroism (CD) spectroscopies show that the degree of helicity is dependent on the physical state of the lipid, the lipid/peptide ratio and the presence of transmembrane potentials (reviewed in (3,4)).
When added to lipid bilayers and natural membranes, the polypeptide exhibits a well defined pattern of successive increases in conductance levels, each of a duration of a few milliseconds (13–15) which resemble those seen in the presence of large voltage- or ligand-gated channel proteins (reviewed in (1–4)). At relatively high concentrations alamethicin is also able to induce a liposomal leakage of carboxyfluorescein-loaded vesicles while at the same time water-membrane partitioning and aggregation phenomena were found to be major determinants of the membrane activity of antimicrobial peptides (16). Even more bulky ions, like Nα-benzoyl-L-arginine-para-nitroanilide were found to pass the membrane in the presence of alamethicin F50/5 (17). The open alamethicin pore has been suggested to consist of “transmembrane helical bundles” or “barrel staves” (2) composed of at least three (18) or four subunits (19). As many as 20 distinguishable conductance states have been described (20) and it has therefore been suggested that these correspond to differently sized bundles of transmembrane alamethicin helices (2). Although self-associated pores are formed above a threshold concentration, the peptides remain associated with the membrane surface at low peptide/lipid ratios (21). Previously, the membrane interactions of alamethicin were investigated by oriented CD (22,23) and by oriented solid-state NMR spectroscopy (24–26). In particular, the transition between the surface-associated state and transmembrane alignment was characterized as a function of peptide/lipid ratio and of membrane phospholipid composition (21–23).
Ampullosporin A (AmpA) is a 15-mer peptaibol whose x-ray structure exhibits a largely regular α-helix starting from the acetylated N-terminus and contains a β-turn at the C-terminus (27). This structure exhibits even more of a bend when compared to the alamethicin x-ray structure. AmpA helices possess a hydrophilic face formed by the polar side chains of Gln-7, Gln-11, and the non-hydrogen bonding carbonyl oxygens of Aib-10 and Gln-11, whereas the hydrophobic face is formed by the bulky side chains of Trp-1, Leu-5, Leu-12 and Leu-15ol. In the crystals, the distance between the Cα of the first residue and the Cα of the last residue is 20.2 Å for AmpA (27), i.e., much shorter than that of antiamoebin (24.4 Å), another 16-mer peptaibol (28). When the structured regions are considered the length of the AmpA helix (residues 1 to 10) is 13.5 Å and that of the antiamoebin helix is 9.4 Å (residues 1 to 7 of unit A pdb: 1JOH).
Ion channel activity of AmpA was demonstrated in phosphatidylcholines (PCs) at P/L ratios as low as 1:3000 (29). However, in comparison to alamethicin, AmpA displays only weak antimicrobial or channel-forming effects (30). However, the peptide induces pigment formation in the fungus Phoma destructiva (31); it also provokes hypothermia and inhibits locomotor activity in mice (30,31).
Whereas the biological activities of several peptaibols, such as the antimicrobial, antimalarial and hemolytic activities, the stimulation of catecholamine secretion, and the uncoupling of mitochondrial oxidative phosphorylation were reported to be generated from their membrane activities, the action mechanisms of the biological activities of ampullosporin A remain unknown although it is suspected that they form channels by related mechanisms (32,33). Furthermore, despite intensive research, our understanding of the detailed mechanisms of voltage-dependent channel-formation by peptaibols remains incomplete. This is in part because of the paucity of information about the structures of the peptides in membranes, which might exhibit different conformational and dynamic properties from their conformations in organic solvents or in the crystal.
Solid-state NMR spectroscopy is a powerful tool for investigating the structure of membrane-active peptides and proteins, because it allows the study of amorphous and partly mobile biological solids directly in the liquid-crystalline lipid bilayers (34–39). Orientation-dependent nuclear spin interactions such as the 15N chemical shift, the 15N-1H dipolar coupling or the 2H quadrupolar splitting provide exquisite probes of the orientation of membrane peptides relative to the bilayer normal (24–26,40,41). Correlating the N-H dipolar coupling and the 15N chemical shift in a two-dimensional solid-state NMR spectrum (42) further allows the determination of the orientational topology of membrane peptides with multiple 15N labels (34,35). In this study we report the 15N and 31P solid-state NMR spectra obtained from mechanically oriented bilayers to investigate the lipid-dependence of AmpA alignment and the membrane-associated structure of both peptaibols when in their transmembrane orientation.
Materials and Methods
Alamethicin F50/7 (Alm), a natural isoform of the F50/5 analog where Ala6 is replaced by Aib, has the sequence Ac-Aib-Pro-Aib-Ala-Aib-Aib-Gln-Aib-Val-Aib-Gly-Leu-Aib-Pro-Val-Aib-Aib-Gln-Gln-Phe-OH (4) and was prepared as previously described and is uniformly labeled with 15N at a level ≥ 92% (43).
15N-ampullosporin A (AmpA), with the sequence Ac-Trp-Ala-Aib-Aib-Leu-Aib-Gln-Aib-Aib-Aib-Gln-Leu-Aib-Gln-Leu-OH was isolated from cultures of Sepedonium ampullosporum HKI-053 grown on 15N enriched medium (Supplementary Material). The strain was cultivated as a surface culture (25 l) at 23°C in 500 mL Erlenmeyer flasks containing 100 mL medium 7∗ (the asterisk indicates uniform labeling with 15N) composed as follows (g/L): maltose 5, 15N-Celtone-Powder 2, KH2PO4 2, MgSO4·7 H2O 0.5, ZnSO4·7 H2O 0.008, pH 5.8 - 6.2. After 34 days of cultivation the culture broth was dispersed and extracted with ethyl acetate. The residue (924 mg) was subjected to silica gel chromatography (silica gel 60, Merck, Darmstadt, Germany) 0.063-0.1 mm, gradient: 100 % CHCl3, 9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 3:7, 100 % methanol). Fractions containing 15N-ampullosporin A were tested by analytical high performance liquid chromatography (HPLC). The combined fractions were subjected to size exclusion chromatography (Sephadex LH-20, methanol) and analyzed by HPLC using a Nucleosil 100 C18 column (100 Å pore size, 3 mm × 125 mm) and an acetonitrile-water/0.1% trifluoro acetic acid gradient at a flow rate of 1 mL/min. The final purification was achieved by preparative HPLC using a Shimadzu LC8A apparatus, equipped with a Eurosher 100 C8-column (5 μm, 250 × 32,5 mm), at a flow rate of 9 mL/min and a gradient 50 to 90% acetonitrile in water/0.1% trifluoro acetic acid. The peptide was detected at 220 nm. The identity of the product was confirmed by mass spectrometry (MS) of the full-length peptide and fragments thereof (Fig. 1).
Figure 1.
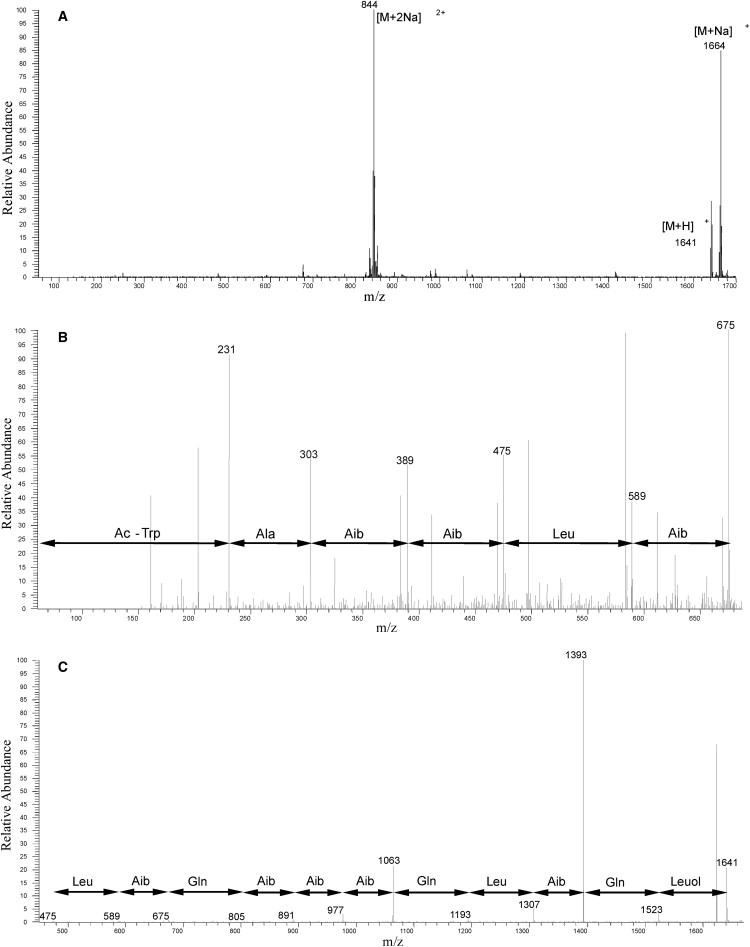
(A) Mass spectrum of 15N-ampullosporin A. (B) Mass spectrometry/mass spectrometry (MS/MS) spectrum of the N-terminal fragment of 15N-ampullosporin A. (C) MS/MS spectrum of the C-terminal fragment of 15N-ampullosporin A.
The high resolution heteronuclear single quantum correlation spectrum of the peptide in CD3OH is shown in the Supplementary Material. Fifteen 15N resonances are observed within the isotropic chemical shift positions of the amides (between 112–135 ppm for 15N and 7.3–8.3 ppm for 1H). An additional 1H resonance at 10.4 ppm was assigned to the tryptophan side chain. Three glutamine side chains exhibit 15N isotropic chemical shift values at ∼97 ppm. The data therefore indicate that all sites were labeled with 15N.
The phospholipids 1,2-didecanoyl-sn-glycero-3-phosphocholine (di-C10:0-PC); 1,2-dilauroyl-sn-glycero-3-phosphocholine (di-C12:0-PC); 1,2-dimyristoleoyl-sn-glycero-3-phosphocholine (di-C14:1-PC); 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC; di-C14:0-PC); and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC; C16:0,C18:1-PC) were from Avanti Polar Lipids(Alabaster, AL). All commercial material was used without further purification.
Sample preparation for NMR
For solid-state NMR measurements 0.75–4 mg of ampullosporin A or alamethicin (both uniformly labeled with 15N) were dissolved in trifluoroethanol and chloroform, respectively, and mixed with the appropriate amount of lipid. The solution was then spread onto 30 ultra-thin cover glasses (9 × 22 mm, Marienfeld, Lauda-Königshofen, Germany) and the samples first dried in air and thereafter in high vacuum overnight to remove all organic solvent. Subsequently, the membranes were equilibrated at 93% relative humidity. For NMR measurements the samples were tightly sealed, inserted into a double resonance flat-coil probe head (44), and introduced into the magnet with the bilayer normal aligned parallel to the magnetic field direction.
Solid-state NMR spectroscopy:
The proton-decoupled 15N cross-polarization (CP) spectra of static aligned samples were acquired at 40.54 MHz on a Bruker Avance wide bore 400 NMR spectrometer. An adiabatic CP pulse sequence was used with a spectral width, acquisition time, CP contact time and recycle delay times of 75 kilohertz (kHz), 3.5 ms, 500 μs and 3 s, respectively. The 1H π/2 pulse and spinal64 heteronuclear decoupling field strengths B1 corresponded to a nutation frequency of 42 kHz. 40 k scans were accumulated and the spectra were zero filled to 4 k points. An exponential line-broadening of 100 Hz was applied before Fourier transformation. Spectra were externally referenced to 15NH4Cl at 41.5 ppm. Samples were cooled with a stream of air at a temperature of 22°C. For the DMPC sample containing alamethicin the temperature of the air was set to 37°C to assure that the lipid bilayers were in their fluid phase.
The two-dimensional PISEMA experiment was used to correlate the 15N-1H dipolar coupling with the 15N chemical shift of the same nitrogen (45). The effective 1H B1 field of the spin-lock amplitude was 45 kHz. During the spin exchange period the amplitude of the 1H B1 field was decreased to 36.8 kHz to maintain the Hartmann– Hahn match condition with an effective field along the magic angle of 45 kHz. For the experimental set-up a fully 15N, 13C labeled N-acetyl leucine single crystal was used.
Proton-decoupled 31P solid-state NMR spectra were acquired at 161.953 MHz on a Bruker Avance widebore 400 NMR spectrometer equipped with a double resonance flat-coil probe (44). Proton decoupled 31P spectra were acquired using a Hahn-echo pulse sequence with a π/2 pulse of 2.5 μs. The spectral sweep width was 75 kHz, the echo- and recycle delays were 40 μs and 3 s, respectively. Samples were cooled with a stream of air at a temperature of 22°C. For DMPC samples the temperature was increased to 37°C to keep the lipid bilayers in their fluid phase. Spectra were referenced externally to 85% H3PO4 at 0 ppm.
Spectral simulations
All numerical simulations were accomplished on a 3.4-GHz Pentium(R) D workstation operating under Windows XP Professional using the SIMPSON/SIMMOL software package (46). The calculated PISEMA spectra were visualized using the GSim software, version 0.12.0. (http://www.dur.ac.uk/vadim.zorin/soft.htm). The 15N-1H dipolar coupling was 9.9 kHz corresponding to a 1.07 Å interspin distance (36) and the amide 1H chemical shift anisotropy tensor δiso = 9.3 ppm, δaniso = 7.7 ppm, η = 0.65 (i.e., σ11 = 2.95, σ22 = 7.95, σ33 = 17; low radio frequency field limit (47)). Whereas magic-angle spinning solid-state NMR spectra of alamethicin and of ampullosporin A show two isotropic signal intensities, the heteronuclear single quantum correlation spectra of peptaibols are indicative of a low-field displacement of the isotropic 15N chemical shifts of Aib when compared to the amide bonds of other amino acid residues (see Fig. S1 in the Supplementary Material). Therefore the following 15N amide chemical shift tensors were used: for Aib δiso = 129.5 ppm, δaniso = 105 ppm, η = 0.2 (i.e., σ11 = 66.5, σ22 = 87.5, σ33 = 234.5) and for all other residues δiso = 117.5 ppm, δaniso = 105 ppm, η = 0.2 (i.e., σ11 = 54.5, σ22 = 75.5, σ33 = 222.5). The experimental evidence that led to this choice will be discussed in detail in another article (E. Salnikov, P. Bertani, J. Raap and B. Bechinger, unpublished). When the spectra are acquired with the external magnetic field direction perpendicular to the bilayer plane this set of tensors accounts for the anisotropic chemical shift or dipolar interactions regardless of motional averaging around the bilayer normal.
To better represent the experimental spectra a line-broadening of 300 Hz was applied in the direct dimension and of 1 kHz in the indirect dimension. To only take the 15N-1H dipolar couplings of the amides into account, the proline residues (numbers 2 and 14 in the alamethicin sequence) were excluded from the simulation.
The root mean-square deviation (RMSD) was calculated from the difference between experimental and simulated spectra in the regions 30–270 ppm and 1–13.7 kHz according to:
where N is the number of data points, and noise is the uncertainty of the experimental spectrum.
where n is the number of data points of a region devoid of signal. As artifacts tend to accumulate in the spectral regions of 0 kHz dipolar, splitting this area was excluded during error analysis. The simulated data were adjusted by a multiplicative factor to give the best overall agreement with the experimental data (i.e., lowest RMSD).The lower the RMSD is the better the simulation fits the experimental data, with values <1 indicative of simulations that deviate from experiment by only the experimental noise level.
Results
To optimize ampullosporin production, Sepedonium ampullosporum HKI-053 was grown under various fermentation conditions (Supplementary Material). The best production was observed in a resting culture at 23°C using medium 7∗ (experimental). Uniform15N labeling was achieved by exchanging peptone for a mixture of 15N-labeled amino acids. The crude extract from a scaled-up culture (24.5 L) was purified by open column chromatography (silica, Sephadex) and semipreparative HPLC, yielding 96 mg of pure 15N-ampullosporin A. The successful isotope labeling was demonstrated by MS (Fig. 1 A). In the mass spectrum only peaks deriving from 15N-ampullosporin A were detected. As expected, the molecular mass (m/z = 1640.9) of the labeled compound was shifted by 19 units as compared to unlabeled ampullosporin A (m/z = 1621.9). In addition, MS/MS analyses of C- and N-terminal fragments indicated that all daughter ions reflect the uniform 15N labeling (Fig. 1, B and C).
Experimental spectra of 15N uniformly labeled AmpA in PCs of different hydrocarbon tail lengths at pH = 4.5 and a peptide/lipid molar ratio of 1:100 are shown in Fig. 2, A–D. The 31P NMR signals of the same samples monitoring the order of the phospholipid headgroups indicate that the membranes are well oriented (Fig. 3, A and B). The 15N signal intensities in the region 65–120 ppm for thick membranes of POPC and di-C14:1-PC are indicative of in-planar alignments of the peptide helices. In contrast, the peptide exhibits 15N chemical shifts in the 200 ppm region, i.e., transmembrane orientations when associated with di-C12:0-PC and di-C10:0-PC. Notably, the 15N chemical shift values are somewhat lower for di-C10:0-PC when compared to di-C12:0-PC indicating that the averaged peptide helix tilt angle is larger for thinner membranes.
Figure 2.
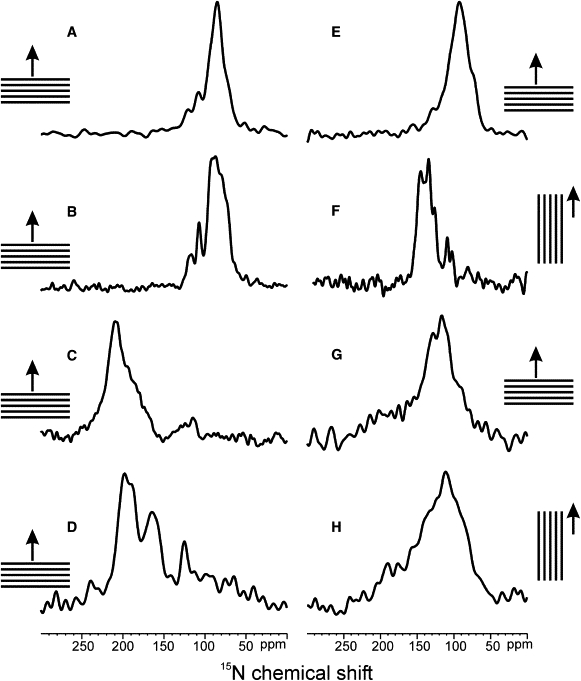
Proton-decoupled 15N solid-state NMR spectra of ampullosporin A labeled uniformly with 15N and reconstituted into oriented PC membranes: (A) POPC (C16:0,C18:1-PC), (B) di-C14:1-PC, (C) di-C12:0-PC, (D) di-C10:0-PC. (E) POPC, P/L ratio 1:100 and pH 7.5. (F) Same sample as A but at a 90° tilted sample alignment. The peptide/lipid molar ratio of samples A–D and F is 1:100 and the pH value is 4.5. (G and H) POPC, pH 7.5, P/L ratio 1:10. The sketches next to the corresponding panels show the membrane/glass plate stacks and the magnetic field direction (arrows). The bilayer normal is parallel to the external magnetic field for A–E and G, and perpendicular for F and H.
Figure 3.
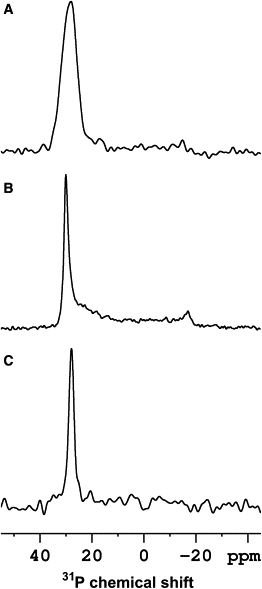
Proton-decoupled 31P spectra of oriented samples of ampullosporin A in PC membranes at the peptide/lipid molar ratios indicated. (A) di-C10:0-PC (1/100), (B) POPC, pH 7.5 (1/100), (C) POPC, pH 7.5 (1/10).
When the oriented AmpA sample in POPC is tilted by 90° relative to the sample alignments shown in Fig. 2 A the resonances are observed in the regions 100–110 ppm and 120–150 ppm (Fig. 2 F). The absence of powder pattern line shapes at this sample alignment is indicative of motional averaging and fast rotational diffusion around the normal of the bilayer (48,49).
Previously it has been observed that the presence of salt and buffer solutions make the preparation of oriented phospholipid bilayers more difficult. Therefore, an AmpA sample was prepared that is identical to that of POPC, P/L 1:100 (Fig. 2 A), except that the pH was adjusted and stabilized at pH 7.5 by the addition of small amounts of 1 N NaOH and 100 μL of 10 mM Tris buffer. The proton-decoupled 31P solid-state NMR spectrum indicates a predominant peak at 30 ppm indicative of well oriented lipid bilayers. However, additional signal intensities at <30 ppm are apparent (Fig. 3 B) and are indicative of some disorder at the level of the phospholipid headgroup due to the presence of salt. Nevertheless, the proton-decoupled 15N spectrum of this sample when aligned with the bilayer normal parallel to the external magnetic field exhibits the same overall features as the one in the absence of salt and at pH 4.5 (Fig. 2, A and E). To optimize sample alignment and resolution for the structural analysis by two-dimensional solid-state NMR, the samples were prepared without the addition of buffer as shown in Fig. 2, A–D and F. It is worth noting that neither peptide contains an ionizable group between pH 0-14.
To increase the signal/noise ratio during the structural analysis of polypeptides by solid-state NMR it is advantageous to work with the highest possible concentration of peptides reconstituted into oriented membranes. However, due to the limited coil volume this also requires an increase in the peptide/lipid ratio. A sample was prepared at pH 7.5 with a peptide/lipid ratio of 1:10 and the proton-decoupled 15N spectrum recorded. The spectrum, obtained with perpendicular bilayer orientation (Fig. 2 G), shows signal intensities in the range of 200–60 ppm when at the same time the 31P NMR spectrum of this preparation is indicative of well oriented membranes (Fig. 3 C). When the sample is tilted by 90° (Fig. 2 H) the changes in the spectral line shape are much less pronounced when compared to the situation at P/L 1/100 (Fig. 2, A and F). Notably, pronounced signal intensities are observed at 110-150 ppm indicative of isotropic realignment of amide resonances or helical tilt angles close to the magic angle. The ensemble of spectra indicates a more heterogeneous conformation and/or alignment of the peptides and an important contribution of tilted conformations and/or of peptide regions undergoing isotropic averaging.
To get a better insight into the secondary structures of the peptides when bound to the lipid bilayer two-dimensional solid-state NMR spectra were recorded where the 15N -1H dipolar coupling was correlated to the 15N chemical shift (45). The PISEMA spectrum of AmpA, labeled uniformly with 15N and reconstituted into oriented di-C10:0-PC, is shown in Fig. 4 A. The intensities exhibiting 15N chemical shifts ≥150 ppm are assigned to transmembrane amide 15N atoms as no other AmpA resonances are expected in this region. The two-dimensional spectrum is characterized by a group of resonances between 185 and 210 ppm as well as between 160 and 170 ppm with signal intensities between 170 and 185 ppm being weak or absent. The resonances at ∼120 ppm appear in all spectra of AmpA bound to PC membranes, regardless of their hydrophobic thickness (Fig. 2), and are therefore tentatively assigned to amino acid side chains of AmpA which carry a 15N label (three Gln and one Trp residues). The isotopic resonances of these residues are indeed expected in this chemical shift range (50) with near zero dipolar couplings.
Figure 4.
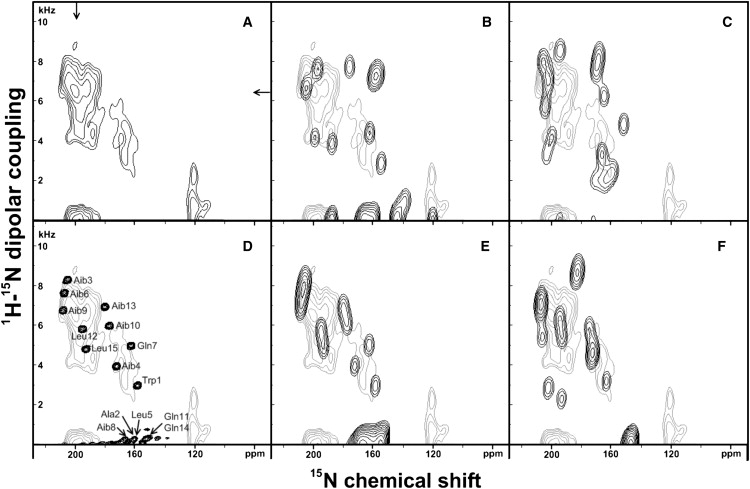
Experimental and simulated PISEMA spectra for ampullosporin A labeled uniformly with 15N and reconstituted into oriented di-C10:0-PC at a peptide/lipid ratio of 1/100. (A) Experimental spectrum. (B) Simulated spectrum of the x-ray diffraction structure of ampullosporin A (pdb: AMPA) superimposed on the experimental spectrum (light gray); (C) Simulation of an ideal α-helix (ϕ = −65°, ψ = −45°). (D) The simulation shown in panel E is represented with reduced line-broadening to resolve the peaks and to monitor the origin of the simulated signal intensities. (E) Simulated spectrum of an ideal 310-helix (ϕ = −50°, ψ = −31°). (F) simulation of a mixed α-/310-helical peptide (the first eight residues folded in α-helical and the last seven residues in 310-helical conformation). The simulation details are summarized in Table 1.
To analyze the experimental PISEMA spectra the expected resonances were simulated for a range of model secondary structures that are likely to represent the conformation of the peptide in its membrane-associated state using the SIMMOL and SIMPSON software (46). First, the spectra resulting from the crystal structure of AmpA were calculated (27). The structure represents an α-helix for the most N-terminal 10 residues and ends with a pronounced kink and a less regular conformation at the C- terminus. The best simulation of the PISEMA experiment was obtained when the AmpA structure (pdb: AMPA) was aligned in such a manner that the N-terminal α-helix was tilted by 36.4° relative to the membrane normal. The alignment of the more C-terminal residues much depends on the azimuthal angle and can vary over almost the whole range of tilt angles. We therefore tested different pitch angles (5° step size with the tilt angle adjusted manually for each pitch angle and 72 different pitch angles in total) and the calculated spectrum which best fit the experimental spectrum is shown in Fig. 4 B. Even after this optimization the model does not explain all of the observed resonances and, in addition, creates intensities which are absent in the experimental spectrum.
Next the PISEMA spectrum of an ideal α-helix (ϕ = −65°, ψ = −45°) was simulated at different orientations. A tilt angle of 32° represents well the range of experimentally observed 15N chemical shift values (160–170 and 185–210 ppm) and results in the spectrum shown in Fig. 4 C. However, this model also fails to explain many details observed in the experiment. It should be noted that due to the differential use of 15N chemical shift tensors for Aib and all other residues the simulated spectrum is the result of two incomplete helical wheels that are slightly shifted relative to each other (Fig. S2). In a third simulation the PISEMA spectra of a 310-helix (ϕ = −50°, ψ = −31°) was calculated at different orientations. The best fit was obtained at a tilt angle of 31.3° (Fig. 4 E). This conformation is consistent with the lack of intensities in the region 170–185 ppm, as observed in the experimental spectrum (Fig. 4, D and E), without creating additional intensities far removed from the experimental peak positions. Finally Fig. 4 F shows a mixed α-/310-helical structure where the first eight residues are α-helical and the last seven are 310-helical. The RMSD values from the simulations of the PISEMA spectrum of AmpA in di-C10:0-PC are summarized in Table 1.
Table 1.
Summary on the simulations of the PISEMA spectra of ampullosporin A in di-C10:0-PC using different structural models
| Model | Tilt angle, degree | RMSD |
|---|---|---|
| XRD structure | 36.4∗ | 1.62 |
| α-helix | 32 | 1.61 |
| 310-helix | 31.3 | 1.49 |
| Mixed α/310-helix | 32 | 1.56 |
XRD: x-ray diffraction. See text for discussion of errors.
Approximate alignment of the helix encompassing residues 1–10.
A remarkable feature of the spectrum shown in Fig. 4 A is the lack of intensities in the chemical shift region 170–185 ppm when AmpA was reconstituted into di-C10:0-PC. Since the PISEMA spectra were recorded using cross-polarization, it remains possible that the efficiency of the magnetization transfer from 1H to 15N is dependent on the resonance position. “Holes” in the spectra around the chemical shift position of the magic angle have been observed previously in cross-polarized 15N or 31P solid-state NMR spectra of membranes (49,51). However, similar experiments performed with ampullosporin A reconstituted into oriented di-C12:0-PC exhibit clear signal intensities in this chemical shift region which is indicative that magnetization transfer works well. Again, the experimental spectrum (Fig. 5 A) was compared to spectral simulations for structural models representing either the crystal structure, a perfect α-helix (ϕ = −65°, ψ = −45°) or a perfect 310-helix (ϕ = −50°, ψ = −31°). Using the crystal structure with the N-terminal helix tilted by 31.4° matches only some of the features of the experimental spectrum (Figs. 5 B). Although a perfect α-helix better represents some of the spectral features it does not provide an explanation for the marked intensity in the center of the helical wheels (Fig. 5 C). Better fits are obtained by 310- (Fig. 5 E) or mixed α-/310-helical conformations at a tilt angle of 31.8° (Fig. 5 F). Again the RMSD values from these simulations are compared to each other (Table 2). The slices from the experimental PISEMA spectra shown in this article are represented in Fig. 6.
Figure 5.
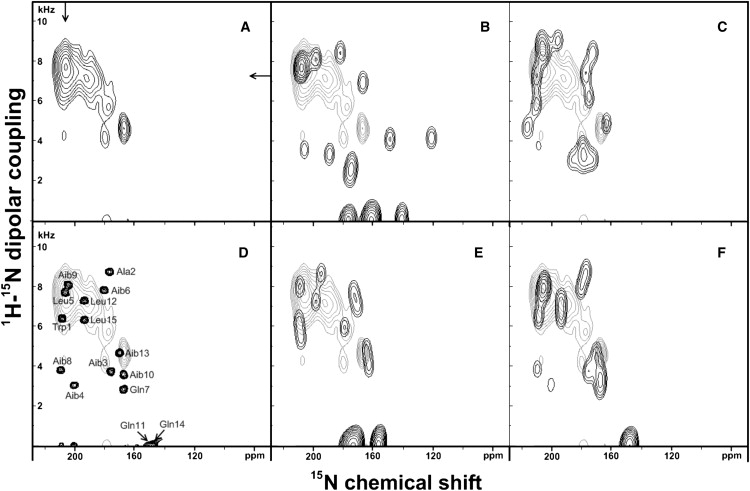
Experimental and simulated PISEMA spectra for 15N uniformly labeled ampullosporin A reconstituted into oriented di-C12:0-PC at a peptide/lipid ratio of 1/75. (A) Experimental spectrum. (B) Simulated spectrum of the x-ray diffraction structure of ampullosporin A (pdb: AMPA) superimposed on the experimental spectrum (light gray); (C) Simulation of an ideal α-helix (ϕ = −65°, ψ = −45°). (D) The simulation shown in panel F is represented with reduced line-broadening to resolve the peaks and to monitor the origin of the simulated signal intensities. (E) Simulated spectrum of an ideal 310-helix (ϕ = −50°, ψ = −31°). (F) simulation of a mixed α-/310-helical peptide (the first eight residues folded in α-helical and the last seven residues in 310-helical conformation). The simulation details are summarized in Table 2.
Table 2.
Summary on the simulation of the PISEMA spectra of ampullosporin A in di-C12:0-PC using different structural models
| Model | Tilt angle, degree | RMSD |
|---|---|---|
| XRD structure, | 31.4∗ | 2.63 |
| α-helix | 28 | 2.58 |
| 310-helix | 34 | 2.40 |
| Mixed α/310-helix | 31.8 | 2.26 |
XRD, x-ray diffraction. See.text for discussion of errors.
Approximate alignment of the helix encompassing residues 1–10.
Figure 6.
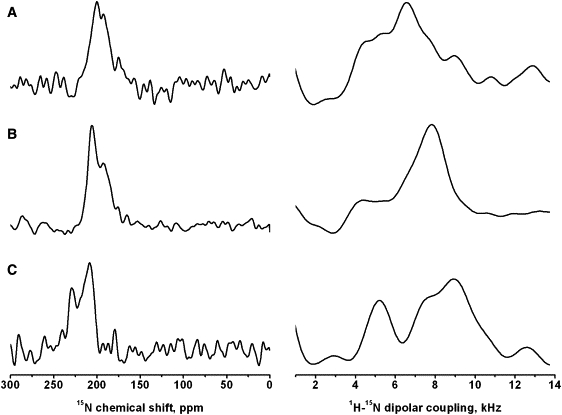
Selected slices of the 15N chemical shift and 1H-15N dipolar coupling dimensions are shown of the two dimensional experimental spectra of AmpA in di-C10:0-PC (A), AmpA in di-C12:0-PC (B), and alamethicin in POPC (C). The frequencies where the slices have been taken are indicated by the arrows on the corresponding two dimensional spectra in Figs. 4A, 5A, and 8A.
Since our data suggest that the secondary structure of the 15-residue transmembrane AmpA peptide encompasses a high degree of 310-helical structures in thin PC membranes, it is of interest to compare this result with the conformation of another peptide from the same family when inserted in membrane environments. Alamethicin is the peptaibol most extensively studied and the membrane alignment of this peptide has been characterized as a function of lipid composition and hydration using oriented CD (22). Furthermore, its membrane interactions have been intensely studied by a variety of techniques including oriented solid-state NMR spectroscopy. Using this latter approach, transmembrane alignments have been observed in POPC using one-dimensional cross-polarization 15N solid-state NMR spectroscopy of uniformly labeled alamethicin at P/L ratios of 1:15 and 1:237 (26). Furthermore, four alamethicin peptides, each labeled at a specific 15N position have been reconstituted into oriented DMPC at P/L ratio of 1:8 and studied using one- and two-dimensional solid-state NMR techniques (24,25). However, in both cases it was assumed that alamethicin adopts an α-helical conformation and the data were analyzed accordingly.
To better compare this with previous data (25), a proton-decoupled 15N spectrum of alamethicin in liquid crystalline DMPC bilayers at a P/L ratio of 1:8 was recorded under similar conditions as the alamethicin sample in POPC membranes (Fig. 7). Although both spectra are indicative of transmembrane alignments, the chemical shift range observed for alamethicin in DMPC (Fig. 7 A) suggests a more tilted helix when compared to that in POPC (Fig. 7 B).
Figure 7.
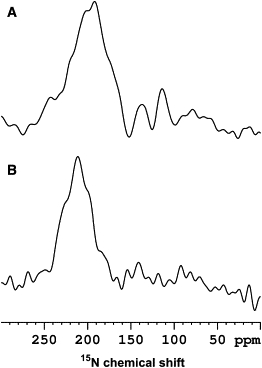
Proton-decoupled 15N cross-polarization NMR spectra of A. alamethicin in DMPC (peptide/lipid molar ratio 1:8 to directly compare to previous work (25)) recorded at 37°C, and (B) in POPC (1/100) recorded at 21°C.
To get insight into the secondary structure of alamethicin inserted into lipid bilayers, two-dimensional 15N-1H dipolar coupling, 15N chemical shift correlation spectra were recorded. The PISEMA spectrum of [U-15N]-alamethicin, reconstituted into POPC bilayers oriented with the normal parallel to the external magnetic field, is shown in Fig. 8 A. The spectrum is characterized by high signal intensities in the 15N chemical shift range of 200–235 ppm, which correlate with 15N-1H dipolar couplings of 4.5–10 kHz. A remarkable feature of this spectrum comes with the resonances at 5 kHz/210 ppm which represents ∼10–20% of the total spectral intensities observed in the amide region (4–11 kHz/195–235 ppm).
Figure 8.
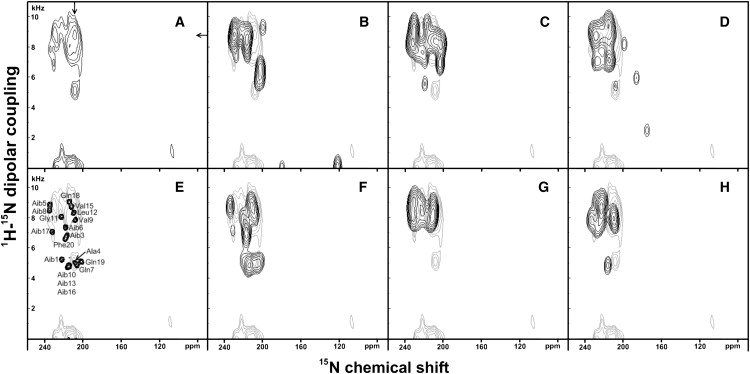
Experimental and simulated PISEMA spectra of 15N uniformly labeled alamethicin reconstituted into oriented POPC at a peptide/lipid ratio of 1/100. (A) Experimental spectrum. (B) Simulated spectrum of the x-ray diffraction (XRD) structure of alamethicin (pdb: 1AMT), unit A; (C) simulation of the XRD structure of alamethicin (pdb: 1AMT), unit B. (D) Simulation of the XRD structure of alamethicin (pdb: 1AMT), unit C. (E) The simulation F at reduced line broadening is shown to resolve the peaks and to monitor the origin of the simulated signal intensities. (F) Simulation of an ideal 310-helix (ϕ = −50°, ψ = −31°); (G) simulation of an ideal α-helix (ϕ = −65°, ψ = −45°), and (H) simulation of a mixed α-/310-helical model peptide (the first ten residues in α-helical and the last ten residues in 310-helical conformation). The simulation details are summarized in Table 3.
The results of the simulations of the PISEMA spectrum using the SIMMOL and SIMPSON (46) software are shown in Fig. 8, B–H. In a first step we compared how the known crystal structure at various alignments relative to the membrane normal fits the experimental spectrum. The unit cell of the alamethicin crystal structure contains three different conformations (5). In all of them the N-terminus is α-helical followed by a less well-ordered secondary structure at the C-terminus. The structures of units A and B exhibit a 25°–35° bent around Pro-14 (27), therefore, residues 1–11 (units A and B) and 1–8 (unit C), respectively, were taken into consideration to describe the alignment of the peptide relative to the membrane normal.
The best simulation is obtained with unit B aligned at 8° relative to the normal bilayer where the simulated intensity of residue 17 occurs in the 5 kHz/220 ppm region (Fig. 8 C) Although the overall fit of unit A seems less suitable to explain the experimental spectrum (Table 3) the simulated resonance of the Phe-20 residues comes very close to the experimental intensity at 1 kHz/107 ppm (Fig. 8 B). Clearly the α-helical model does not explain many of the features of the experimental spectrum such as the pronounced extension at 5 kHz/210 ppm (Fig. 8 G) which is, however, easily explained by 310 helical contributions (Fig. 8, E, F, and H). Therefore, the α-helical structure consistently gives the highest RMSD when the simulated and experimental spectra are compared to each other (Table 3). The 310-helical model simulation (Fig. 8 F) is of similar quality to the one obtained from the unit B of the crystal structure. The peak assignment for the 310-helical model simulation is shown in Fig. 8 E.
Table 3.
Summary on the simulations of the PISEMA spectra of alamethicin in POPC using different structural models
| Model | Tilt angle, degree | RMSD |
|---|---|---|
| XRD structure, unit A | 6/20∗ | 1.34 |
| XRD structure, unit B | 8/20∗ | 1.29 |
| XRD structure, unit C | 8.4† | 1.30 |
| α-helix | 6.3 | 1.32 |
| 310-helix | 14 | 1.29 |
| Mixed α/310-helix | 8.7 | 1.32 |
XRD: x-ray diffraction. See text for discussion of errors.
Approximate alignment of the helix encompassing residues 1–11/residues 12–19.
Approximate alignment of the helix encompassing the eight most N-terminal residues.
Discussion
Here we have investigated the interactions, topologies and structural details of ampullosporin A and alamethicin F50/7 (4), two 15- and 20-residue peptaibols. This class of peptides exhibits a high propensity to adopt helical conformations in the crystal, in solution or in membrane environments (http://www.cryst.bbk.ac.uk/peptaibol/structure.htm). Both peptides are hydrophobic, devoid of charged amino acid side chains, and carry an acetyl group at the N-terminus and a 1,2-amino alcohol at the C-terminus. A small number of polar side chains (Gln) and the slight perturbation of the regular polypeptide backbone due to the presence of prolines and glycine add some hydrophilic character to the peptide creating an amphipathic structure. However, the hydrophobic moments of these peptides are much lower than those of cationic peptide antibiotics such as the magainins or cecropins. Predicting the membrane alignment of peptaibols is less obvious, although these latter sequences have been experimentally observed to be located at the bilayer interface with stable helix orientations parallel to the membrane surface.
Therefore, in the past topological phase diagrams were established experimentally for alamethicin in bilayers of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC; di-C18:1-PC); 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC; di-(CH3)4C16:0-PC); or mixtures with 1,2-diphytanoyl-sn-glycero-3-phosphoethanolamine (DPhPE) bilayers as a function of lipid composition, hydration and peptide/lipid ratio using oriented CD spectroscopy (22,23). These studies show that both the reduction of membrane hydration and the increase of peptide concentration augment the tendency of alamethicin to adopt a membrane-inserted state. At concentrations below a lipid-dependent threshold the peptides have been found to be oriented parallel to the membrane surface whereas transmembrane alignments are observed at higher concentrations. This equilibrium has been explained with a membrane thinning effect when peptides penetrate the membrane interface at orientations parallel to the surface (52,53).
The membrane alignments of AmpA have so far not been investigated experimentally. In this study we therefore tested the alignment of AmpA as a function of membrane composition by reconstituting the 15N-labeled peptides into oriented PC bilayers of variable thickness and by measuring the resulting 15N chemical shift solid-state NMR spectra. Fig. 2, A–D illustrate that the peptide adopts in-plane alignments in liquid crystalline bilayers characterized by a hydrophobic thickness of >20 Å, but transmembrane alignments in bilayers of di-C10:0-PC or di-C12:0-PC. In the absence of peptides, the hydrophobic layer of these latter membranes amounts to 15.5 Å and 19.5 Å in width, respectively (54).
In the crystal structure the distance between Trp1 Cα and Aib10 Cα, i.e., the helical length of the peptide, is only 13.5 Å and therefore the peptide is too short to completely span even the thin membranes. The experimental spectra shown in Fig. 2, C and D indicates a transmembrane alignment. However, Figs. 4 and 5 strongly suggest that the AmpA helix in di-C10:0- and di-C12:0-PC encompass a high degree of 310-helical conformation, possibly mixed with α-helical contributions, which augments the length by a few Å, an effect that is partially compensated by the tilted alignment (ca. 30-35° in di-C12:0- and di-C10:0-PC membranes, Tables 1 and 2). It is estimated that at the given tilt angles a 310-helix of ∼8–12 residues is required to cross these di-C10:0 or di-C12:0-PC membranes.
A helical structure encompassing all AmpA residues should be long enough to even cross the di-C14:1-PC bilayer (20 Å (54)). The in-plane alignment obvious from Fig. 2 B suggests that the helix does not encompass the full length of the sequence and/or carries a bend somewhere in the central portion of the polypeptide chain. Such structural features would also explain some of the deviations between the simulated and the experimental spectra recorded in the thin membranes (Figs. 4 and 5). Furthermore, although the hydrophobic mismatch seems a major determinant, other energy contributions such as the preferential interactions with the aqueous environment of polar side chains (three glutamines) and exposure of the polypeptide backbone hold the AmpA helix in a surface alignment. Similar observations were made with the 16-residue zervamicin IIA peptide that exhibited a transition between transmembrane and in-plane alignments when the bilayer thickness was increased beyond di-C12:0-PC (26). In this case, structural studies later indicated that the peptide helix is bent by ∼20° at residue 10 and that this helix exhibits a considerably amphipathic character (55).
The mismatch is even more pronounced when AmpA is inserted into the POPC membranes. Notably, the fatty acyl chain composition of this lipid is considered to represent the average thickness of biological membranes and is characterized by a hydrophobic thickness of 26–27 Å (54). This exceeds even the overall length of AmpA in the crystal, as well as that of an idealized 15-residue α-helical conformation (22 Å in both cases).
Related observations were made when alamethicins were investigated by oriented solid-state NMR spectroscopy, where the peptides adopted transmembrane alignments in liquid crystalline POPC (26), DMPC (Fig. 7 A, (24,25)), or 1,2-dipalmitoyl-sn-glycero-3-phosphocholine membranes, but in-plane orientations in gel phase 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (56). In the crystal the length of the alamethicin helix, as estimated from the distance between Cα1 and Cα15, is ∼20 Å and falls short when compared to the hydrophobic thickness of POPC (∼26–27 Å). The helix can extend and include some of the more C-terminal Alm residues. It is also possible that the bilayer locally adapts its thickness (57), and/or the peptides elongate by changing conformation (58). Indeed, our data suggest that Alm in POPC adopts a mixed α-/310 helical conformation which is tilted by ∼10° (Fig. 8, Table 3). For 20 residues this translates into a hydrophobic length of ∼28–29 Å, and therefore it should match well with the hydrophobic thickness of the POPC or DMPC bilayers permitting the stable transmembrane insertion observed for this sequence (24–26). In a related manner, α-helical model peptides exhibit stable transmembrane alignments when the hydrophobic lengths of the helices are within ≤3 Å too short or ≤14 Å too long (54). Furthermore, when the membrane insertion of helical signal sequences was investigated, their tilt angles were a function of the bilayer hydrophobic thickness (39). The solid-state NMR data shown here do not provide direct information on the membrane penetration depth. However, such information has been obtained for alamethicin using electron paramagnetic resonance and fluorescence quenching techniques (59,60). In egg PC the C-terminus of Alm derivatives seems to reside in the aqueous phase ∼3–4 Å from the interface. The distance of the N-terminus was estimated to be ∼16 Å from the opposing interface using the first approach.
The one-dimensional 15N solid-state NMR spectra shown in Figs. 2 and 7 provide information on the membrane alignments of the helices but they do not offer sufficient details to analyze the structural features of the membrane-associated peptides. We therefore also recorded two-dimensional spectra revealing the correlations between the 15N amide chemical shift and 15N-1H the dipolar coupling. When uniformly labeled peptides are investigated by this technique, the tilt angles of α-helices can be determined with higher accuracy than from the chemical shift measurements alone by fitting the resulting “helical wheels” (Fig. S2). However, the experimental spectra of the peptaibols investigated here do not show the typical wheel-like features expected for an α-helix. We have therefore tested the structural constraints obtained from the separated local field spectra shown in Figs. 4, 5, and 8 by comparing them to several models of peptaibols when inserted in a transmembrane fashion in PC bilayers. The models include the crystal structures, ideal α- and 310-helices, as well as mixtures thereof. In a first step the helical tilt and pitch angles of each of the models was adjusted to obtain the best fit possible, then the spectral pattern was compared to the simulated spectra (Tables 1–3). None of the idealized models represents the experimental features completely and it becomes clear from this quantitative analysis that neither the crystal nor the pure α-helical structures can fit the experimental data well (Tables 1–3; RMSD > 1). However, for the AmpA experimental spectra the addition of 310-helical contributions improves the fit, and in the case of Alm this conformation together with one of the x-ray structures provide the best explanations for the data. The 310-helical conformations explain certain features of the spectra such as the lack of intensity at 170–185 ppm chemical shifts and in the region 6–9 kHz/150–180 ppm when AmpA is reconstituted into C10:0-PC (Fig. 4) or the unique intensity at 5kHz/210 ppm for alamethicin in POPC (Fig. 8). Based on the structural data, which indicate a rather stable α-helical N-terminus and a more flexible C-terminus (5–7,9,10), we have calculated the spectra of ideal mixed α-/310-helical structures where the secondary structures occur in separated domains (Fig. 8 H). Clearly many other combinations could explain the Alm spectrum as long as 2-4 residues in a 310 helical conformation are positioned in such a pitch angle to create the 5 kHz/210 ppm intensity and the remaining residues adopt a mixture of α- and 310-helical conformations to represent the intensity in the 6–10 kHz/200–230 ppm region.
In structural databases the 310-helical motif occurs at a frequency of only 10% within all helical regions and its average lengths encompasses in most cases only 3–4 residues (61). However, in a few protein structures 310-helices of up to ten residues in length have also been found (61). Furthermore, it is known that the α,α-tetrasubstituted Aib residues, of which alamethicin F50/7 carries nine and AmpA seven, promote this structural feature as the 310-helix provides additional space to accommodate the bulky side chains (reviewed in Crisma et al. (62)).
Although in micellar environments the N-terminal half of alamethicin is predominantly characterized by α-helical conformations (7), NMR structural studies and molecular modeling calculations suggest that in methanol or DOPC membranes several residues of alamethicin are involved in the formation of 310-helical conformations (11,63,64). In another NMR study in the same solvent alamethicin exhibits 310-helical conformations involving the very N-terminal residues (6), a structural feature that has also been identified for the nonadeca peptaibol trichorzianin TAVII (65).
The structure of the C-terminus of alamethicin is even more difficult to confirm from NMR experiments in sodium dodecyl sulfate micelles. For this region of the peptide a more unordered or flexible conformation was observed (7). But careful inspection of the nuclear Overhauser effect spectra leaves the possibility that a 310-helical conformation exists. It is interesting to note that x-ray grazing incidence scattering experiments indicate that alamethicins in di-C12:0-PC membranes exhibits a high degree of conformational and topological heterogeneity (66), where the diffraction patterns could be explained by a wide range of tilt angles of the x-ray crystal structure or by a mixture of helical conformations, or both (T. Salditt, Univeristy of Göttingen, personal communication, 2008). A mixed α-/310-helical structure of the N- and C-terminal domains have been observed when zervamicin IIB was investigated in the presence of DPC micelles (67). In addition to the mixed α-/310-structure, 7–8% of the more extended 27-structure was found in membrane mimicking solvents by pulsed electron-electron double resonance and double spin labeled zervamicin (68). For the 16-mer antiamoebin I peptaibol in methanol the first six residues and Aib8 have been found to be in fast exchange between right- and left-handed 310-helical conformations (69). The propensity of Aib to promote 310-helices has also been observed when the dodecameric Aib20 peptide has been studied by molecular modeling simulations (70).
The structural data for peptaibols in solvent or micellar systems indicate that the peptides exhibit highly dynamic regions where conformational exchange occurs. These conformational equilibriums could be shifted by additional interactions that favor the 310-helical structures such as hydrophobic mismatch in which the peptide is too short to span the hydrophobic bilayer thickness. In such cases an α- to 310-helix transition increases the length of the peptide by ∼0.5 Å per residue. Such a mechanism has been postulated (58) and, to our knowledge, the data presented in this study present the first experimental indication for such a structural transition in lipid bilayers.
Notably, the 15N NMR line widths are rather broad (Figs. 4, 5, and 8) in particular as the 31P solid-state NMR spectra indicate that the samples are well oriented (Fig. 3, A and B). This observation is suggestive that the peptaibols investigated here exhibit a high degree of conformational exchange. It is well possible that under the conditions tested several peptide conformations and aggregation states co-exist in the membrane. In fact several conformers have been revealed for alamethicin also by x-ray analysis and in NMR structural investigations in a variety of environments, including variable kink angles around Pro-14 or a highly flexible C-terminus (5–7,9,10). This might explain why a single model cannot explain all the spectral features consistently and why in all our simulations considerable deviations remain.
During the analysis of PISEMA spectra, potential errors may arise from the use of averaged 15N chemical shift tensors for all the residues although this is a common approach used in oriented solid-state NMR spectroscopy. Indeed it has been shown that the tensor elements of the 15N amide bond vary within a narrow range (<±10 ppm) when the standard amino acids are investigated (71–76) and a more systematic recent investigation illustrates how the 15N chemical shift tensor of the peptide bond varies with side chain structure, hydrogen bonding and secondary structure (77). Glycine is an exception as this amino acid exhibits an isotropic chemical shift position ∼10 ppm upfield due to its lack of an extended side chain (71,78). A similar effect is observed when the 15N tensor of α-aminobutyric acid (Aib), an unusual amino-acid produced by fungi, is analyzed. This amino acid carries an additional methyl group at the Cα carbon and its amide tensor is shifted downfield with regard to alanine, leucine or other “conventional” amino acids. The investigations of [U-15N]-alamethicin by fast magic angle spinning solid-state NMR spectroscopy indicate that the isotropic chemical shift of the 15N amide is different by ∼10–15 ppm (data not shown). Furthermore, the static powder pattern line shape of [U-15N]-alamethicin exhibits discontinuities at 51.3, 78.8, and 230 ppm (i.e., δiso = 110 ppm, δaniso = 105 ppm and η = 0.25; data not shown) which represents contributions from all residues. Therefore, during the simulations shown in this work different tensor values have been taken into account for Aib compared to those for the “standard” residues. This has allowed us to refine the analysis of the two-dimensional correlation spectra (Tables 1–3, Figs. 4, 5, and 8).
In the case of transmembrane peptides the most pronounced errors would occur from changes in the σ33 static tensor element, whereas variations of σ11 and σ22 have relatively little influence on the 15N chemical shift spectrum (see Fig. 3 in Bechinger and Sizun (36)). We therefore investigated the effects on our simulation results of changing σ33 within a 10 ppm range. Systematic changes in the size of the tensor elements have a predominant effect on the calculated tilt angles and to a lesser extent on the secondary structure analysis. For example, when the σ33 component is increased by 10 ppm the best fit for alamethicin, assuming an ideal α-helix, is obtained when the helix tilt angle is increased by 6.5°. On the other hand the σ33 value cannot be smaller than the highest experimental intensity observed limiting the tilt angle deviations in the other direction. For AmpA in di-C10:0-PC a decrease of σ33 by 10 ppm gives a 28° tilt for the 310-helix, while an increase of σ33 by 10 ppm leads to a 31°–35° tilt angle, a result quite similar to what we have observed when using the “standard values” for σ33. The situation would be slightly more complex if the σ33 tensor elements varied significantly among the Aibs or among the “conventional” amino acids of the peptide sequences, which could be an indication of strong conformational variations within the primary structure.
It should also be mentioned that in the crystal structures the peptide bonds show deviations from the planar conformation, therefore, considerable uncertainty exists about how to align the 15N amide tensor within the molecular frame. As a 10° shift of the angle between the least shielded axis of the chemical shift anisotropy tensor and the N-H bond results in a change of helix tilt angles by ∼7° this uncertainty can have an effect on the outcome of the simulation.
From these considerations it becomes clear that the choice of the tensor details can have a pronounced effect on the simulation details and the small differences in the RMSD values observed for the three Alm x-ray structures can become inverted when other parameters are used in the simulation. We have therefore based our conclusions on a number of unique spectral features rather than a solely automated selection procedure. As for the membrane-associated peptides, none of the models tested fits perfectly in the spectrum. Conformational exchange should be considered and conformation details remain to be adjusted. Specific and/or selective labeling of amino acids, which would permit the unambiguous assignment of peak intensities as well as for an in depth tensor analysis for each site, would allow for a detailed structure calculation. Such an extensive study requires new labeling strategies using the peptaibol producing fungi or laborious peptide synthesis and is beyond the scope of this initial structural analysis.
The data presented in this study support the concepts of channel formation and antimicrobial action of alamethicin through the oligomerization of transmembrane helical bundles, the helices being likely of mixed α-/310-helical conformation. In contrast, our data suggest that the AmpA peptides will reside at the membrane surface in PC bilayers representing the thickness of natural membranes (Fig. 2, A and B). However, the 15N solid-state NMR data also show that this latter peptide exhibits a sensitive topological equilibrium with transmembrane alignments which become obvious when the membrane thickness is varied (Fig. 2, C and D). It is therefore possible that environmental changes, such as the application of a transmembrane electric potential, induce realignment of AmpA at least for some of the helices, and thereby cause channel and antimicrobial activities through the formation of transmembrane helical bundles. Notably, one might speculate that the lower biological activities of the shorter peptaibols when compared to the dodecameric alamethicin are a result of the lower concentration in active transmembrane peptide configurations (30). Alternatively, the shorter peptaibols may disrupt the bilayer packing in a more detergent-like fashion similar to the propositions made for linear cationic peptide antibiotics or other mechanisms of antimicrobial action (79). However, at low peptide concentrations membrane openings through such mechanisms might be less likely to occur when compared to peptides that are characterized by stable transmembrane insertion.
Supplementary Material
Two figures and one table are available at www.biophys.org/biophysj/supplemental/S0006-3495(08)00003-9.
Supplementary Material
Acknowledgments
E.S.S. acknowledges the PhD fellowship from the French Government (BGF). Exchange visits were supported by the Groupement de Recherche Suprachem France-Russia. We are grateful to G.-M. Schwinger and A. Perner (HKI) for technical assistance and Tim Salditt for discussion. We thank the Institute for Supramolecular Chemistry, Strasbourg to host the Bechinger laboratory.
The financial support by The Netherlands Organization of Scientific Research, project NWO 047.017.034 to J.R. and the Agence Nationale de Recherche to B.B. is acknowledged. J.O. was supported by the National Sciences and Engineering Research Council of Canada.
References
- 1.Woolley G.A., Wallace B.A. Model ion channels: gramicidin and alamethicin. J. Membr. Biol. 1992;129:109–136. doi: 10.1007/BF00219508. [DOI] [PubMed] [Google Scholar]
- 2.Sansom M.S. Alamethicin and related peptaibols–model ion channels. Eur. Biophys. J. 1993;22:105–124. doi: 10.1007/BF00196915. [DOI] [PubMed] [Google Scholar]
- 3.Bechinger B. Structure and Functions of Channel-Forming Polypeptides: Magainins, Cecropins, Melittin and Alamethicin. J. Membr. Biol. 1997;156:197–211. doi: 10.1007/s002329900201. [DOI] [PubMed] [Google Scholar]
- 4.Leitgeb B., Szekeres A., Manczinger L., Vagvolgyi C., Kredics L. The history of alamethicin: a review of the most extensively studied peptaibol. Chem. Biodivers. 2007;4:1027–1051. doi: 10.1002/cbdv.200790095. [DOI] [PubMed] [Google Scholar]
- 5.Fox R.O., Richards F.M. A voltage-gated ion channel model inferred from the crystal structure of alamethicin at 1.5-A resolution. Nature. 1982;300:325–330. doi: 10.1038/300325a0. [DOI] [PubMed] [Google Scholar]
- 6.Yee A.A., Babiuk R., O'Neil J.D.J. The conformation of an alamethicin in metahnol by multinuclear NMR spectroscopy and distance geometry simulated annealing. Biopolymers. 1995;36:781–792. [Google Scholar]
- 7.Franklin J.C., Ellena J.F., Jayasinghe S., Kelsh L.P., Cafiso D.S. Structure of micelle-associated alamethicin from 1H NMR. Evidence for conformational heterogeneity in a voltage-gated peptide. Biochemistry. 1994;33:4036–4045. doi: 10.1021/bi00179a032. [DOI] [PubMed] [Google Scholar]
- 8.Spyracopoulos L., Yee A.A., O'Neil J.D.J. Backbone dynamics of an alamethicin in methanol and aqueous detergent solution determined by heteronuclear H-1-N-15 NMR spectrsocopy. J. Biomol. NMR. 1996;7:283–294. doi: 10.1007/BF00200430. [DOI] [PubMed] [Google Scholar]
- 9.North C.L., Franklin J.C., Bryant R.G., Cafiso D.S. Molecular flexibility demonstrated by paramagnetic enhancements of nuclear relaxation. Application to alamethicin: a voltage- gated peptide channel. Biophys. J. 1994;67:1861–1866. doi: 10.1016/S0006-3495(94)80667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yee A., Szymczyna B., O'Neil J.D. Backbone dynamics of detergent-solubilized alamethicin from amide hydrogen exchange measurements. Biochemistry. 1999;38:6489–6498. doi: 10.1021/bi982959t. [DOI] [PubMed] [Google Scholar]
- 11.Sessions R.B., Gibbs N., Dempsey C.E. Hydrogen bonding in helical polypeptides from molecular dynamics simulations and amide hydrogen exchange analysis: alamethicin and melittin in methanol. Biophys. J. 1998;74:138–152. doi: 10.1016/S0006-3495(98)77775-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tieleman D.P., Hess B., Sansom M.S. Analysis and evaluation of channel models: simulations of alamethicin. Biophys. J. 2002;83:2393–2407. doi: 10.1016/s0006-3495(02)75253-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon L.G., Haydon D.A. The unit conductance channel of alamethicin. Biochim. Biophys. Acta. 1972;255:1014–1018. doi: 10.1016/0005-2736(72)90415-4. [DOI] [PubMed] [Google Scholar]
- 14.Boheim G. Statistical analysis of alamethicin channels in black lipid membranes. J. Membr. Biol. 1974;19:277–303. doi: 10.1007/BF01869983. [DOI] [PubMed] [Google Scholar]
- 15.Vedovato N., Rispoli G. A novel technique to study pore-forming peptides in a natural membrane. Eur. Biophys. J. 2007;36:771–778. doi: 10.1007/s00249-007-0152-4. [DOI] [PubMed] [Google Scholar]
- 16.Pispisa B., Stella L., Mazzuca C., Venanzi M. Trichogin topology and activity in model membranes as determined by fluorescence spectroscopy. In: Geddes C.D., Lakowicz J.R., editors. Reviews in Fluorescence. Springer; New York: 2006. pp. 47–70. [Google Scholar]
- 17.Kropacheva T.N., Raap J. Enzymatic reaction in a vesicular microreactor: peptaibol-facilitated substrate transport. Chem. Biodivers. 2007;4:1388–1394. doi: 10.1002/cbdv.200790118. [DOI] [PubMed] [Google Scholar]
- 18.Hanke W., Boheim G. The lowest conductance state of the alamethicin pore. Biochim. Biophys. Acta. 1980;596:456–462. doi: 10.1016/0005-2736(80)90134-0. [DOI] [PubMed] [Google Scholar]
- 19.Sansom M.S.P. The Biophysics of Peptide Models of Ion Channels. Prog. Biophys. Mol. Biol. 1991;55:139–235. doi: 10.1016/0079-6107(91)90004-c. [DOI] [PubMed] [Google Scholar]
- 20.Taylor R.J., de Levie R. “Reversed” alamethicin conductance in lipid bilayers. Biophys. J. 1991;59:873–879. doi: 10.1016/S0006-3495(91)82299-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He K., Ludtke S.J., Heller W.T., Huang H.W. Mechnaism of alamethicin insertion into lipid bilayers. Biophys. J. 1996;71:2669–2679. doi: 10.1016/S0006-3495(96)79458-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang H.W., Wu Y. Lipid-alamethicin interactions influence alamethicin orientation. Biophys. J. 1991;60:1079–1087. doi: 10.1016/S0006-3495(91)82144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen F.Y., Lee M.T., Huang H.W. Sigmoidal concentration dependence of antimicrobial peptide activities: a case study on alamethicin. Biophys. J. 2002;82:908–914. doi: 10.1016/S0006-3495(02)75452-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.North C.L., Barranger-Mathys M., Cafiso D.S. Membrane orientation of the N-terminal segment of alamethicin determined by solid-state 15N NMR. Biophys. J. 1995;69:2392–2397. doi: 10.1016/S0006-3495(95)80108-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bak M., Bywater R.P., Hohwy M., Thomsen J.K., Adelhorst K. Conformation of alamethicin in oriented phospholipid bilayers determined by N-15 solid-state nuclear magnetic resonance. Biophys. J. 2001;81:1684–1698. doi: 10.1016/S0006-3495(01)75822-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bechinger B., Skladnev D.A., Ogrel A., Li X., Swischewa N.V. 15N and 31P solid-state NMR investigations on the orientation of zervamicin II and alamethicin in phosphatidylcholine membranes. Biochemistry. 2001;40:9428–9437. doi: 10.1021/bi010162n. [DOI] [PubMed] [Google Scholar]
- 27.Kronen M., Gorls H., Nguyen H.H., Reissmann S., Bohl M. Crystal structure and conformational analysis of ampullosporin A. J. Pept. Sci. 2003;9:729–744. doi: 10.1002/psc.495. [DOI] [PubMed] [Google Scholar]
- 28.Snook C.F., Woolley G.A., Oliva G., Pattabhi V., Wood S.F. The structure and function of antiamoebin I, a proline-rich membrane-active polypeptide. Structure. 1998;6:783–792. doi: 10.1016/s0969-2126(98)00079-3. [DOI] [PubMed] [Google Scholar]
- 29.Kropacheva T.N., Salnikov E.S., Nguyen H.H., Reissmann S., Yakimenko Z.A. Membrane association and activity of 15/16-membered peptide antibiotics: zervamicin IIB, ampullosporin A and antiamoebin I. Biochim. Biophys. Acta. 2005;1715:6–18. doi: 10.1016/j.bbamem.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 30.Ritzau M., Heinze S., Dornberger K., Berg A., Fleck W. Ampullosporin, a new peptaibol-type antibiotic from Sepedonium ampullosporum HKI-0053 with neuroleptic activity in mice. J. Antibiot. (Tokyo) 1997;50:722–728. doi: 10.7164/antibiotics.50.722. [DOI] [PubMed] [Google Scholar]
- 31.Kronen M., Kleinwachter P., Schlegel B., Hartl A., Grafe U. Ampullosporines B, C,D,E1,E2,E3 and E4 from Sepedonium ampullosporum HKI-0053: structures and biological activities. J. Antibiot. (Tokyo) 2001;54:175–178. doi: 10.7164/antibiotics.54.175. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen H.-H., Imhof D., Kronen M., Schlegel B., Hörtl U. Synthesis and biological evaluation of analogues of the peptaibol ampullosporin A. J. Med. Chem. 2002;45:2781–2787. doi: 10.1021/jm0208018. [DOI] [PubMed] [Google Scholar]
- 33.Chugh J.K., Wallace B.A. Peptaibols: models for ion channels. Biochem. Soc. Trans. 2001;29:565–570. doi: 10.1042/bst0290565. [DOI] [PubMed] [Google Scholar]
- 34.Wang J., Denny J., Tian C., Kim S., Mo Y. Imaging membrane protein helical wheels. J. Magn. Reson. 2000;144:162–167. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
- 35.Marassi F.M., Opella S.J. A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Reson. 2000;144:150–155. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bechinger B., Sizun C. Alignment and structural analysis of membrane polypeptides by 15N and 31P solid-state NMR spectroscopy. Concepts Magn. Reson. 2003;18A:130–145. [Google Scholar]
- 37.Aisenbrey C., Sizun C., Koch J., Herget M., Abele U. Structure and dynamics of membrane-associated ICP47, a viral inhibitor of the MHC I antigen-processing machinery. J. Biol. Chem. 2006;281:30365–30372. doi: 10.1074/jbc.M603000200. [DOI] [PubMed] [Google Scholar]
- 38.Ramamoorthy A., Thennarasu S., Lee D.K., Tan A., Maloy L. Solid-state NMR investigation of the membrane-disrupting mechanism of antimicrobial peptides MSI-78 and MSI-594 derived from magainin 2 and melittin. Biophys. J. 2006;91:206–216. doi: 10.1529/biophysj.105.073890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramamoorthy A., Kandasamy S.K., Lee D.K., Kidambi S., Larson R.G. Structure, topology, and tilt of cell-signaling peptides containing nuclear localization sequences in membrane bilayers determined by solid-state NMR and molecular dynamics simulation studies. Biochemistry. 2007;46:965–975. doi: 10.1021/bi061895g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aisenbrey C., Bechinger B. Tilt and rotational pitch angles of membrane-inserted polypeptides from combined 15N and 2H solid-state NMR spectroscopy. Biochemistry. 2004;43:10502–10512. doi: 10.1021/bi049409h. [DOI] [PubMed] [Google Scholar]
- 41.Bertelsen K., Pedersen J.M., Rasmussen B.S., Skrydstrup T., Nielsen N.C. Membrane-bound conformation of peptaibols with methyl-deuterated alpha-amino isobutyric acids by 2H magic angle spinning solid-state NMR spectroscopy. J. Am. Chem. Soc. 2007;129:14717–14723. doi: 10.1021/ja0749690. [DOI] [PubMed] [Google Scholar]
- 42.Ramamoorthy A., Wei Y., Lee D. PISEMA Solid-State NMR Spectroscopy. Annual Reports on NMR Spectroscopy. 2004;52:1–52. [Google Scholar]
- 43.Yee A.A., O'Neil J.D. Uniform 15N labeling of a fungal peptide: the structure and dynamics of an alamethicin by 15N and 1H NMR spectroscopy. Biochemistry. 1992;31:3135–3143. doi: 10.1021/bi00127a014. [DOI] [PubMed] [Google Scholar]
- 44.Bechinger B., Opella S.J. Flat-Coil Probe for NMR Spectroscopy of Oriented Membrane Samples. J. Magn. Reson. 1991;95:585–588. [Google Scholar]
- 45.Wu C.H., Ramamoorthy A., Opella S.J. High-resolution heteronuclear dipolar solid-state NMR spectroscopy. J. Magn. Reson. 1994;109:270–272. [Google Scholar]
- 46.Bak M., Rasmussen J.T., Nielsen N.C. SIMPSON: A general simulation program for solid-state NMR spectroscopy. J. Magn. Reson. 2000;147:296–330. doi: 10.1006/jmre.2000.2179. [DOI] [PubMed] [Google Scholar]
- 47.Wu C.H., Ramamoorthy A., Gierasch L.M., Opella S.J. Simultaneous Characterization of the Amide 1H Chemical Shift, 1H-15N Dipolar, and 15N Chemical Shift Interaction Tensors in a Peptide Bond by Three-Dimensional Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 1995;117:6148–6149. [Google Scholar]
- 48.Aisenbrey C., Bechinger B. Investigations of peptide rotational diffusion in aligned membranes by 2H and 15N solid-state NMR spectroscopy. J. Am. Chem. Soc. 2004;126:16676–16683. doi: 10.1021/ja0468675. [DOI] [PubMed] [Google Scholar]
- 49.Prongidi-Fix L., Bertani P., Bechinger B. The membrane alignment of helical peptides from non-oriented 15N chemical shift solid-state NMR spectroscopy. J. Am. Chem. Soc. 2007;129:8430–8431. doi: 10.1021/ja072668k. [DOI] [PubMed] [Google Scholar]
- 50.Lambotte S., Jasperse P., Bechinger B. Orientational distribution of alpha-helices in the colicin B and E1 channel domains: A one- and two dimensional 15N solid-state NMR investigation in uniaxially aligned phospholipid bilayers. Biochemistry. 1998;37:16–22. doi: 10.1021/bi9724671. [DOI] [PubMed] [Google Scholar]
- 51.Frye J., Albert A.D., Selinsky B.S., Yeagle P.L. Cross Polarization P-31 Nuclear Magnetic Resonance of Phospholipids. Biophys. J. 1985;48:547–552. doi: 10.1016/S0006-3495(85)83812-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang H.W. Molecular mechanism of antimicrobial peptides: The origin of cooperativity. Biochim. Biophys. Acta. 2006;1758:1292–1302. doi: 10.1016/j.bbamem.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 53.Salditt T., Li C., Spaar A. Structure of antimicrobial peptides and lipid membranes probed by interface-sensitive X-ray scattering. Biochim. Biophys. Acta. 2006;1758:1483–1498. doi: 10.1016/j.bbamem.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 54.Harzer U., Bechinger B. The alignment of lysine-anchored membrane peptides under conditions of hydrophobic mismatch: A CD, 15N and 31P solid-state NMR spectroscopy investigation. Biochemistry. 2000;39:13106–13114. doi: 10.1021/bi000770n. [DOI] [PubMed] [Google Scholar]
- 55.Shenkarev Z.O., Paramonov A.S., Balashova T.A., Yakimenko Z.A., Baru M.B. High stability of the hinge region in the membrane-active peptide helix of zervamicin: paramagnetic relaxation enhancement studies. Biochem. Biophys. Res. Commun. 2004;325:1099–1105. doi: 10.1016/j.bbrc.2004.10.115. [DOI] [PubMed] [Google Scholar]
- 56.Salnikov E.S. University Louis Pasteur Strasbourg and University of Novosibirsk; 2007. Structural studies of membrane-modifying peptaibol antibiotic peptides by Pulsed EPR and Solid State NMR spectroscopy. [Google Scholar]
- 57.Periole X., Huber T., Marrink S.J., Sakmar T.P. G protein-coupled receptors self-assemble in dynamics simulations of model bilayers. J. Am. Chem. Soc. 2007;129:10126–10132. doi: 10.1021/ja0706246. [DOI] [PubMed] [Google Scholar]
- 58.Killian J.A. Hydrophobic mismatch between proteins and lipids in membranes. Biochim. Biophys. Acta. 1998;1376:401–415. doi: 10.1016/s0304-4157(98)00017-3. [DOI] [PubMed] [Google Scholar]
- 59.Barranger-Mathys M., Cafiso D.S. Membrane structure of voltage-gated channel forming peptides by site-directed spin-labeling. Biochemistry. 1996;35:498–505. doi: 10.1021/bi951985d. [DOI] [PubMed] [Google Scholar]
- 60.Stella L., Burattini M., Mazzuca C., Palleschi A., Venanzi M. Alamethicin interaction with lipid membranes: a spectroscopic study on synthetic analogues. Chem. Biodivers. 2007;4:1299–1312. doi: 10.1002/cbdv.200790111. [DOI] [PubMed] [Google Scholar]
- 61.Pal L., Basu G. Novel protein structural motifs containing two-turn and longer 3.10 helices. Protein Eng. 1999;12:811–814. doi: 10.1093/protein/12.10.811. [DOI] [PubMed] [Google Scholar]
- 62.Crisma M., Formaggio F., Moretto A., Toniolo C. Peptide helices based on alpha-amino acids. Biopolymers. 2006;84:3–12. doi: 10.1002/bip.20357. [DOI] [PubMed] [Google Scholar]
- 63.Dempsey C.E., Handcock L.J. Hydrogen bond stabilities in membrane-reconstituted alamethicin from amide-resolved hydrogen-exchange measurements. Biophys. J. 1996;70:1777–1788. doi: 10.1016/S0006-3495(96)79741-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gibbs N., Sessions R.B., Williams P.B., Dempsey C.E. Helix bending in alamethicin: molecular dynamics simulations and amide hydrogen exchange in methanol. Biophys. J. 1997;72:2490–2495. doi: 10.1016/S0006-3495(97)78893-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Condamine E., Rebuffat S., Prigent Y., Segalas I., Bodo B. Three-dimensional structure of the ion-channel forming peptide trichorzianin TA VII bound to sodium dodecyl sulfate micelles. Biopolymers. 1998;46:75–88. doi: 10.1002/(SICI)1097-0282(199808)46:2<75::AID-BIP3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 66.Spaar A., Munster C., Salditt T. Conformation of peptides in lipid membranes studied by x-ray grazing incidence scattering. Biophys. J. 2004;87:396–407. doi: 10.1529/biophysj.104.040667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shenkarev Z.O., Balashova T.A., Efremov R.G., Yakimenko Z.A., Ovchinnikova T.V. Spatial structure of zervamicin IIB bound to DPC micelles: implications for voltage-gating. Biophys. J. 2002;82:762–771. doi: 10.1016/S0006-3495(02)75438-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Milov A.D., Samoilova M.I., Tsvetkov Y.D., Jost M., Peggion C. Supramolecular structure of self-assembling alamethicin analog studied by ESR and PELDOR. Chem. Biodivers. 2007;4:1275–1298. doi: 10.1002/cbdv.200790110. [DOI] [PubMed] [Google Scholar]
- 69.Shenkarev Z.O., Paramonov A.S., Nadezhdin K.D., Bocharov E.V., Kudelina I.A. Antiamoebin I in methanol solution: rapid exchange between right-handed and left-handed 3(10)-helical conformations. Chem. Biodivers. 2007;4:1219–1242. doi: 10.1002/cbdv.200790106. [DOI] [PubMed] [Google Scholar]
- 70.Breed J., Kerr I.D., Sankararamakrishnan R., Sansom M.S. Packing interactions of Aib-containing helices: molecular modeling of parallel dimers of simple hydrophobic helices and of alamethicin. Biopolymers. 1995;35:639–655. doi: 10.1002/bip.360350610. [DOI] [PubMed] [Google Scholar]
- 71.Oas T.G., Hartzell C.J., Dahlquist F.W., Drobny G.P. The Amide 15N Chemical Shift Tensors of Four Peptides Determined from 13C Dipole-Coupled Chemical Shift Powder Patterns. J. Am. Chem. Soc. 1987;109:5962–5966. [Google Scholar]
- 72.Hartzell C.J., Whitfield M., Oas T.G., Drobny G.P. Determination of the 15N and 13C Chemical Shift Tensors of L- [13C]Alanyl-L-[15N]alanine from the Dipole-Coupled Powder Patterns. J. Am. Chem. Soc. 1987;109:5966–5969. [Google Scholar]
- 73.Bechinger B., Kim Y., Chirlian L.E., Gesell J. Orientations of amphipathic helical peptides in membrane bilayers determined by solid- state NMR spectroscopy. J. Biomol. NMR. 1991;1:167–173. doi: 10.1007/BF01877228. [DOI] [PubMed] [Google Scholar]
- 74.Lazo N.D., Hu W., Cross T.A. Low-temperature solid-state 15N NMR characterization of polypeptide backbone librations. J. Magn. Reson. 1995;107:43–50. doi: 10.1006/jmrb.1995.1056. [DOI] [PubMed] [Google Scholar]
- 75.Lee D.K., Ramamoorthy A. A simple one-dimensional solid-state NMR method to characterize the nuclear spin interaction tensors associated with the peptide bond. J. Magn. Reson. 1998;133:204–206. doi: 10.1006/jmre.1998.1442. [DOI] [PubMed] [Google Scholar]
- 76.Lee D.K., Wei Y., Ramamoorthy A. A two-dimensional magic-angle decoupling and magic-angle turning solid-state NMR method: An application to study chemical shift tensors from peptides that are nonselectively labeled with 15N isotope. J. Phys. Chem. B. 2001;105:4752–4762. [Google Scholar]
- 77.Poon A., Birn J., Ramamoorthy A. How does an amide-N-15 chemical shift tensor vary in peptides? J. Phys. Chem. B. 2004;108:16577–16585. doi: 10.1021/jp0471913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shoji A., Ozaki T., Fujito T., Deguchi K., Ando I. 15N chemical shift tensors and conformation of solid polypeptides containing 15N-labeled glycine residue by 15N NMR. J. Mol. Struct. 1998;441:251–266. [Google Scholar]
- 79.Bechinger B., Lohner K. Detergent-like action of linear cationic membrane-active antibiotic peptides. Biochim. Biophys. Acta. 2006;1758:1529–1539. doi: 10.1016/j.bbamem.2006.07.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.