

Abstract
Postreplication DNA mismatch repair is initiated by the eukaryotic protein MSH2-MSH6 or the prokaryotic protein MutS, both showing overall conserved structure and functionality. Crystal structures of MSH2-MSH6 and MutS bound to the mismatch DNA reveal a closed architecture of the clamp and the lever domains exhibiting strong contacts with the bent DNA backbone. Long molecular dynamics simulations of the human MSH2-MSH6 protein in the absence of a DNA show an altered conformation of the protein that reflects the protein's state before binding to DNA. The clamp and the lever domains of both MSH6 and MSH2 open in an asymmetric and dramatic fashion. The opening of the clamp and the lever domains in the absence of DNA is coupled to changes in the ATPase domains, which explains the experimentally observed diminished ATPase activity in DNA-free MSH2-MSH6 and illustrates the allosteric coupling between DNA binding and ATPase activity.
In postreplication DNA repair, eukaryotic MSH2-MSH6 and prokaryotic MutS are responsible for recognizing DNA base-base mismatches or base insertions/deletions, and initiating repair (1,2). Throughout the mismatch recognition cycle, MSH2-MSH6 and MutS adopt different functional conformations, out of which only the mismatch-bound form has been solved in recent crystal structures (3,4). All of these structures have indicated that the protein binds to the mismatch site through a conserved motif of domain I of chain MSH6 or MutS S1, whereas the clamps (domain IV) interact with the DNA backbone to stabilize a highly bent DNA structure. Several kinetics studies and a recent normal-mode-analysis-based study have indicated the existence of other conformational states before and after mismatch binding (5–7). In particular, the structure of MSH2-MSH6 or MutS in the absence of any DNA is presumed to be different from the known mismatch-bound structure, as evidenced by data from small-angle x-ray scattering (8).
Here, the atomic details of the structural change upon DNA dissociation from MSH2-MSH6 are described, based on long molecular dynamics (MD) simulations of MSH2(1-855)-MSH6(362-1335) with and without mismatch DNA. One simulation over 170 ns was started from the crystal structure 2O8B (4) with completed missing fragments but without the DNA present in the crystal structure. For comparison, a second simulation over 140 ns was carried out with the DNA from the crystal structure containing a G:T mismatch. In both simulations, full explicit solvent and neutralizing counterions were present, but nucleotides bound to the ATPase domains were not included. Further simulation details are given as Supporting Material.
Fig. 1 compares the final conformations of the DNA-free and mismatch-bound MSH2-MSH6 structures at the end of their respective simulations. Fig. 2 shows the Cα root mean-square deviation for the whole protein and selected domains. It can be seen that although the mismatch-bound protein retains a conformation close to the crystal structure, the DNA-free structure undergoes a drastic conformational change. The main result is an opening of the cavity where the DNA is bound by the clamp domains in the crystal structure. The opening is achieved by a large conformational change in the clamp and lever domains. The opening motion of the clamp and lever domains proceeds in opposite directions in MSH6 and MSH2 and leads to an asymmetric open cavity (domains indicated by shades of red and cyan in Fig. 1, a and b). The opening of the MSH6 clamp is accompanied by a partial loss of secondary structure elements (in particular, the β-sheet and -turn involving residues 968–980), which may require the presence of DNA to be stabilized. In the MSH2 clamp, the two β-sheets are maintained intact but assume a different relative orientation in the absence of the DNA than that observed in the G:T-bound protein. The clamps do not move significantly until after 40 ns but then quickly open up from an initial separation distance (between the center of geometry of the two clamps) of ∼17 Å to 60 Å at 100 ns. The lever domains also separate significantly along with the clamps but they do not lose their secondary structure, which is an indication of the remarkable plasticity of the MSH2-MSH6 structure. Smaller changes are seen in other domains as a result of DNA removal from the complex. In particular, the second cavity visible in the DNA-bound structure becomes mostly occluded as a result of a contraction involving domain II of both chains and domain I of MSH2 (Fig. 1).
Figure 1.
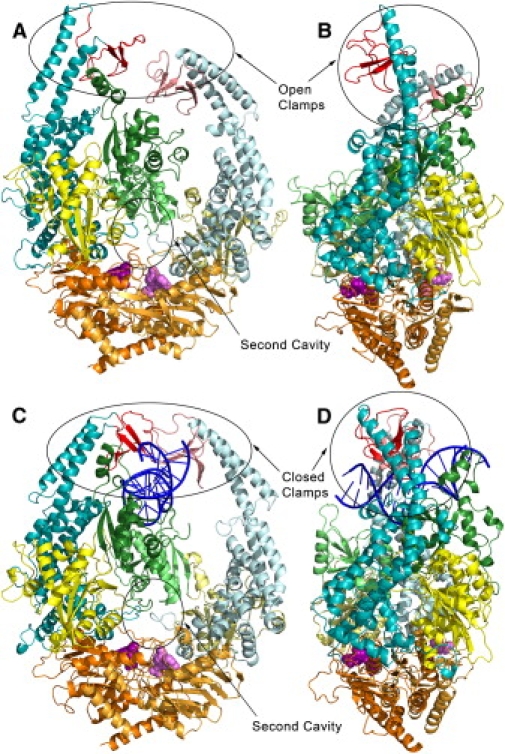
Final simulated conformations of DNA-free (A and B) and G:T mismatch-bound MSH2-MSH6 (C and D) aligned to crystal structure. Protein domains are in green (I), yellow (II), cyan (III), red (IV) and orange (V). DNA and ADP projected from the cystal structure are shown in blue and magenta, respectively. Darker shades correspond to MSH6 and lighter shades to MSH2.
Figure 2.
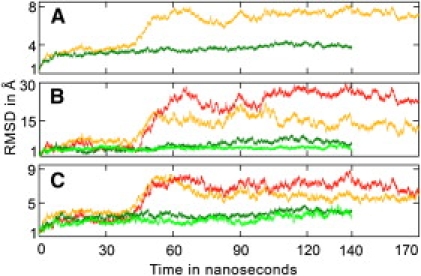
Root mean-square deviation (RMSD) of Cα atoms of (A) DNA-free (orange) and G:T-bound MSH2-MSH6 (dark green); (B) only clamp and; (C) only lever domain of DNA-free MSH6 (orange), DNA-free MSH2 (red), G:T-bound MSH6 (dark green) and G:T-bound MSH2 (light green).
Two ATPase sites, one situated in the domain V of MSH2 and one in that of MSH6, play a key role in mismatch recognition and repair initiation. These ATPase sites show a sequential pattern of ATP binding and hydrolysis (9) and variations of nucleotide binding affinities and hydrolysis rates according to the state of MSH2-MSH6 enzyme. It is, therefore, especially interesting how interactions with DNA are coupled to changes in the C-terminal ATPase sites (5,6). MSH2-MSH6 shows overall low ATPase activity in the absence of any DNA, but a burst of ATP hydrolysis upon DNA binding, apparently caused by MSH6 ATPase activity (5). Before mismatch recognition, MSH2 has a high affinity for ADP and low ATPase activity (5). Upon mismatch recognition, there are further coupled changes in the ATPase sites that stall ATP hydrolysis in MSH6 and increase ATP-binding affinity in the MSH2 ATPase domain (1,5–7,10). All of these observations suggest strong long-range allostery between the N-terminal DNA binding domains and the C-terminal ATPase sites.
The MD simulations reported here allow the effect of DNA binding on the ATPase sites to be examined. The simulations do indeed reveal subtle conformational changes in the ATPase sites that appear to be correlated to the removal of the DNA. Each ATPase site consists of a Walker A motif that binds the nucleotide phosphates and a Walker B motif that coordinates the interaction of the nucleotide γ-phosphate and a divalent cation, typically Mg2+, to catalyze the hydrolysis reaction (11). The simulations did not contain nucleotides, so conformational changes as a result of DNA dissociation can be observed more clearly. However, the approximate positions of bound nucleotides and the Mg2+ ion can be obtained from superposition of the ATPase domains to the crystal structure. Fig. 3 shows the comparison between the final conformation of the mismatch-bound and the DNA-free simulations, along with the aligned ADP-Mg+2 ligand complex. In the DNA-free MSH6 ATPase structure, the carboxylic group of the Walker B E1214 residue that is involved in coordinating Mg+2 and is assumed to play a key role during hydrolysis has moved away from the nucleotide-Mg+2 complex, which would suggest a molecular interpretation of the experimentally observed diminished ATP hydrolysis rate in the absence of DNA. The motion of E1214 is the direct result of rotation and translation of the entire β-sheet preceding the Walker B loop, which can be traced back to changes in the lever arm that communicate the opening of the clamp domains along the MSH2-MSH6 complex in the absence of DNA.
Figure 3.
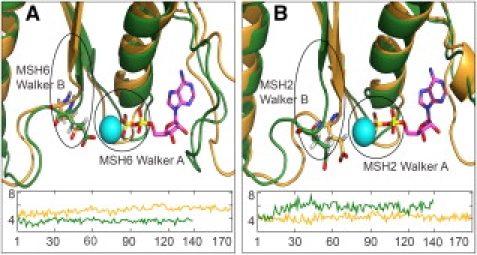
Close-up view of the aligned ADP-Mg+2 complex in the MSH2 (A) and MSH6 (B) ATPase sites of the final simulated conformation in the absence of DNA (orange cartoon) and with G:T mismatch DNA (green cartoon). Walker B loop residue E1214 in MSH6 and E749 in MSH2 are shown as sticks. Distance between Cδ of E1214/749 and projected Mg+2 versus simulation time is shown in the inset for DNA-free (orange) and G:T-bound (dark green) protein.
In the MSH2 ATPase domain, the effect of clamp domain opening appears to be asymmetrically correlated to the changes in the MSH6 ATPase site. The analogous glutamate E749 actually moves closer to the projected Mg2+ binding site in the absence of DNA but moves away in the presence of DNA (Fig. 3). The latter observation is consistent with the experimentally observed lack of ATPase activity in MSH2 until after repair has been initiated. However, the average distance of E749 to Mg2+ in the absence of DNA remains longer than the corresponding distance in MSH6 when bound to DNA, suggesting that MSH2 may also have reduced ATPase activity in the DNA-free state. Furthermore, the flexible loop that interacts with the adenine ring of the nucleotides occludes the binding site for most of the time in MSH2, which would disfavor ATP or ADP binding in the MSH2 ATPase site of the DNA-free enzyme.
The MD simulations presented here offer first atomic-level insight into the structural changes in MSH2-MSH6 upon DNA binding and suggest, in particular, how such changes may result in the functionally important differential modulation of MSH2 and MSH6 ATPase activity. In particular, the simulations suggest that only the MSH6 ATPase becomes catalytically active upon DNA binding as a result of subtle conformational changes in the Walker B motif. The observation of the enzyme opening without DNA after tens of nanoseconds underscores the need for simulations on at least 100 ns timescales.
It is our hope that the results reported here will stimulate further experimental studies to confirm our findings. At the same time, the simulations reported here provide a template for additional computational studies on the coupling between DNA mismatch binding and ATPase activity during the repair cycle of MSH2-MSH6 and MutS.
Acknowledgments
The authors thank Sean M. Law for valuable discussions.
Financial support from National Science Foundation CAREER grant 0447799 and the Alfred P. Sloan Foundation is acknowledged, as well as computational resources through Teragrid (TG-MCB090003) and the High Performance Computing Center at Michigan State University.
Supporting Material
References and Footnotes
- 1.Kunkel T.A., Erie D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 2.Modrich P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006;281:30305–30309. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamers M.H., Perrakis A., Enzlin J.H., Winterwerp H.H.K., de Wind N. The crystal structure of DNA mismatch repair protein MutS binding to a G center dot T mismatch. Nature. 2000;407:711–717. doi: 10.1038/35037523. [DOI] [PubMed] [Google Scholar]
- 4.Warren J.J., Pohlhaus T.J., Changela A., Iyer R.R., Modrich P.L. Structure of the human MutS alpha DNA lesion recognition complex. Mol. Cell. 2007;26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 5.Mazur D.J., Mendillo M.L., Kolodner R.D. Inhibition of Msh6 ATPase activity by mispaired DNA induces a Msh2(ATP)-Msh6(ATP) state capable of hydrolysis-independent movement along DNA. Mol. Cell. 2006;22:39–49. doi: 10.1016/j.molcel.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 6.Jacobs-Palmer E., Hingorani M.M. The effects of nucleotides on MutS-DNA binding kinetics clarify the role of MutS ATPase activity in mismatch repair. J. Mol. Biol. 2007;366:1087–1098. doi: 10.1016/j.jmb.2006.11.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukherjee S., Law S.M., Feig M. Deciphering the Mismatch Recognition Cycle in MutS and MSH2–MSH6 using Normal-Mode Analysis. Biophys. J. 2009;96:1707–1720. doi: 10.1016/j.bpj.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kato R., Kataoka M., Kamikubo H., Kuramitsu S. Direct observation of three conformations of MutS protein regulated by adenine nucleotides. J. Mol. Biol. 2001;309:227–238. doi: 10.1006/jmbi.2001.4752. [DOI] [PubMed] [Google Scholar]
- 9.Lamers M.H., Winterwerp H.H.K., Sixma T.K. The alternating ATPase domains of MutS control DNA mismatch repair. EMBO J. 2003;22:746–756. doi: 10.1093/emboj/cdg064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iyer R.R., Pluciennik A., Burdett V., Modrich P.L. DNA mismatch repair: Functions and mechanisms. Chem. Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 11.Antony E., Khubchandani S., Chen S.Y., Hingorani M.M. Contribution of Msh2 and Msh6 subunits to the asymmetric ATPase and DNA mismatch binding activities of Saccharomyces cerevisiae Msh2-Msh6 mismatch repair protein. DNA Repair (Amst.) 2006;5:153–162. doi: 10.1016/j.dnarep.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.