

Abstract
Endothelin-1 (ET-1), a potent vasoactive peptide, plays an important role in the pathogenesis of liver disease and portal hypertension. Two major endothelin receptors (ET-A and ET-B) mediate biological effects, largely on the basis of their known downstream signaling pathways. We hypothesized that the different receptors are likely to mediate divergent effects in portal hypertensive mice. Liver fibrosis and cirrhosis and portal hypertension were induced in 8-wk-old male BALB/c mice by gavage with carbon tetrachloride (CCl4). Portal pressure was recorded acutely during intravenous infusion of endothelin receptor antagonists in normal or portal hypertensive mice. In vivo microscopy was used to monitor sinusoidal dynamics. Additionally, the effect of chronic exposure to endothelin antagonists was assessed in mice during induction of fibrosis and cirrhosis with CCl4 for 8 wk. Intravenous infusion of ET-A receptor antagonists into normal and cirrhotic mice reduced portal pressure whereas ET-B receptor antagonism increased portal pressure. A mixed endothelin receptor antagonist also significantly reduced portal pressure. Additionally, the ET-A receptor antagonist caused sinusoidal dilation, whereas the ET-B receptor antagonist caused sinusoidal constriction. Chronic administration of each the endothelin receptor antagonists during the induction of fibrosis and portal hypertension led to reduced fibrosis, a significant reduction in portal pressure, and altered sinusoidal dynamics relative to controls. Acute effects of endothelin receptor antagonists are likely directly on the hepatic and sinusoidal vasculature, whereas chronic endothelin receptor antagonism appears to be more complicated, likely affecting fibrogenesis and the hepatic microcirculation. The data imply a relationship between hepatic fibrogenesis or fibrosis and vasomotor responses.
Keywords: fibrosis, cirrhosis, stellate cell, endothelial cell, portal pressure, in vivo microscopy, endothelin A receptor, endothelin B receptor, mixed endothelin receptor antagonist
cirrhosis is the 12th leading cause of death in the United States (7). Much of the mortality from cirrhosis is attributable specifically to complications resulting from portal hypertension, such as bleeding esophageal varices, hepatic coma, and spontaneous bacterial peritonitis. Endothelin-1 (ET-1) is a potent endothelium-derived vasoactive peptide that plays a central role in regulating hepatic vascular tone in healthy individuals but has multiple other actions important in disease, including stimulation of cell growth and in the wound healing response and tissue fibrogenesis (12, 17, 22). ET-1 has received considerable interest in the area of the pathogenesis of cirrhosis, its contribution to portal hypertension, and the possibility that endothelin antagonists might be used in the treatment of portal hypertension (16).
It has been established that levels of ET-1 are increased in the liver after liver injury, and that the cellular source of enhanced ET-1 production is the hepatic stellate cell (13, 19). ET-1 binds to one of two major ET receptors, known as ET-A and ET-B. In the liver, ET-A receptors are found on vascular smooth muscle cells and hepatic stellate cells whereas ET-B receptors are found on sinusoidal endothelial cells and hepatic stellate cells. These G protein-coupled receptors signal to a cell contraction mechanism in some cells (4), whereas in endothelial cells ET-1 binding to the ET-B receptor leads to Akt activation and subsequently endothelial nitric oxide synthase (eNOS) activation (10).
In the liver, both endothelin receptor A and B are upregulated after liver injury, the increase in expression appears attributable primarily to hepatic stellate cells (6, 36). Importantly, endothelin induces contraction of each stellate cells (20) and the hepatic sinusoid (38) and cell contraction is enhanced in stellate cells from cirrhotic rat livers and in the intact liver (21). Thus it has been proposed that increased hepatosplenic production of endothelin contributes to portal hypertension by mediating intrahepatic stellate cell contraction and an increase in hepatic sinusoidal tone. This concept has been supported by studies in animal models of portal hypertension that have shown that administration of endothelin antagonists reduces portal pressure (21, 25).
Although present data emphasize the importance of endothelin in the liver, the physiological function of each the ET-A and ET-B receptors in the liver remains unclear. Given what is known about the functional effects of the different endothelin receptors, we have hypothesized that ET-A and ET-B receptors are likely to mediate divergent physiological effects and may therefore have differing effects in the liver. Thus we have aimed to examine the effect of antagonism of specific endothelin receptors in normal and in portal hypertensive mice, both acutely and chronically.
MATERIALS AND METHODS
Animal model of cirrhosis.
Carbon tetrachloride (CCl4, 2 ml/kg in a 1:1 corn oil mix) was administered to 8-wk-old male BALB/c mice by gavage for 8 wk (2 doses/wk). Age-matched mice given corn oil alone served as controls. All animals received humane care according to National Institutes of Health (NIH) guidelines; studies were performed after approval by the University of Texas Southwestern Institutional Animal Care and Use Committee.
Acute endothelin antagonism.
To further understand the endothelin system in liver disease, we studied acute and chronic effects of endothelin antagonists. Part of our rationale for study with the specific endothelin nonpeptide antagonists chosen is that their pharmacological characteristics have been extensively studied (5, 29–34).
For determining the effect of acute exposure to endothelin antagonism, an ET-A receptor-selective antagonist (A-147627, also ABT-627, Abbott Laboratories, Abbott Park, IL), an ET-B receptor-selective antagonist (A-192621, Abbott Laboratories), and a mixed ET receptor antagonist (A-182086, Abbott Laboratories) were administered in a final concentration of 2 μM in saline intravenously (0.1 ml iv) to cirrhotic and control animals. Compounds were administered in a slow continuous bolus (given over 1 min). We performed several dose-finding experiments, including doses of all compounds from 2 mM to 2 nM. A final concentration of ultimately 2 μM was chosen based on previously published IC50 data (Table 1) and the predicted final concentrations of 0.32, 32, and 3.2 nM for A-147627, A-192621, and A-182086, respectively.
Table 1.
Pharmacological features of ET receptor antagonists
Chronic endothelin antagonism.
In chronic endothelin antagonism experiments, animals were divided into five groups as follows: 1) corn oil control, 2) CCl4-induced cirrhosis control, 3) CCl4 + ET-A receptor antagonist (A-147627 was given daily by gavage at a dose of 50 mg/kg), 4) CCl4 + ET-B receptor antagonist (A-192621 was given daily by gavage at a dose of 40 mg/kg), and 5) CCl4 + mixed ET receptor antagonist (A-182086 was given daily by gavage at a dose of 30 mg/kg).
Doses given were determined on the basis of previous reports of in vivo pharmacological half-life measurements and of recommendations by the manufacturer, also based on their experience with the compounds (5, 9, 27, 29, 30, 32, 33). Half-life characteristics after oral dosing have been reported in a variety of mammals including human, monkey, dog, rat, and/or mouse (approximate plasma half-life in rodents is as shown in Table 1). Previous reports indicate that the compounds have pharmacological activity with once daily dosing, presumably a result of trough concentrations of endothelin antagonists in the nanomolar range.
PVP measurement.
Mice were anesthetized (Nembutal, 50 mg/kg ip) and the portal vein was catheterized with a 24-gauge intravenous catheter (Becton Dickinson). A low-pressure transducer (LPA, Micro-Med, Louisville, KY) was then connected to the catheter via polyethylene tubing (Clay Adams). Portal vein pressure (PVP) measurements were recorded by use of Digi-Med System Integrator software (Micro-Med).
In vivo microscopy.
Mice were anesthetized and the jugular vein was catheterized for administration of experimental substances. The liver was freed and displaced onto a glass coverslip embedded in a petri dish in which the animal was placed. The liver was immobilized by placing another glass coverslip on top of it. The liver vasculature was visualized by use of an inverted microscope (Nikon Eclipse TE 300) equipped with a ×10 plan apo objective and a charge-coupled device camera (Nikon DXM 1200). Time-lapse video recordings of liver sinusoids were created during intravenous administration of specific agents (i.e., 0.9% saline, ET receptor antagonists), then analyzed with image analysis software (MetaVue, Universal Imaging). Random fields were chosen, and then a threshold level of gray corresponding with sinusoids was assigned. The thresholded area was systematically quantitated as described (3).
Morphometry.
Livers were fixed in 10% phosphate-buffered formalin for 48 h at 4°C, washed twice with water, stored in 70% ethanol at 4°C, and embedded in paraffin. Five-micrometer sections were stained with picrosirius red (Sigma) and counterstained with methyl green (Sigma). The proportion of tissue stained with picrosirius red content was assessed by morphometric analysis with MetaView software (Universal Imaging, Downingtown, PA) as described (17), and collagen stained with picrosirius red was quantitated in the sections that were randomly chosen (under ×10 magnification, 10 fields each from a sample).
Immunohistochemistry.
Livers were fixed as above, and 5-μm sections were deparaffinized and incubated with blocking mixture containing PBS and BSA. Sections were incubated with Cy3-conjugated monoclonal anti-smooth muscle α-actin antibody (1:200, Sigma) overnight at 4°C. Sections were washed, mounted, and photographed (Nikon Eclipse TE 300, equipped with epifluorescence).
Statistics.
Data are expressed as means ± SE. Statistical analysis was conducted by use of Microsoft Excel and SigmaStat (SPSS). The Mann-Whitney test was used for statistical comparisons among groups. The Student's t-test was used for statistical comparisons between two groups. For calculation of mean values and statistical variation, n indicates the number of experiments. Error bars depict the standard error. P values less than 0.05 were considered statistically significant.
RESULTS
Continued CCl4 treatment resulted in grossly nodular livers as described (3), which upon histological examination demonstrated bridging fibrosis. In normal animals (i.e., those exposed to corn oil alone), basal mean PVP was 2.99 mmHg (Fig. 1A), compared with 4.01 in CCl4-treated mice (Fig. 1B) (n = 6, P < 0.01 for normal compared with CCl4-treated mice).
Fig. 1.
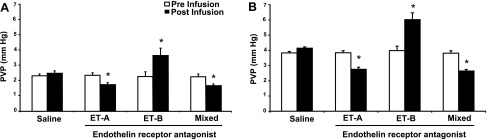
Effect of acute administration of endothelin receptor antagonists on portal vein pressure (PVP) in normal and cirrhotic mice. Mice were exposed to either corn oil alone (control, A) or carbon tetrachloride (CCl4, B) as described in methods. After anesthesia, portal pressure was measured continuously, also as in methods. Specific antagonists to endothelin receptors ET-A and ET-B as well as a mixed receptor antagonist were injected intravenously (100 μl of 2 μmol compound). The maximum change in portal pressure is shown (after a maximum observation period of 15 min) (n = 6, *P < 0.01 for postinfusion pressure compared with preinfusion pressure).
Effect of acute intravenous administration of endothelin receptor antagonists on PVP and sinusoidal volume.
Intravenous infusion of saline had no effect on PVP in either normal or CCl4-treated mice (Fig. 1). Infusion of the ET-A receptor antagonist led to a small but significant reduction in PVP in normal mice (Fig. 1A). In contrast, infusion of the ET-B receptor antagonist caused a significant increase in PVP. The mixed ET receptor antagonist also caused a reduction in PVP. In cirrhotic mice, basal PVP was higher at baseline than in normal mice (Fig. 1). Furthermore, infusion the ET-A receptor antagonist and the mixed antagonist led to a reduction in PVP whereas infusion of the ET-B receptor antagonist caused a significant increase in PVP (Fig. 1B). Of note, neither the ET-A receptor antagonist nor the mixed ET receptor antagonist reduced PVP in cirrhotic mice to normal levels.
We next measured acute changes in sinusoidal diameter in response to administration of the ET receptor antagonists by in vivo microscopy (Fig. 2). In normal mice, sinusoidal features were readily visible and exhibited characteristic features (3) (Fig. 2A). In contrast, in CCl4-induced injury, fibrosis, cirrhosis, and portal hypertension, the sinusoids appeared markedly attenuated (Fig. 2B).
Fig. 2.

Dynamic effect of acute administration of endothelin receptor antagonists on hepatic sinusoids in normal and cirrhotic mice. Mice and infusion of endothelin receptor antagonists were as in Fig. 1. Microscopic images were obtained by in vivo microscopy as described in methods. Images depict sinusoids from normal mice (A), at baseline (top row) or after receptor antagonist infusion (bottom row) and sinusoids from cirrhotic mice (B), at baseline (top row) or after receptor antagonist infusion (bottom row). As a control, the same volume of vehicle was infused (saline) as in endothelin antagonist experiments. Please also see supplemental data (movies) at the American Journal of Physiology Gastrointestinal and Liver Physiology website highlighting dynamic sinusoidal effects.
In normal mice, saline infusion increased sinusoid area by ∼10%, consistent with an increase in total intravascular volume during the infusion and consistent with previous reports (3) (Fig. 3A). Also, in normal mice, intravenous administration of the ET-A receptor antagonist led to a significant increase in sinusoidal volume, whereas infusion of the ET-B receptor antagonist decreased sinusoidal volume compared with the saline control (Fig. 3A). The effect of the mixed ET antagonist was similar to that of the ET-A receptor antagonist. Of note, we have previously validated flow during sinusoidal volume changes; in this work, as sinusoidal volume increases, so does sinusoidal blood flow (3).
Fig. 3.
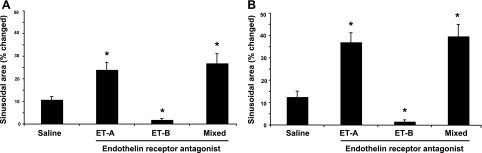
Endothelin receptor antagonists acutely modulate hepatic sinusoidal diameter in normal and cirrhotic mice. Mice and infusion of endothelin receptor antagonists were as in Fig. 2. Microscopic images were obtained by in vivo microscopy as described in methods; in brief, time-lapse video recordings of liver sinusoids were created and sinusoidal volume was quantitated as described in methods (the maximum change in sinusoidal diameter over 15 min was used in evaluation of the quantitative effect of antagonists). As a control, the same volume of vehicle was infused (saline) as in endothelin antagonist experiments. A: change in sinusoidal diameter in normal mice (n = 6, *P < 0.01 for postinfusion sinusoidal diameter compared with sinusoidal diameter after saline infusion). B: change in sinusoidal diameter in cirrhotic mice (n = 6, *P < 0.01 for postinfusion sinusoidal diameter compared with sinusoidal diameter after saline infusion).
Chronic liver injury resulted in reduction of the numbers of sinusoids (Fig. 2); the sinusoids per high-power field (HPF) in normal livers was 21.1 ± 5.4 (n = 8 random fields from 8 different livers) compared with 7.1 ± 2.2 (n = 8 random fields from 8 different livers; P < 0.01 for normal vs. injured) in injured livers. After liver injury, the patterns of change in sinusoidal volume in response to acute administration of endothelin receptor antagonists were similar to those in normal mice, including the fact that the ET-A receptor antagonist and the mixed antagonist led to an increase in sinusoidal volume whereas infusion of the ET-B receptor antagonist caused a significant decrease in sinusoidal volume (Fig. 3B). The changes in sinusoidal volume both for normal mice and in injured mice were inversely related to portal pressure (Figs. 1 and 3).
Finally, since changes in sinusoidal diameter could theoretically result from changes induced simply by portal vein dilation (as a result of endothelin antagonism), we specifically asked whether we could detect visual changes (using electronic calipers and a dissecting microscope/objective) in the portal vein during in vivo microscopy. When observed by in vivo microscopy, we did not observe changes in portal vein size after acute infusion of any of the endothelin antagonists (n = 3 for each).
Effect of chronic administration of ET receptor antagonists on PVP and liver fibrogenesis.
We next assessed the effect of concomitant ET receptor antagonism during liver injury on PVP. As before, chronic administration of CCl4 led to a significant increase in PVP compared with control (Fig. 4). Each of the endothelin receptor antagonists led to a reduction in PVP, and all to approximately the same degree, although the effect of dual receptor antagonism was slightly, though not statistically significantly, greater (Fig. 4). Chronic administration of ET receptor antagonists did not appear to affect the above-alluded-to reduction in the numbers of sinusoids per HPF in injured livers.
Fig. 4.

Endothelin receptor antagonism during liver injury progression leads to reduced portal pressure. As in methods, mice were exposed to either corn oil alone (sham treatment), CCl4 (to induce cirrhosis), or CCl4 and a specific endothelin receptor antagonist as follows: A-147627 (ET-A receptor antagonist) at 50 mg/kg by gavage, once daily; A-192621 (ET-B receptor antagonist) at 40 mg/kg by gavage, once daily; A-182086 (mixed ET receptor antagonist) at 30 mg/kg by gavage, once daily. After a total of 8 doses of CCl4, mice were anesthetized and portal vein pressure was measured as in methods. The highest portal pressure measurement obtained over a recording period of 10 min is shown (n = 6, *P < 0.01 compared with normal control mice; #P < 0.01, compared with CCl4-only mice).
Figure 5 shows the morphological response to CCl4 administration. Picrosirius red staining of the liver demonstrated a substantial increase in fibrosis in CCl4-treated livers, in a pattern of portal-portal and portal-central bridging, as previously described (24) (Fig. 5A). Smooth muscle α-actin expression in the liver mirrored that of collagen stained by picrosirius red (Fig. 5B).
Fig. 5.
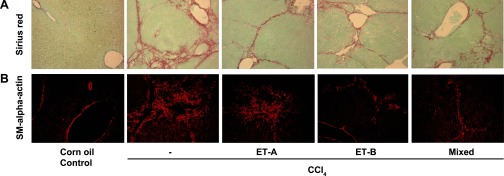
Endothelin receptor antagonism during liver injury progression leads to reduced expression of hepatic myofibroblasts and reduced collagen synthesis (morphological assessment). Mice and endothelin receptor antagonists were as in Fig. 4. A: representative (of 6 mice livers) immunohistochemical expression of smooth muscle α-actin in liver sections. B: representative (of 6 mice livers) picrosirius red staining of liver sections.
When fibrosis was quantified by morphometric methods, each of the endothelin receptor antagonists caused a reduction in fibrosis and smooth muscle α-actin expression (Fig. 6). Interestingly, the ET-B receptor antagonist alone caused the greatest reduction in fibrosis (Fig. 6B), although this difference was not statistically different from that of either the ET-A receptor antagonist alone or the mixed receptor antagonist. Expression of smooth muscle α-actin was similar to that of picrosirius red with the exception of that for the ET-A receptor antagonist, in which the expression of smooth muscle α-actin was slightly lower than for picrosirius red (Fig. 6). Type I collagen mRNA expression was measured in the mixed receptor model and was found to be significantly reduced after endothelin receptor antagonism, similar to that with picrosirius red and similar to previous work (17). This result is consistent with previous data showing that type I collagen mRNA correlates closely with type I collagen mRNA and hydroxyproline expression in rodent models of fibrogenesis (35).
Fig. 6.
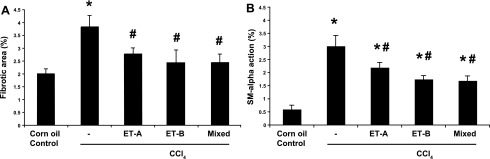
Endothelin receptor antagonism during liver injury progression leads to reduced expression of collagen and smooth muscle α-actin (quantitative assessment). Mice and endothelin receptor antagonists were as in Fig. 4. Morphometric analysis of the areas stained with picrosirius red (A) or the immunofluorescent signal (B) was performed as in methods (n = 6, *P < 0.01 compared with normal control mice; #P < 0.01 compared with CCl4-only mice).
DISCUSSION
In this study, we have provided further evidence to support the importance of the endothelin system in liver injury and portal hypertension. An advance of the work is to demonstrate in a mouse model of liver injury, fibrogenesis, and portal hypertension the relative effects of the major endothelin receptor antagonists. A further advance is that we have demonstrated an inverse relationship between portal pressure and sinusoidal volume, consistent with changes in flow as previously described (3). We have specifically shown that endothelin receptor antagonists reduce portal pressure, both in the acute situation and when administered chronically. Finally, the work suggests that there may be endothelin receptor specificity in biological outcomes.
We suspect that the cellular targets of ET-1 in the liver are likely to be diverse. ET-A receptors are present on both hepatic stellate cells and vascular smooth muscle cells (21), whereas ET-B receptors appear to be present on both hepatic stellate cells and sinusoidal endothelial cells (18). The ET-A receptor is well accepted to mediate cellular contraction, and our results demonstrating that the acute affect of ET-A receptor antagonism was to increase portal pressure and cause “constriction” of hepatic sinusoids is highly consistent with this concept.
Although our findings after acute blockade of ET-A receptors appear to be relatively straightforward, the effect of acute blockade of ET-B receptors appeared to be more complicated. We found that acute administration of the ET-B receptor antagonist caused an increase in portal pressure. One speculative mechanistic explanation is that the ET-B receptor is expressed on sinusoidal endothelial cells (18), and it is known that sinusoidal endothelial cell production of nitric oxide (NO) in the liver is an important regulator of intrahepatic hepatic resistance (11, 23). Thus it is possible that at a cellular level, paracrine NO (i.e., from endothelial cell to stellate or smooth muscle cell) was inhibited, facilitating cellular contraction. Although hepatic stellate cells, thought to be important regulators of sinusoidal tone by virtue of their contractile phenotype (37), possess ET-B receptors (15) that may mediate cellular contraction, our data raise the possibility that the balance of cellular contraction and relaxation in the sinusoid may favor a critical role and effect of NO. Furthermore, the data have important translational implications, since acute blockade of ET-B generated NO production in the liver is likely to raise portal pressure.
Our work also is notable for the finding that after chronic endothelin receptor antagonism, there was a reduction in fibrosis and portal pressure. Compared with other studies, our results were both similar and different (1, 14, 17, 28). On one hand, mixed endothelin receptor antagonists, bosentan and TAK-044, significantly reduced fibrosis in rat models (17, 28). Another study using an ET-A receptor antagonist found that chronic blockade of the A receptor led to a markedly reduced fibrosis in a bile duct ligation model (1). However, another study showed that rats undergoing CCl4 induced-liver injury and receiving a mixed endothelin antagonist, Ro 48-5695, had higher hepatic hydroxyproline content and procollagen type I mRNA expression than cirrhotic animals receiving vehicle (14). Additionally, rats receiving the endothelin antagonist exhibited increased portal pressure compared with controls. On the basis of our data and those of other studies examining the hepatic endothelin system in chronic liver disease, we are not able to provide a clear mechanistic explanation for the differences in outcomes of these studies. However, it is noteworthy that the study evaluating Ro 48-5695 had a unique study design in that animals were dosed with CCl4 for a long time (17 wk) with very high doses of CCl4, and moreover the endothelin antagonist was begun substantially after injury was initiated. Furthermore, in this study, it appeared that there was a high mortality rate associated with induction of liver injury. Finally, it is possible that interrupting endothelin signaling after injury has begun (i.e., after the endothelin system has been activated) has different cell biological effects than interrupting the endothelin system early in the liver injury process.
Our data also raise the possibility that the “function” of ET receptors may vary in the normal liver and in chronic injury. For example, ET-B antagonism acutely caused an increase in portal pressure, whereas its chronic blockade in an ongoing liver injury model led to reduced portal pressure. The mechanism underlying this finding is unclear. One possibility is that the ET-B receptor has different functions in cells from the normal and injured liver. For example, in the normal liver, the ET-B receptor is found predominantly on endothelial cells, where it mediates NO release, presumably in an autocrine fashion. On the other hand, the ET-B receptor is clearly upregulated on stellate cells after liver injury, and in this setting, it has been proposed that it may in part mediate the wound healing response (i.e., via stimulating fibrogenesis and cell contraction) (8, 26). Additionally, the ET-B receptor also appears to mediate proliferation (12), emphasizing its diverse functional capabilities. It is further important to emphasize that endothelin synthesis is increased during hepatic wound healing (19). Thus, in the normal liver, the data suggest that the ET-B receptor mediates NO production, which is presumably important in maintenance of sinusoidal relaxation, whereas in the injured liver the ET-B receptor mediates the fibrogenic and contractile response by hepatic stellate cells; in this setting, blocking this effect leads to specific antifibrogenic and portal hypertensive responses.
Although we believe that a primary site of action of the endothelin antagonists used in this study was in the liver itself, we cannot exclude the possibility that the effects of the endothelin antagonists were outside of the liver. For example, in rats with cirrhosis, it has been reported that a mixed ET receptor antagonist decreased portal pressure in vivo by reducing hepatocollateral vascular resistance (2).
Finally, our findings have several therapeutic implications. For example, acute administration of an endothelin receptor antagonist could acutely reduce portal pressure (for example in patients with esophageal variceal hemorrhage). In such a setting, the selectivity of the antagonist is likely to be important (i.e., blockade of the ET-A receptor would appear to be more desirable than of the ET-B receptor). The other area in which there are therapeutic implications of the work is in chronic liver disease. In this setting, chronic blockade of endothelin receptors may lead to reduced fibrogenesis, and perhaps portal hypertension. Here, blockade of either ET-A or ET-B receptor may be advantageous, and in the context of the concept that the ET-B receptor may be a key mediator of fibrogenesis and fibrosis, it is possible that antagonism of either or both of the endothelin receptors may be beneficial. Thus, to move this field forward, it will be critically important to consider the biological effects of each receptor subtype when considering therapeutic intervention.
GRANTS
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (Grant R01 DK 50344 to D. C. Rockey).
Supplementary Material
Acknowledgments
We thank Che-Chang Chan for helpful discussion regarding in vivo microscopy. We thank Abbott Laboratories for provision of endothelin antagonists and advice regarding appropriate oral dosing.
Supplemental material for this article is available online at the American Journal of Physiology Gastrointestinal and Liver Physiology website.
REFERENCES
- 1.Cho JJ, Hocher B, Herbst H, Jia J, Ruehl M, Hahn EG, Riecken EO, Schuppan D. An oral endothelin A receptor antagonist blocks collagen synthesis and deposition in advanced rat liver fibrosis. Gastroenterology 118: 1169–1178, 2000. [DOI] [PubMed] [Google Scholar]
- 2.De Gottardi A, Shaw S, Sagesser H, Reichen J. Type A, but not type B, endothelin receptor antagonists significantly decrease portal pressure in portal hypertensive rats. J Hepatol 33: 733–737, 2000. [DOI] [PubMed] [Google Scholar]
- 3.Edwards C, Feng HQ, Reynolds C, Mao L, Rockey DC. Effect of the nitric oxide donor V-PYRRO/NO on portal pressure and sinusoidal dynamics in normal and cirrhotic mice. Am J Physiol Gastrointest Liver Physiol 294: G1311–G1317, 2008. [DOI] [PubMed] [Google Scholar]
- 4.Goto K, Kasuya Y, Matsuki N, Takuwa Y, Kurihara H, Ishikawa T, Kimura S, Yanagisawa M, Masaki T. Endothelin activates the dihydropyridine-sensitive, voltage-dependent Ca2+ channel in vascular smooth muscle. Proc Natl Acad Sci USA 86: 3915–3918, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guimaraes CL, Trentin PG, Rae GA. Endothelin ET(B) receptor-mediated mechanisms involved in oleic acid-induced acute lung injury in mice. Clin Sci (Lond) 103, Suppl 48: 340S–344S, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Housset C, Rockey DC, Bissell DM. Endothelin receptors in rat liver: lipocytes as a contractile target for endothelin 1. Proc Natl Acad Sci USA 90: 9266–9270, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoyert DL, Heron MP, Murphy SL, Kung HC. Deaths: final data for 2003. Natl Vital Stat Rep 54: 1–120, 2006. [PubMed] [Google Scholar]
- 8.Karam H, Heudes D, Bruneval P, Gonzales MF, Loffler BM, Clozel M, Clozel JP. Endothelin antagonism in end-organ damage of spontaneously hypertensive rats. Comparison with angiotensin-converting enzyme inhibition and calcium antagonism. Hypertension 28: 379–385, 1996. [DOI] [PubMed] [Google Scholar]
- 9.Kavanagh M, Battistini B, Jean S, Crochetiere J, Fournier L, Wessale J, Opgenorth TJ, Cloutier R, Major D. Effect of ABT-627 (A-147627), a potent selective ET(A) receptor antagonist, on the cardiopulmonary profile of newborn lambs with surgically-induced diaphragmatic hernia. Br J Pharmacol 134: 1679–1688, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu S, Premont RT, Kontos CD, Huang J, Rockey DC. Endothelin-1 activates endothelial cell nitric-oxide synthase via heterotrimeric G-protein betagamma subunit signaling to protein kinase B/Akt. J Biol Chem 278: 49929–49935, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Liu S, Premont RT, Kontos CD, Zhu S, Rockey DC. A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nat Med 11: 952–958, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Nelson J, Bagnato A, Battistini B, Nisen P. The endothelin axis: emerging role in cancer. Nat Rev Cancer 3: 110–116, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Pinzani M, Milani S, De Franco R, Grappone C, Caligiuri A, Gentilini A, Tosti-Guerra C, Maggi M, Failli P, Ruocco C, Gentilini P. Endothelin 1 is overexpressed in human cirrhotic liver and exerts multiple effects on activated hepatic stellate cells. Gastroenterology 110: 534–548, 1996. [DOI] [PubMed] [Google Scholar]
- 14.Poo JL, Jimenez W, Maria Munoz R, Bosch-Marce M, Bordas N, Morales-Ruiz M, Perez M, Deulofeu R, Sole M, Arroyo V, Rodes J. Chronic blockade of endothelin receptors in cirrhotic rats: hepatic and hemodynamic effects. Gastroenterology 116: 161–167, 1999. [DOI] [PubMed] [Google Scholar]
- 15.Rockey DC Characterization of endothelin receptors mediating rat hepatic stellate cell contraction. Biochem Biophys Res Commun 207: 725–731, 1995. [DOI] [PubMed] [Google Scholar]
- 16.Rockey DC Vascular mediators in the injured liver. Hepatology 37: 4–12, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Rockey DC, Chung JJ. Endothelin antagonism in experimental hepatic fibrosis. Implications for endothelin in the pathogenesis of wound healing. J Clin Invest 98: 1381–1388, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rockey DC, Chung JJ. Reduced nitric oxide production by endothelial cells in cirrhotic rat liver: endothelial dysfunction in portal hypertension. Gastroenterology 114: 344–351, 1998. [DOI] [PubMed] [Google Scholar]
- 19.Rockey DC, Fouassier L, Chung JJ, Carayon A, Vallee P, Rey C, Housset C. Cellular localization of endothelin-1 and increased production in liver injury in the rat: potential for autocrine and paracrine effects on stellate cells. Hepatology 27: 472–480, 1998. [DOI] [PubMed] [Google Scholar]
- 20.Rockey DC, Housset CN, Friedman SL. Activation-dependent contractility of rat hepatic lipocytes in culture and in vivo. J Clin Invest 92: 1795–1804, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rockey DC, Weisiger RA. Endothelin induced contractility of stellate cells from normal and cirrhotic rat liver: implications for regulation of portal pressure and resistance. Hepatology 24: 233–240, 1996. [DOI] [PubMed] [Google Scholar]
- 22.Rubanyi GM, Botelho LH. Endothelins. FASEB J 5: 2713–2720, 1991. [DOI] [PubMed] [Google Scholar]
- 23.Shah V, Haddad FG, Garcia-Cardena G, Frangos JA, Mennone A, Groszmann RJ, Sessa WC. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J Clin Invest 100: 2923–2930, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi Z, Wakil AE, Rockey DC. Strain-specific differences in mouse hepatic wound healing are mediated by divergent T helper cytokine responses. Proc Natl Acad Sci USA 94: 10663–10668, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sogni P, Moreau R, Gomola A, Gadano A, Cailmail S, Calmus Y, Clozel M, Lebrec D. Beneficial hemodynamic effects of bosentan, a mixed ET(A) and ET(B) receptor antagonist, in portal hypertensive rats. Hepatology 28: 655–659, 1998. [DOI] [PubMed] [Google Scholar]
- 26.Sticherling M The role of endothelin in connective tissue diseases. Rheumatology (Oxford) 45, Suppl 3: iii8–iii10, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Taylor TA, Gariepy CE, Pollock DM, Pollock JS. Unique endothelin receptor binding in kidneys of ETB receptor deficient rats. Am J Physiol Regul Integr Comp Physiol 284: R674–R681, 2003. [DOI] [PubMed] [Google Scholar]
- 28.Thirunavukkarasu C, Yang Y, Subbotin VM, Harvey SA, Fung J, Gandhi CR. Endothelin receptor antagonist TAK-044 arrests and reverses the development of carbon tetrachloride induced cirrhosis in rats. Gut 53: 1010–1019, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vassileva I, Mountain C, Pollock DM. Functional role of ETB receptors in the renal medulla. Hypertension 41: 1359–1363, 2003. [DOI] [PubMed] [Google Scholar]
- 30.Verhaar MC, Grahn AY, Van Weerdt AW, Honing ML, Morrison PJ, Yang YP, Padley RJ, Rabelink TJ. Pharmacokinetics and pharmacodynamic effects of ABT-627, an oral ETA selective endothelin antagonist, in humans. Br J Clin Pharmacol 49: 562–573, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Von Geldern TW, Tasker AS, Sorensen BK, Winn M, Szczepankiewicz BG, Dixon DB, Chiou WJ, Wang L, Wessale JL, Adler A, Marsh KC, Nguyen B, Opgenorth TJ. Pyrrolidine-3-carboxylic acids as endothelin antagonists. 4. Side chain conformational restriction leads to ET(B) selectivity. J Med Chem 42: 3668–3678, 1999. [DOI] [PubMed] [Google Scholar]
- 32.Wessale JL, Adler AL, Novosad EI, Calzadilla SV, Dayton BD, Marsh KC, Winn M, Jae HS, von Geldern TW, Opgenorth TJ, Wu-Wong JR. Pharmacology of endothelin receptor antagonists ABT-627, ABT-546, A-182086 and A-192621: ex vivo and in vivo studies. Clin Sci (Lond) 103, Suppl 48: 112S–117S, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Winn M, von Geldern TW, Opgenorth TJ, Jae HS, Tasker AS, Boyd SA, Kester JA, Mantei RA, Bal R, Sorensen BK, Wu-Wong JR, Chiou WJ, Dixon DB, Novosad EI, Hernandez L, Marsh KC. 2,4-Diarylpyrrolidine-3-carboxylic acids—potent ETA selective endothelin receptor antagonists. 1. Discovery of A-127722. J Med Chem 39: 1039–1048, 1996. [DOI] [PubMed] [Google Scholar]
- 34.Wu-Wong JR, Dixon DB, Chiou WJ, Sorensen BK, Liu G, Jae HS, Tasker A, von Geldern TW, Winn M, Opgenorth TJ. Pharmacology of endothelin receptor antagonists ABT-627, ABT-546, A-182086 and A-192621: in vitro studies. Clin Sci (Lond) 103, Suppl 48: 107S–111S, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Yang L, Chan CC, Kwon OS, Liu S, McGhee J, Stimpson SA, Chen LZ, Harrington WW, Symonds WT, Rockey DC. Regulation of peroxisome proliferator-activated receptor-gamma in liver fibrosis. Am J Physiol Gastrointest Liver Physiol 291: G902–G911, 2006. [DOI] [PubMed] [Google Scholar]
- 36.Yokomori H, Oda M, Ogi M, Kamegaya Y, Tsukada N, Nakamura M, Ishii H. Enhanced expression of endothelin receptor subtypes in cirrhotic rat liver. Liver 21: 114–122, 2001. [DOI] [PubMed] [Google Scholar]
- 37.Zhang B, Calmus Y, Wen L, Sogni P, Lotersztajn S, Houssin D, Weill B. Endothelin-1 induces liver vasoconstriction through both ETA and ETB receptors. J Hepatol 26: 1104–1110, 1997. [DOI] [PubMed] [Google Scholar]
- 38.Zhang JX, Pegoli WJ, Clemens MG. Endothelin-1 induces direct constriction of hepatic sinusoids. Am J Physiol Gastrointest Liver Physiol 266: G624–G632, 1994. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.