

Abstract
Nitrogenase catalyzes the biological reduction of N2 to ammonia (nitrogen fixation), as well as the two-electron reduction of the non-physiological alkyne substrate acetylene (HC≡CH). A complex metallo-organic species called FeMo-cofactor provides the site of substrate reduction within the MoFe protein, but exactly where and how substrates interact with FeMo-cofactor remains unknown. Recent results have shown that the MoFe protein α-70Val residue, whose side-chain approaches one Fe-S face of FeMo-cofactor, plays a significant role in defining substrate access to the active site. For example, substitution of α-70Val by alanine results in an increased capacity for the reduction of the larger alkyne propyne (HC≡C-CH3), whereas substitution by isoleucine at this position nearly eliminates the capacity for the reduction of acetylene. These and complementary spectroscopic studies led us to propose that binding of short chain alkynes occurs with side-on binding to Fe atom 6 within FeMo-cofactor. In the present work, the α-70Val residue was substituted by glycine and this MoFe protein variant shows an increased capacity for reduction of the terminal alkyne, 1-butyne (HC≡C-CH2-CH3). This protein shows no detectable reduction of the internal alkyne 2-butyne (H3C-C≡C-CH3). In contrast, substitution of the nearby α-191Gln residue by alanine, in combination with the α-70Ala substitution, does result in significant reduction 2-butyne, with the exclusive product being 2-cis-butene. These results indicate that the reduction of alkynes by nitrogenases involves side-on binding of the alkyne to Fe6 within FeMo-cofactor, and that a terminal acidic proton is not required for reduction. The successful design of amino acid substitutions that permit the targeted accommodation of an alkyne that otherwise is not a nitrogenase substrate provides evidence to support the current model for alkyne interaction within the nitrogenase MoFe protein.
Introduction
The reduction of dinitrogen (N2) to two ammonia molecules (2 NH3) catalyzed by the molybdenum-dependent nitrogenase requires: (i) two nitrogenase component proteins (the Fe protein and the MoFe protein), (ii) electrons, protons, and (iii) the hydrolysis of MgATP [1–4]. The Fe protein is a homodimer that contains a [4Fe-4S] cluster bridging the two subunits [5], with each subunit having a site for MgATP binding. During turnover, the reduced Fe protein transfers one electron at a time from its [4Fe-4S] cluster to the MoFe protein in a reaction coupled to the hydrolysis of two MgATP molecules [3, 6]. The MoFe protein is an α2β2 heterotetramer that contains two unique types of [Fe-S] clusters [7, 8]. An [8Fe-7S] cluster (designated the P-cluster) is located at each αβ subunit interface and is proposed to serve as an intermediary in the electron transfer process. The FeMo-cofactor is a [7Fe-9S-X-Mo-homocitrate] cluster [9] located entirely within each α subunit and it has been identified as providing the substrate reduction site [10]. Nitrogenase also reduces protons forming H2. In addition to these physiological substrates, a number of small, triple or double bonded compounds, for example, acetylene (HC≡CH), azide (N3−), and nitrous oxide (N2O), have also been shown to be substrates for reduction by nitrogenase [1, 11].
A significant challenge in nitrogenase research involves identification of the exact location and nature of substrate interaction with FeMo-cofactor (Figure 1). A number of different interaction sites can be considered upon inspection of the structure of FeMo-cofactor and calculations have been reported that support a number of different binding sites [4, 12]. In our earlier studies, we began to localize the site of alkyne substrate interactions with FeMo-cofactor to one Fe-S face that includes Fe atoms numbered 2, 3, 6 and 7 [2, 13–20]. Analysis of the capacity of MoFe proteins having amino acid substitutions for residues that approach this Fe-S face to reduce a range of substrates has indicated that this face might provide the only site for substrate binding and reduction. For example, it was found that substitution of the α-70Val residue by alanine results in a MoFe protein that can effectively reduce propyne (HC≡C-CH3) and propargyl alcohol (HC≡C-CH2OH) to their corresponding alkenes, whereas these alkynes are only very poor substrates for the wild-type MoFe protein [19]. In contrast, substitution of the α-70Val residue by isoleucine prevents the effective reduction of all substrates except protons [18]. By using the amino acid substitution approach, conditions were also developed that allowed trapping and characterization of an intermediate that accumulates during the reduction of propargyl alcohol by the α-70Ala-substituted MoFe protein [18]. Characterization of this intermediate by EPR and 13C- and 1/2H-electron nuclear double resonance (ENDOR) spectroscopies were interpreted to indicate side-on binding of a 2e−/2H+ reduced allyl alcohol species (H2C=CH-CH2OH) that is bound to FeMo-cofactor through a single Fe atom [17]. The effect of pH dependence on the capacity of the α-70Ala MoFe protein to reduce either propargyl alcohol or propargyl amine (HC≡C-CH2NH2) further localized the proposed binding site to Fe atom 6 [16] within FeMo-cofactor (Figure 1). In the present work, we have further explored the nitrogenase substrate binding site by characterizing a MoFe protein for which the α-70Val residue has been substituted by glycine, and also by characterizing a doubly-substituted MoFe protein for which both the α-70Val residue and the α-191Gln residue have been replaced by alanine.
Figure 1. FeMo-cofactor and its environment.

(panel A) A stereoview of FeMo-cofactor and the side chains of α-70Val and α-191Gln is shown. Fe atoms 2, 3, 6, and 7 are labeled. R-homocitrate is shown in stick format at the back of the view bound to Mo. (panel B) Same view as panel A with 2-butyne (cyan) placed into position for interaction with Fe atom 6. Van der Waals surfaces are shown around the 2-butyne and the side chain of α-191Gln. The steric overlap between the 2-butyne and the side chains of α-70Val and α-191Gln is apparent. Coordinates are from the protein database file 1M1N.pdb with carbon shown in gray, oxygen in red, iron in rust, sulfur in yellow, molybdenum in magenta, and nitrogen in dark blue. The atom of unknown identity (X) at the center of FeMo-cofactor is shown in green.
Experimental Procedures
Strain construction, cell growth, and purification
Strains of Azotobacter vinelandii containing amino acid substitutions at the α-70Val and α-191Gln positions were constructed using site directed mutagenesis and gene replacement techniques previously described [21, 22]. Wild-type MoFe protein was isolated from A. vinelandii strain DJ995 [23], α-70Ala MoFe protein from DJ1310 [19], α-70Gly MoFe protein from DJ1313, α-191Ala MoFe protein from DJ1242, and α-70Ala/α-191Ala MoFe protein from DJ1495. All MoFe proteins in this study contained a poly-histidine insertion near the carboxyl terminus of each α-subunit. A. vinelandii strains DJ1242, DJ1313, and DJ1495 were constructed for this study, and a detailed description of strain construction follows. DJ1313 was constructed by transforming DJ995 [23] with pDB1133 (SstI ‘nifH-nifD’fragment containing GGG as codon 70 of nifD). The transformants were selected by loss of the ability to grow under nitrogen fixing conditions. DJ1242 was constructed by transforming DJ1238 × pDB753. DJ1238 carries a deletion between codons 188–196 of nifD gene, and pDB 753 is a ‘nifD’-KpnI fragment carrying a codon change (GCC) at position 191. DJ1242 was selected by rescuing the ability to grow under nitrogen fixing conditions. Lastly, DJ1495 was constructed by transforming DJ1310 [19] with pDB753. This strain was selected by a slower growth rate under nitrogen fixing conditions.
For protein purification, cells were grown at 30°C in 120 L culture as described previously [23]. Cell-free extracts were prepared by an osmotic shock method and the MoFe protein was purified by an immobilized metal-affinity chelation chromatography (IMAC) protocol [23]. The Fe protein used in all experiments was purified from wild-type cells and did not contain a histidine tag. All solutions were made anaerobic by repeated evacuation and flushing with Ar that had been scrubbed with a heated BASF catalyst tower to remove oxygen. Protein concentration was determined by the biuret method using bovine serum albumin as the standard [23]. Protein purity was estimated by polyacrylamide gel electrophoresis with Coomassie blue staining.
Cell growth
Cells were grown on solid agar medium with additions according to the method of Burk [24]. Where indicated, 100 μl of 2.5 M freshly prepared 2-butyne-1-ol (Fluka, Milwaukee, WI) or 2-butyne-1,4-diol (Sigma, St. Louis, MO) were spread on each plate (10 mM final concentration) before the cells were added. Plates were incubated at 30°C for 4 days.
In vitro assays
The methods and reaction mixture compositions for substrate reduction activities have been described elsewhere [19]. Here, each assay contained 0.05 mg of MoFe protein and 0.45 mg of Fe protein. All reactions were carried out under an argon atmosphere in 9.2 ml sealed septum vials. The reactions were initiated by the addition of Fe protein and reactions were allowed to proceed for 8 min while shaking in a 30°C water bath. Each reaction was terminated by the addition of 250 μL of a 0.4 M EDTA solution, pH 7.4. Acetylene was freshly prepared for each experiment by the reaction of calcium carbide (Aldrich, St. Louis, MO) in water. Propyne (98% pure) was purchased from Aldrich and 1-butyne (95% pure) was purchased from Pfaltz and Bauer (Chicago, IL).
H2 was quantified by injection of 200 μL from the gas phase of a reaction vial into a Shimadzu GC-14 gas chromatograph equipped with a Supelco 80/100 molecular sieve 5A column and product quantified using a thermal conductivity detector. Ethylene, propene and 1-butene were quantified by gas chromatography using a Hewlett-Packard 5890A instrument equipped with an Al2O3 capillary column and product quantified by flame ionization detection. Quantification of propene and 1-butene was performed using an ethylene standard and applying a correction factor to the peak area taken from the measurement of a 2–6 carbon olefin standard (MG Scientific Gases, Somerville, NJ) containing equimolar mixtures of these gases.
Initial assays of 2-butyne reduction were uninformative because the sample received from the manufacturer (Aldrich) was slightly contaminated by propyne. Contaminating propyne was essentially eliminated by cycling 2-butyne several times through a series of dry ice/ethanol traps. Under these conditions, 2-butyne could be collected as a solid (mp −32°C), while the contaminating propyne (mp −103°C) could be eliminated in the gas phase. This procedure resulted in a 2-butyne sample having less than 0.03% propyne contamination as estimated by analysis using gas chromatography. Under the conditions used, cis-2-butene and trans-2-butene resolved at different retention times during gas chromatography, 1.93 min and 1.68 min, respectively, providing a simple means of determining isomer composition. For kinetic analyses, initial velocity rates were plotted against the partial pressure of the substrates and the data fit to the Michaelis-Menten equation.
Results
Expanding the capacity of nitrogenase to reduce short chain alkynes
The genotypes and corresponding phenotypes of A. vinelandii strains examined in the present work are listed in Table 1. These data show that all strains examined, with the exception of the one that produces a MoFe protein having the α-70Val residue substituted by glycine, can grow without the addition of a fixed source of nitrogen to the growth medium. Consistent with this observation, all of the purified MoFe proteins, except the α-70Gly variant, showed high levels of N2 reduction activity. The α-70Gly MoFe protein exhibited no detectable capacity for N2 reduction, although it did retain high levels of activity for the reduction of acetylene and protons. Because all nitrogenase substrates compete for the same electron pool [25], and possibly the same binding site [14], their capacity for interaction with the active site can be indirectly evaluated by their ability to inhibit the reduction of other substrates. In the case of the α-70Gly-substituted MoFe protein, N2 is neither a substrate nor an inhibitor of proton reduction (Table 1), indicating that this substitution prevents the productive interaction of N2 with the active site. Clearly, this feature cannot reflect an inability of N2 to gain access to the active site of the α-70Gly-substituted MoFe protein because other larger substrates retain their capacity to serve as substrates. The specific defect in N2 reduction associated with the α-70Gly-substituted MoFe is, therefore, more likely to reflect an inability of this protein to achieve the redox state necessary for N2 binding [1]. This aspect of the α-70Gly MoFe protein was not further explored in the present work.
Table 1.
Features of MoFe Protein Variants
|
A. vinelandii Strain |
Amino Acid | Phenotypea | Inhibition of H2 evolution by N2 (%)b |
|---|---|---|---|
| DJ995 (WT) | α-70Val | Nif+ | 63 |
| DJ1313 | α-70Gly | Nif− | 0 |
| DJ1310 | α-70Ala | Nif+ | 37 |
| DJ1242 | α-191Ala | Nif+ | 38 |
| DJ1495 | α-70Ala/α-191Ala | Nif+ | 31 |
Nif+ is defined as able to grow under diazotrophic conditions, while Nif-as not able to grow under diazotrophic conditions.
for purified proteins in a standard H2 evolution assay under 1 atm of N2
Although the α-70Gly MoFe protein does not reduce the natural nitrogenase substrate N2, its ability to reduce acetylene (Figure 2) permitted an extension of previous work [19] on the effect of the size of different side chains at the α-70 residue position within the MoFe protein on the capacity for reduction of different short chain alkynes. MoFe proteins having this position occupied by α-70Val, α-70Ala, or α-70Gly were examined for their ability to reduce the progressively larger alkynes acetylene, propyne and 1-butyne (Figure 2). All three proteins exhibit similar rates for the reduction of acetylene but are differentiated by their respective abilities to reduce propyne or 1-butyne (Figure 3). The Km and Vmax values for acetylene reduction were found to be 0.016 atm and 2380 nmol/min/mg for the wild-type, 0.007 atm and 2040 nmol/min/mg for the α-70Ala, and 0.014 atm and 1800 nmol/min/mg for the α-70Gly MoFe proteins. The wild-type MoFe protein showed very low rates of propyne reduction even at the highest concentrations of propyne, whereas the α-70Ala- and α-70Gly-substituted MoFe proteins showed significant rates of propyne reduction. Fitting the propyne reduction data for the α-70Ala and α-70Gly substituted MoFe proteins to the Michaelis-Menten equation reveals a Km for propyne reduction of 0.02 atm for the α-70Ala MoFe protein and 0.001 atm for the α-70Gly MoFe protein, while the corresponding Vmax values were 1150 and 1400 nmol propene/min/mg MoFe protein, respectively. Reduction of 1-butyne showed the same trend with the exception that it is reduced at lower rates in all cases and, consequently, conditions of substrate saturation could not be achieved experimentally.
Figure 2.

Structures of substrates used in this study.
Figure 3. Reduction of short chain alkynes by α–70 MoFe protein variants.

The specific activity (nmol product/min/mg MoFe protein) is plotted against the partial pressure of the indicated substrate. Argon makes up the remaining gas to 1 atm total pressure. Substrates: (panel A) acetylene; (panel B) propyne; and (panel C) 1-butyne. The MoFe protein variants are: α-70Val wild-type (-■-), α-70Ala (-▲-) and α-70Gly (-●-). Fits of the data to the Michaelis-Menten equation are shown (lines).
A genetic screen for A. vinelandii strains that can reduce internal short-chain alkynes
Decreasing the size of the side chain at the α-70Val position only increases the size of short chain alkynes that can be reduced by nitrogenase if the triple bond is terminal. Namely, we found that short chain alkynes, such as 2-butyne, cannot serve as nitrogenase substrates even when the normal side chain located at the α-70Val-position is shortened. This observation can be explained in one of two different ways. According to our model, alkyne binding within the MoFe protein occurs through exclusive coordination with Fe6 of FeMo-cofactor in a side-on fashion [15, 16]. Thus, steric constraints imposed by side-chains other than α-70 could prevent the productive interaction with nitrogenase by alkynes that have an internal triple bond. A second possibility is that a terminal acidic proton is required for productive interaction of alkynes with the nitrogenase active site. To differentiate between these possibilities, we sought to determine whether or not strategic replacement of amino acids within the MoFe protein could be used to expand its substrate reduction capacity to include internal alkynes.
Because the biochemical analysis of nitrogenase is laborious, we previously developed a genetic and physiological strategy to assess the effect of amino acid substitutions within the MoFe protein on the capacity for reduction of short chain alkynes [26]. The experimental rationale of this approach is that different nitrogenase substrates compete for the same pool of available reducing equivalents. Consequently, any substrate that can compete with N2 for such reducing equivalents should inhibit growth on N2, providing the inhibitor is added to the growth medium at sufficient levels. This strategy, however, is complicated because most nitrogenase substrates are gases, and many alternative nitrogenase substrates are also explosive at concentrations required for physiological inhibition of nitrogenase. We previously solved these problems by using alcohol-substituted alkynes as potential inhibitors because many of them are liquids at ambient temperature and can, therefore, be added to the growth media at high concentrations. This approach was previously applied in experiments that were used to identify the role of the α-70Val residue in controlling the size of alkyne substrates that can access the active site. Namely, it was shown that addition of propargyl alcohol to the growth medium of nitrogen fixing cells of wild-type A. vinelandii has no effect on growth because propargyl alcohol is normally unable to interact with the nitrogenase active site [18, 19]. However, nitrogen fixation-dependent growth of a mutant strain of A. vinelandii for which the α-70Val residue is substituted by alanine is severely impaired when propargyl alcohol is added to the growth medium.
In the current work, we used a similar genetic approach to ask whether or not strains could be isolated whose ability to fix nitrogen is inhibited by the addition of internal alkyne alcohols to the growth medium. This strategy was guided by inspection of the polypeptide environment that surrounds the previously identified alkyne binding region (Figure 1). If our hypothesis that Fe6 within FeMo-cofactor provides the unique site for alkyne substrate binding is correct, then van der Waals contact with the side chains of α-70Val and α-191Gln are predicted to control access of any internal alkyne to the active site. For example, in the case of 2-butyne binding, the methyl group located at one end of the alkyne would prevent binding owing to interference by the α-70Val side chain and the methyl group located at the other end of the alkyne would prevent binding owing to interference by the α-191Gln side chain. Consistent with this prediction, the addition of neither 2-butyne-1-ol (HOCH2-C≡C-CH3) nor 2-butyne-1,4-diol (HOCH2-C≡C-CH2OH) to the growth medium has any effect on the capacity of wild-type A. vinelandii to sustain diazotrophic growth. Similarly, the addition of these same alkynes to strains that produce MoFe proteins having either the α-70Val residue or the α-191Gln residue, individually substituted by alanine, has no affect on the capacity for diazotrophic growth (Figure 4). In contrast, when these amino acid substitutions are combined within the same MoFe protein (α-70Ala/α-191Ala), both 2-butyne-1-ol and 2-butyne-1,4-diol inhibit the capacity for diazotrophic growth when added to the culture medium (Figure 4). Based on these results, the doubly-substituted protein was isolated and analyzed for its ability to catalyze reduction of 2-butyne (Figure 5). In addition to confirming the capacity for the doubly-substituted MoFe protein to reduce 2-butyne, it was also observed that 2-cis-butene is the exclusive reduction product.
Figure 4. The effect of 2-butyne-1-ol and 2-butyne-1,4-diol on diazotrophic growth of A. vinelandii expressing MoFe protein variants.

Cells expressing the MoFe protein variants include: α-191Ala, α-70Ala, and α-191Ala/α-70Ala. The indicated strain was cultured on agar plates made with Burk’s media lacking a fixed nitrogen source and including the following additions: no addition; + 2-butyne-1-ol, 10 mM 2-butyne-1-ol; and + 2-butyne-1,4 diol, 10 mM 2-butyne-1,4 diol.
Figure 5. Reduction of 2-butyne by α.
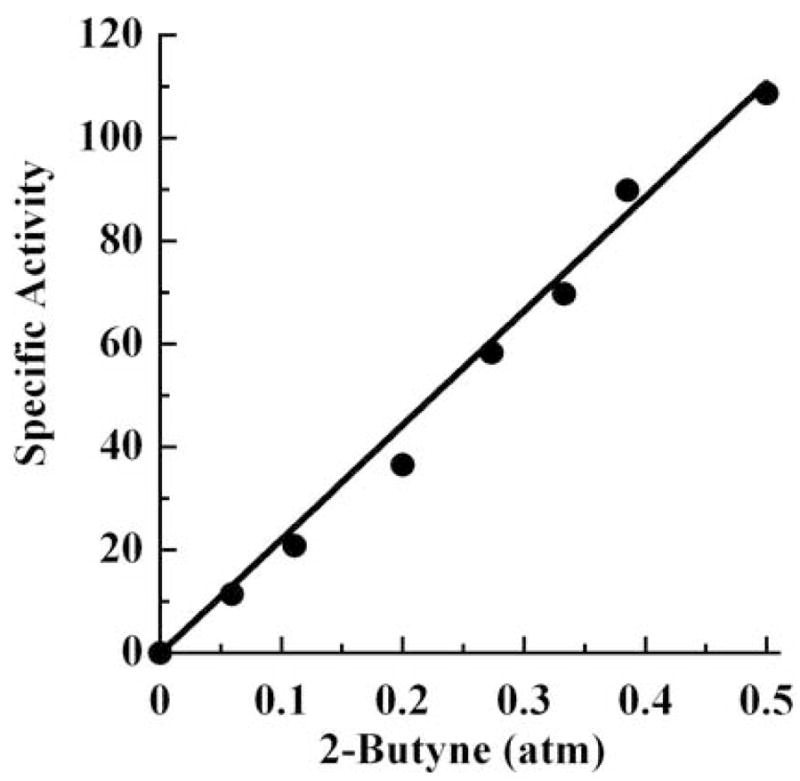
-70Ala/α-191Ala MoFe protein. The specific activity for 2-butyne reduction (nmol of cis-2-butene produced/min/mg MoFe protein) is plotted against the partial pressure of the substrate, 2-butyne. The remaining gas is argon with a final total pressure of 1 atm. The data are fit to a straight line.
Discussion
The most significant observation to emerge from this work is that the polypeptide environment immediately surrounding FeMo-cofactor can be manipulated in a predictable way such that binding and reduction of different size alkyne substrates can be accommodated. Most importantly, a working model for where and how alkyne substrates interact with the FeMo-cofactor, at the unique Fe6 site, was used for the rational design of a doubly-substituted MoFe protein that becomes endowed with the capacity to reduce 2-butyne, which otherwise is not a nitrogenase substrate. The fact that 2-butyne can be reduced by the doubly-substituted MoFe protein α-70Ala/α-191Ala also provides evidence that a terminal acidic proton is not necessary for the activation of alkyne reduction catalyzed by nitrogenase.
Also of mechanistic importance is the observation that reduction of 2-butyne by the doubly-substituted MoFe protein results in the exclusive production 2-cis-butene with no detectable 2-trans-butene. This product profile is consistent with side-on binding of 2-butyne, with proton addition occurring at one face. Earlier work on wild-type nitrogenase revealed that proton addition to acetylene during its reduction to ethylene occurs with stereospecificity [27–29]. The product of acetylene (HC≡CH) reduction in the presence of D2O is 96 % cis-ethylene (HDC=CDH), with only 4 % of the trans-isomer [30]. This stereospecificity would be consistent with side-on binding of acetylene to one or more Fe atoms of FeMo-cofactor, with both protons (or deuterons) added to one side of the bound acetylene. Ed Stiefel suggested a concerted proton and electron addition mechanism to explain this stereospecificity of proton addition to acetylene [31]. Interestingly, it was later found that lowering the electron flux through nitrogenase or substituting certain amino acids around FeMo-cofactor, can relax this stereospecificity, with up to 50 % of the trans-isomer being observed [30, 32]. One explanation for this higher level of trans addition of protons is a rearrangement of a semi-reduced intermediate species bound to FeMo-cofactor during catalysis. This model is supported by an increased level of trans-isomer product under conditions of lower electron flux, where a semi-reduced intermediate would have more time for rearrangement [32]. Given the relatively larger size of 2-butyne and the steric constraints that must be associated with its interaction within the substrate binding pocket of the MoFe protein, it seems unlikely that a semi-reduced 2-butyne intermediate bound to FeMo-cofactor could rearrange, thus greatly limiting the production of 2-trans-butene.
In summary, considering the work presented here along with earlier studies, the site of alkyne substrate, and likely all substrates, interactions with FeMo-cofactor appears to be localized to a specific Fe atom (number 6) on FeMo-cofactor, and the reduction of a range of alkynes with both terminal and internal triple bonds can be rationally controlled by changes in the sizes of specific amino acid side chains that surround this specific face of FeMo-cofactor.
Acknowledgments
This work was supported by NIH grant R01-GM59087 to LCS and DRD. PCD acknowledges the Advance-VT for a postdoctoral fellowship.
Footnotes
This work is dedicated in memory of Dr. Edward I. Stiefel, who made many contributions to our understanding of molybdenum-containing enzymes, including nitrogenase, over his distinguished career.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Burgess BK, Lowe DJ. Chem Rev. 1996;96:2983–3011. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- 2.Dos Santos PC, Igarashi RY, Lee HI, Hoffman BM, Seefeldt LC, Dean DR. Acc Chem Res. 2005;38:208–214. doi: 10.1021/ar040050z. [DOI] [PubMed] [Google Scholar]
- 3.Howard JB, Rees DC. Annu Rev Biochem. 1994;63:235–24. doi: 10.1146/annurev.bi.63.070194.001315. [DOI] [PubMed] [Google Scholar]
- 4.Seefeldt LC, Dance I, Dean DR. Biochemistry. 2004;43:1401–1409. doi: 10.1021/bi036038g. [DOI] [PubMed] [Google Scholar]
- 5.Georgiadis MM, Komiya H, Chakrabarti P, Woo D, Kornuc JJ, Rees DC. Science. 1992;257:1653–1659. doi: 10.1126/science.1529353. [DOI] [PubMed] [Google Scholar]
- 6.Seefeldt LC, Dean DR. Acc Chem Res. 1997;30:260–266. [Google Scholar]
- 7.Kim J, Rees DC. Nature. 1992;360:553–560. doi: 10.1038/360553a0. [DOI] [PubMed] [Google Scholar]
- 8.Kim J, Rees DC. Science. 1992;257:1677–1682. doi: 10.1126/science.1529354. [DOI] [PubMed] [Google Scholar]
- 9.Einsle O, Tezcan FA, Andrade SLA, Schmid B, Yoshida M, Howard JB, Rees DC. Science. 2002;297:1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 10.Shah VK, Brill WJ. Proc Natl Acad Sci USA. 1977;74:3249–3253. doi: 10.1073/pnas.74.8.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burgess BK. In: Metal Ions in Biology: Molybdenum Enzymes. Spiro TG, editor. John Wiley and Sons; New York: 1985. pp. 161–220. [Google Scholar]
- 12.Dance I. J Am Chem Soc. 2004;126:11852–11863. doi: 10.1021/ja0481070. [DOI] [PubMed] [Google Scholar]
- 13.Barney BM, Yang TC, Igarashi RY, Dos Santos PC, Laryukhin M, Lee HI, Hoffman BM, Dean DR, Seefeldt LC. J Am Chem Soc. 2005;127:14960–14961. doi: 10.1021/ja0539342. [DOI] [PubMed] [Google Scholar]
- 14.Barney BM, Laryukhin M, Igarashi RY, Lee HI, Dos Santos PC, Yang TC, Hoffman BM, Dean DR, Seefeldt LC. Biochemistry. 2005;44:8030–8037. doi: 10.1021/bi0504409. [DOI] [PubMed] [Google Scholar]
- 15.Lee HI, Igarashi RY, Laryukhin M, Doan PE, Dos Santos PC, Dean DR, Seefeldt LC, Hoffman BM. J Am Chem Soc. 2004;126:9563–9569. doi: 10.1021/ja048714n. [DOI] [PubMed] [Google Scholar]
- 16.Igarashi RY, Dos Santos PC, Niehaus WG, Dance IG, Dean DR, Seefeldt LC. J Biol Chem. 2004;279:34770–34775. doi: 10.1074/jbc.M403194200. [DOI] [PubMed] [Google Scholar]
- 17.Barney BM, Igarashi RY, Dos Santos PC, Dean DR, Seefeldt LC. J Biol Chem. 2004;279:53621–53624. doi: 10.1074/jbc.M410247200. [DOI] [PubMed] [Google Scholar]
- 18.Benton PMC, Laryukhin M, Mayer SM, Hoffman BM, Dean DR, Seefeldt LC. Biochemistry. 2003;42:9102–9109. doi: 10.1021/bi034595x. [DOI] [PubMed] [Google Scholar]
- 19.Mayer SM, Niehaus WG, Dean DR. J Chem Soc, Dalton Trans. 2002;5:802–807. [Google Scholar]
- 20.Christiansen J, Seefeldt LC, Dean DR. J Biol Chem. 2000;275:36104–36107. doi: 10.1074/jbc.M004889200. [DOI] [PubMed] [Google Scholar]
- 21.Robinson AC, Burgess BK, Dean DR. J Bacteriol. 1986;166:180–186. doi: 10.1128/jb.166.1.180-186.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobson MR, Brigle KE, Bennett LT, Setterquist RA, Wilson MS, Cash VL, Beynon J, Newton WE, Dean DR. J Bacteriol. 1989;171:1017–1027. doi: 10.1128/jb.171.2.1017-1027.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christiansen J, Goodwin PJ, Lanzilotta WN, Seefeldt LC, Dean DR. Biochemistry. 1998;37:12611–12623. doi: 10.1021/bi981165b. [DOI] [PubMed] [Google Scholar]
- 24.Strandberg GW, Wilson PW. Proc Natl Acad Sci U S A. 1967;58:1404–1409. doi: 10.1073/pnas.58.4.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burris RH. J Biol Chem. 1991;266:9339–9342. [PubMed] [Google Scholar]
- 26.Christiansen J, Cash VL, Seefeldt LC, Dean DR. J Biol Chem. 2000;275:11459–11464. doi: 10.1074/jbc.275.15.11459. [DOI] [PubMed] [Google Scholar]
- 27.Dilworth MJ. Biochim Biophys Acta. 1966;127:285–294. doi: 10.1016/0304-4165(66)90383-7. [DOI] [PubMed] [Google Scholar]
- 28.Hardy RWF, Holsten RD, Jackson EK, Burns RC. Plant Physiol. 1968;43:1185–1207. doi: 10.1104/pp.43.8.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelly M. Biochim Biophys Acta. 1969;191:527–540. doi: 10.1016/0005-2744(69)90346-5. [DOI] [PubMed] [Google Scholar]
- 30.Fisher K, Dilworth MJ, Kim CH, Newton WE. Biochemistry. 2000;39:2970–2979. doi: 10.1021/bi992092e. [DOI] [PubMed] [Google Scholar]
- 31.Stiefel EI. Proc Natl Acad Sci U S A. 1973;70:988–992. doi: 10.1073/pnas.70.4.988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benton PMC, Christiansen J, Dean DR, Seefeldt LC. J Am Chem Soc. 2001;123:1822–1827. doi: 10.1021/ja003662x. [DOI] [PubMed] [Google Scholar]