

Abstract
Objectives
In this study we will determine the function of the interaction between AT2R and ACE, and AT1R and ACE in the control of mesenteric resistance artery (MRA) tone from normotensive and Angiotensin II (AII)-dependent hypertensive mice.
Methods-results
Hypertension (HT) was induced by infusion of Ang-II (200ng/kg/day) for 3 weeks. Freshly MRA (100–120μm) were isolated from HT and normotensive (NT) mice and mounted in an arteriograph. Dose-response of Ang-I induced a similar contraction of MRA from NT and HT mice, which was increased after endothelium removal. AT2R antagonist (PD123319, 1μM) significantly increased Ang-I-induced contraction of MRA from NT but not from HT mice. In addition, PD123319 significantly increased in vivo blood pressure in response to Ang-I. Luminal incubation with ACE-antibody (50ng/mL) to block only endothelial ACE function significantly enhanced Ang-I-induced contraction of MRA from NT mice. ACE inhibitor (captopril, 10μM) completely blocked Ang-I-induced contraction of MRA from both animals and prevented the increased blood pressure. Freshly isolated MRA subjected to immunoprecipitation, western blot analysis and RT-PCR revealed AT1R/ACE and AT2R/ACE complexes formation, and similar AT1R, AT2R, and ACE expression level in both groups.
Conclusion
The present findings show the existence of ACE/AT2R and ACE/AT1R complexes on endothelial cells and VSMC respectively. ACE/AT2R complex plays a modulator effect on ACE/AT1R-SMC-induced contraction of MRA, which is altered in hypertension.
Keywords: Angiotensin II, angiotensin I, AT1R, AT2R, ACE, resistance artery, contraction, hypertension
INTRODUCTION
The renin-angiotensin system (RAS) is a major hormonal system involved in the regulation of cardiovascular function (1, 2). Indeed, increased RAS stimulation, especially at the tissue level, seems to be an initiating event in several pathophysiological processes including arterial dysfunction and structural remodeling in hypertension.
RAS plays a crucial role in the homeostasis of resistance artery tone and blood pressure (3, 4). The angiotensin-converting enzyme (ACE) is a major link between RAS and kinin systems (5). Ang-II activates at least two receptors type: the Ang-II type 1 receptors (AT1R) and the Ang-II type 2 receptors (AT2R) (6, 7). In resistance arteries, the stimulation of AT2R induces relaxation via a nitric oxide-dependent pathway (8–10).
It is well known that Ang-II formation in vivo is highly dependent on ACE localization and activity (11, 12). Interestingly, it has been documented that ACE is expressed on both artery endothelial and smooth muscle cells. The endothelium has a key role in vascular homeostasis through the release of relaxing factors to modulate contraction of SMC. It is well established that AT2R and AT1R are involved in SMC relaxation and contraction respectively. Previously, it has been reported that the AT2R binds directly to the AT1R and thereby antagonizes the function of the AT1R (13). Therefore, the fundamental localization of AT2R, ACE and AT1R on endothelial and SMC plays an important role in the rapid initiation and control of resistance artery tone in response to Ang I. Thus, in this study we investigated the interaction between ACE, AT1R, AT2R and their effect on resistance artery tone in normotensive and hypertensive mice. We hypothesized that endothelial modulator effect of ACE/AT2R complex on ACE/AT1R complexes-induced contraction of RA in response to Ang I was altered in hypertension.
METHODS
Animal Model
C57BL/6J male adult mice were obtained from Jackson Laboratory and separated into 2 groups. The first group was considered as control (normotensive) and the second group (hypertensive) received Ang-II (200 ng/kg/day)(14) for 3 weeks using mini-osmotic pump. These studies are conformed to the principles of the National Institute of Health “Guide for the Care and Use of Laboratory Animals”, and were approved by the LSU Institutional Animal Care and Use Committee.
Mean arterial pressure measurement
Mice were anesthetized with ketamine/xylazine (45/2.5 mg/kg i.p.) A catheter connected to a pressure transducer and recording system (www.livingsys.com) was placed in the left carotid of mouse. The animal was placed on thermo pad at 37°C. After 30 min recovery time, blood pressure was then measured. All measurements of blood pressure were performed under ketamine/xylazine-anesthetized mice.
Isolated mesenteric resistance artery
Freshly isolated mesenteric resistance arteries (mean diameter 120±5 μm at 50 mmHg of intraluminal pressure) were mounted onto two glass micropipettes in a vessel chamber and slowly pressurized to 100 mmHg using a pressure-servo-control perfusion (Living Systems Instruments, www.livingsys.com, livingsys, USA) in order to stretch the artery and set a constant artery length. Vessel diameter was continuously monitored by a video image analyzer as previously described.(15) Cannulated arterial segments were submerged in 2 ml of physiological salt solution (PSS) was used (mmol/L): NaCl 130, NaHCO3 14.9, KCl 3.7, CaCl2 · 2H2O 1.6, KH2PO4 1.2, MgSO4 · 7H2O 1.2, glucose 11, and HEPES 10, and albumin 4%. Temperature was 37°C to 38°C, pH 7.4, oxygenated with (10%O2- 5%CO2 and 85%N2) (15, 16). The functional integrity of the endothelial cell layer was assessed by endothelium-dependent vasodilation in response to acetylcholine (1 μM) after precontraction with phenylephrine (1 μM).
For pharmacological studies, mesenteric resistance arteries from normotensive and hypertensive mice were equilibrated at 50 mmHg of intraluminal pressure for 45 minutes. Then dose-response Ang-I and Ang-II contraction were performed. To avoid the desensitization, Ang-I and Ang-II were used in separate resistance arteries. Dose-responses were performed in the presence of AT2R inhibitor (PD123319, 1 μM), ACE inhibitor (captopril, 10 μM), specific ACE antibody (50 ng/ml) and without endothelium.
Endothelium Removal
The endothelial layer was removed as previously described.(17) Briefly, the mesenteric network was perfused with 1 mL carbon dioxide for 30 seconds. We verified that topically applied acetylcholine (10 μmol/L) had no more dilating effect after preconstriction with phenylephrine (10 μmol/L).
Immunoprecipitation and western blot analysis
Freshly isolated mesenteric resistance arteries from normotensive and hypertensive mice were immediately snap-frozen in liquid nitrogen. Frozen vessel segments were pulverized and re-suspended in ice-cold lysis buffer as described.(15) Each sample was immunoprecipitated with AT1R, AT2R or ACE antibodies and then subjected to immunoblotting with AT1R, AT2R or ACE antibodies (1:1000, Cell Signaling Technology). Blots were stripped and reprobed with the AT1R, AT2R or ACE antibodies used for immunoprecipitation to verify the interaction and the gel loading.
RT-PCR
RT-PCR analysis—Resistance arteries Total RNA was isolated from normotensive and hypertensive mice with the use of an RNeasy Mini kit (Qiagen, Velencia, CA), and 1 μg portions were subjected to reverse transcription (RT) with HotStarTaq (Qiagen, Velencia, CA). One-tenth of the resulting cDNA was amplified by PCR with the following primers (sense and antisense, respectively): mouse AT1R, 5′-Forward CCA TTG TCC ACC CGA TGA AG-3′, 5′-Reverse TGC AGG TGA CTT TGG CCAC-3′; mouse AT2R, 5′-Forward CAG CAG CCG TCC TTT TGA TAA-3′, 5′-Reverse TTA TCT GAT GGT TTG TGT GAG CAA-3′; mouse actin, 5′-Forward AGA GGG AAA TCG TGC TGT AC-3′ and 5′-Reverse CAA TAG TGA TGA CCT GGC CGT-3′. Amplification was performed for 30 cycles of denaturation for 30 s at 94°C, annealing for 15 s at 63°C, and primer extension for 60 s at 72°C for AT1R or for 30 cycles of denaturation for 45 s at 94°C, annealing for 45 s at 53°C, and primer extension for 1.5 min at 72°C for AT2R. The resulting products were analyzed by electrophoresis using 2% agarose gels and staining with ethidium bromide.
Statistical analysis
Results are expressed as mean±sem, where n is the number of arterial segments studied. Significance of the differences between groups was determined by 1- or 2-factor ANOVA, where appropriate. Differences were considered significant at P<0.05.
RESULTS
Mean arterial pressure (MAP) was significantly increased in mice treated with Ang-II for 3 weeks compared to control (87.3±2 vs. 106±2 mmHg, p<0.05), indicating that Ang II treatment was sufficient to induce increase blood pressure in normal mice.
Freshly isolated resistance arteries were mounted in an arteriograph under 50 mmHg and were subjected to Ang-I dose-response (10−10 to 10−7 M). The application of Ang-I was associated with a similar degree of contraction in both groups indicating a local and functional presence of ACE of mesenteric resistance artery (Figure 1, left panel). Pretreatment of resistance arteries with the ACE inhibitor (captopril, 10 μM) completely blocked the contraction to Ang-I in both groups (Figure 1, left panel). Contraction and relaxation induced by phenylephrine and acetylcholine respectively were not affected with ACE inhibitor indicating the specificity of captopril (data not shown). Intraluminal incubation with specific ACE antibodies (to block only endothelial ACE) significantly enhanced Ang I-induced contraction of resistance artery from normotensive mice but no effect was observed in hypertensive mice (Figure 1, right panel). Intraluminal incubation with non-immune IgG antibody had no effect on the contraction induced by Ang-I (data not shown) indicating the specificity of ACE antibody. We previously have shown than AT2R stimulation induced resistance artery relaxation (16, 18). Thus, the inhibition of AT2R with PD123319 (1 μM) significantly increased Ang I-induced contraction of resistance artery from normotensive but no effect was observed in hypertensive mice (Figure 2, left panel). In both groups, endothelium removal significantly increased Ang I-induced contraction of resistance artery (Figure 2, right panel).
Figure 1.

Left panel: Dose-response to Ang-I (10−10 to 10−7 mol/L) and diameter decrease relationship in normotensive (NT) and hypertensive (HT) mouse mesenteric resistance artery held at 50 mmHg without and with ACE inhibitor (captopril, 10 μM), n=6 per group. *P<0.05, statistically significant, NT vs. NT+captopril; HTN vs. HTN+captopril; Right panel: Dose-response to Ang-I (10−10 to 10−7 mol/L) and diameter decrease relationship in normotensive (NT) and hypertensive (HT) mouse mesenteric resistance artery held at 50 mmHg without and with intraluminal specific ACE antibody (50 ng/ml), n=6 per group. *P<0.05, statistically significant, NT vs. NT+ACE antibody at 10−7 of Ang-I
Figure 2.

Left panel: Dose-response to Ang-I (10−10 to 10−7 mol/L) and diameter decrease relationship in normotensive (NT) and hypertensive (HT) mouse mesenteric resistance artery held at 50 mmHg without and with AT2R inhibitor (PD123, 1 μM), n=6 per group. *P<0.05, statistically significant, NT vs. NT+PD123; Right panel: Dose-response to Ang-I (10−10 to 10−7 mol/L) and diameter decrease relationship in normotensive (NT) and hypertensive (HT) mouse mesenteric resistance artery held at 50 mmHg without and with endothelium, n=6 per group. *P<0.05, statistically significant, NT vs. NT without endothelium; HTN vs. HTN without endothelium
In normotensive mice, Ang-II dose-response induced an elevated contraction compared to contraction induced by Ang-I (Figure 1), which was not affected by AT2R inhibition (Figure 3).
Figure 3.

Dose-response to Ang-II (10−10 to 10−7 mol/L) and diameter decrease relationship in normotensive (NT) mouse mesenteric resistance artery held at 50 mmHg without and with AT2R inhibitor (PD123, 1 μM), n=6 per group.
Freshly in vivo isolated mesenteric resistance arteries were subjected to immunoprecipitation, western blot analysis and RT-PCR. Data showed that AT1R and AT2R protein and mRNA were expressed to a similar level in both groups (Figure 4A–B). Co-immunoprecipitation analysis of arterial lysates revealed complexes formation AT1R/ACE and AT2R/ACE (Figure 4C). Non-immune IgG was used as control and no effect was observed indicating the specificity of immunoprecipitation approach (data not shown).
Figure 4.
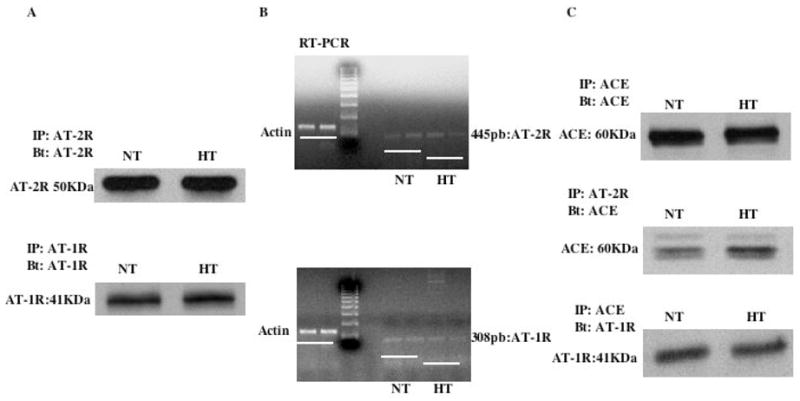
4A: Western blot analysis showing the presence of AT2R (upper panel) and AT1R (Lower panel) protein expression in normotensive (NT) and hypertensive (HT) mouse resistance arteries; 4B: RT-PCR showing no difference of AT2R (upper panel) and AT1R (Lower panel) mRNA level in resistance arteries from normotensive (NT) and hypertensive (HT) mouse; 4C: immunoprecipitation and western blot analysis showing the presence of ACE in both groups (Upper panel), the interaction between ACE and AT2R (Middle panel) and between ACE and AT1R (Lower panel) in resistance arteries from both groups. n=6 per group.
We measured in vivo blood pressure in response to acute Ang-I vein injection in normotensive mice. Ang-I (100 nM) significantly increased blood pressure, which was enhanced in the presence of AT2R inhibition (Figure 5). However, increased blood pressure in response to acute Ang-I perfusion was completely blocked in the presence of ACE inhibitor (Figure 5).
Figure 5.

Effect of acute injection of Ang I on in vivo blood pressure in the absence or presence of AT2R antagonist (PD123) or ACE inhibitor (captopril); n=6 per group. *P<0.05, statistically significant, Ang I vs. Ang I+PD123; Ang I vs Ang I+captopril.
DISCUSSION
In this study, the new findings consist of the existence of local and functional complexes interaction between ACE and AT2R on endothelial cells and, ACE and AT1R on smooth muscle cells dictating mouse resistance artery tone in response to Ang-I. The pattern of this distribution and interaction is physiologically very important since in vivo endothelial cells and VSMC are in contact with Ang I first and then Ang-II derived from Ang-I through ACE activation. Thus, the AT2R vasodilatory effect is observed in the presence of Ang-I but not under Ang-II, and is altered in Ang II-dependent hypertension.
Resistance arteries play a crucial role in the regulation of blood pressure and tissue perfusion. Resistance artery tone is mainly regulated by mechanical (pressure and shear stress) and humoral factors such as Ang-II and bradykinin (4, 17, 19, 20). The two mechanical factors antagonize each other. Thus, increased pressure induces contraction (myogenic tone) and increased shear stress induces relaxation (15, 21, 22). This balance between contraction/relaxation in response to mechanical factors is very crucial in the regulation of resistance artery tone and subsequently blood pressure and tissue perfusion. A perturbation of this balance (generally more contraction and less relaxation) is a hallmark of cardiovascular and metabolic disease such as hypertension and diabetes. Humoral factors are also involved in the control resistance artery tone. Thus, Ang-II acts through two different receptors: AT1R and AT2R. The activation of AT1R induces resistance artery contraction, while the activation of AT2R induces relaxation (16). It is worthwhile to note that AT2R activation does not fully relax artery contraction induced by phenylephrine compared to acetylcholine effect (18). Others and we have shown that AT2R activation induces artery relaxation of resistance artery from normotensive rat (16, 18).
Functionally, the endothelium removal significantly enhanced resistance artery contraction in response to Ang-I. This observation is in agreement with our previous study showing the presence of AT2R on the endothelium of resistance artery (16). In Ang-II-dependent hypertensive mice, Ang-I induced a similar contraction of resistance artery to that observed in normotensive mice. These data indicate that hypertension Ang-II-dependent does not affect the contraction induced by Ang-I, which is in concordance with previous studies showing that hypertension is associated with increased peripheral resistance but not contraction to vasoconstrictors (23, 24). The mechanism responsible for these observations is unclear, but may reflect compensatory vasodilation elicited by other vasorelaxing receptors. Our data showed AT2R dysfunction of resistance artery from hypertensive mice are in agreement with previous studies (25). Surprisingly, endothelium removal developed a similar contraction of resistance artery from hypertensive and normotensive mice in response to Ang I. To extend the local interaction between ACE and Ang-II receptors, we first used a functional approach with different pharmacological inhibitors and function-blocking using ACE antibodies. AT2R inhibition significantly enhanced contraction of resistance artery from normotensive but not from hypertensive mice. In addition, the intraluminal incubation with specific ACE antibody (which blocks only ACE on endothelial cell) significantly potentiated contraction of resistance artery in response to Ang-I in normotensive mice, but no from hypertensive mice. Together, these data corroborate physiological and functional interactions between AT2R and ACE in a complex on endothelial cell membrane. To strengthen our data, we performed immunoprecipitation and western blot analysis on freshly isolated mesenteric resistance arteries, and found an interaction between AT2R and ACE, and AT1R and ACE. In addition, our data revealed a similar expression level of AT2R, AT1R and ACE mRNA and protein expression of resistance artery from normotensive and hypertensive mice. These data are not in agreement with a previous study showing a decrease of AT2R protein in hypertensive rat (25). This difference could be related to both the species used and the hypertension model. To strengthen our in vitro data, we measured in vivo blood pressure after acute venous injection of Ang-I with and without AT2R or ACE inhibition. We observed a significant enhance blood pressure in response to Ang-I upon AT2R inhibition. On the other hand, the increased blood pressure in response to Ang-I was completely prevented in the presence of ACE inhibitor. These data indicate that AT2R plays a role in the regulation of resistance arteries tone and subsequently blood pressure, and only ACE is involved in Ang-I conversion to Ang-II in mouse resistance arteries. Our data are not in agreement with previous studies showing a role of chymase in the conversion of Ang I to Ang II of mesenteric artery (not mesenteric resistance artery) (26). Our data are supported by in vitro and in vivo studies indicating that only ACE is involved in Ang-I conversion to Ang-II in mouse mesenteric resistance artery. In figure 6, we established a model of complexes formation ACE/AT2R and ACE/AT1R on endothelial and smooth muscle cells, respectively.
Figure 6.

This model shows the physiological role of ACE/AT2R and ACE/AT1R complexes on endothelial and smooth muscle cells respectively in the regulation of resistance artery tone. This model illustrates the access and the activation of AT2R and AT1R under Ang I leading to a module contraction. On the other hand, under Ang II, there is a fast access of Ang II to AT1R and less to AT2R leading to a high contraction.
Thus, our data provide evidence regarding the existence of interaction between ACE and AT2R on endothelial cells of MRA modulating the AT1R-ACE-activation-dependent contraction. Since resistance artery should respond instantly to any humoral factors changes (such Ang I) compared to large artery, it is such crucial of an important distribution of ACE, AT1R and AT2R in order to regulate the contraction induced by Ang II formation. In vitro the Ang I is formed before Ang II through ACE activation. Thus, when Ang I is distributed close to endothelial and smooth muscle cells, the conversion of Ang I to Ang II will simultaneously distributed to AT2R and AT1R. Endothelial ACE/AT2R complex may facilitate for a fast diffusion of Ang-II to AT2R and AT1R leading to intermediate tone in the presence of Ang I. This conclusion was confirmed by a great contraction of MRA in response to exogenous Ang II compared to Ang I. The modulator effect of endothelial ACE/AT2R complex was altered in Ang II-dependent hypertension.
Acknowledgments
This work was supported by American Heart Association SDG grant (0430278N, PI: Khalid Matrougui) and NIH-HL072889 (PI, Hamid Boulares).
References
- 1.Wang DH, Du Y, Yao A. Regulation of the gene-encoding angiotensin II receptor in vascular tissue. Microcirculation. 1996;3:237–239. doi: 10.3109/10739689609148295. [DOI] [PubMed] [Google Scholar]
- 2.Dzau VJ. Molecular and physiological aspects of tissue renin-angiotensin system: emphasis on cardiovascular control. J Hypertens Suppl. 1988;6:S7–12. [PubMed] [Google Scholar]
- 3.Hall JE. Control of blood pressure by the renin-angiotensin-aldosterone system. Clin Cardiol. 1991;14:IV6-21. doi: 10.1002/clc.4960141802. discussion IV51–25. [DOI] [PubMed] [Google Scholar]
- 4.Matrougui K, Levy BI, Henrion D. Tissue angiotensin II and endothelin-1 modulate differently the response to flow in mesenteric resistance arteries of normotensive and spontaneously hypertensive rats. Br J Pharmacol. 2000;130:521–526. doi: 10.1038/sj.bjp.0703371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waeber B, Juillerat-Jeanneret L, Aubert JF, Schapira M, Nussberger J, Brunner HR. Involvement of the kallikrein-kinin system in the antihypertensive effect of the angiotensin converting enzyme inhibitors. Br J Clin Pharmacol. 1989;27(Suppl 2):175S–180S. doi: 10.1111/j.1365-2125.1989.tb03479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheuer DA, Perrone MH. Angiotensin type 2 receptors mediate depressor phase of biphasic pressure response to angiotensin. Am J Physiol. 1993;264:R917–923. doi: 10.1152/ajpregu.1993.264.5.R917. [DOI] [PubMed] [Google Scholar]
- 7.Wiemer G, Scholkens BA, Wagner A, Heitsch H, Linz W. The possible role of angiotensin II subtype AT2 receptors in endothelial cells and isolated ischemic rat hearts. J Hypertens Suppl. 1993;11:S234–235. [PubMed] [Google Scholar]
- 8.Matrougui K, Loufrani L, Levy BI, Henrion D. High NaCl intake decreases both flow-induced dilation and pressure-induced myogenic tone in resistance arteries from normotensive rats: involvement of cyclooxygenase-2. Pharmacol Toxicol. 2001;89:183–187. doi: 10.1111/j.0901-9928.2001.890407.x. [DOI] [PubMed] [Google Scholar]
- 9.Siragy HM, Carey RM. The subtype-2 (AT2) angiotensin receptor regulates renal cyclic guanosine 3′, 5′-monophosphate and AT1 receptor-mediated prostaglandin E2 production in conscious rats. J Clin Invest. 1996;97:1978–1982. doi: 10.1172/JCI118630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ichiki T, Labosky PA, Shiota C, et al. Effects on blood pressure and exploratory behaviour of mice lacking angiotensin II type-2 receptor. Nature. 1995;377:748–750. doi: 10.1038/377748a0. [DOI] [PubMed] [Google Scholar]
- 11.Robertson JI, Tillman DM. Converting enzyme inhibitors in the treatment of hypertension. J Cardiovasc Pharmacol. 1987;10(Suppl 7):S43–48. doi: 10.1097/00005344-198706107-00007. [DOI] [PubMed] [Google Scholar]
- 12.Robertson JI, Tillman DM, Herd GW. The clinical use of angiotensin converting enzyme inhibitors in hypertension and cardiac failure. Clin Exp Hypertens A. 1987;9:489–511. doi: 10.3109/10641968709164218. [DOI] [PubMed] [Google Scholar]
- 13.AbdAlla S, Lother H, Abdel-tawab AM, Quitterer U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J Biol Chem. 2001;276:39721–39726. doi: 10.1074/jbc.M105253200. [DOI] [PubMed] [Google Scholar]
- 14.Izumiya Y, Kim S, Izumi Y, et al. Apoptosis signal-regulating kinase 1 plays a pivotal role in angiotensin II-induced cardiac hypertrophy and remodeling. Circ Res. 2003;93:874–883. doi: 10.1161/01.RES.0000100665.67510.F5. [DOI] [PubMed] [Google Scholar]
- 15.Lucchesi PA, Sabri A, Belmadani S, Matrougui K. Involvement of metalloproteinases 2/9 in epidermal growth factor receptor transactivation in pressure-induced myogenic tone in mouse mesenteric resistance arteries. Circulation. 2004;110:3587–3593. doi: 10.1161/01.CIR.0000148780.36121.47. [DOI] [PubMed] [Google Scholar]
- 16.Matrougui K, Loufrani L, Heymes C, Levy BI, Henrion D. Activation of AT(2) receptors by endogenous angiotensin II is involved in flow-induced dilation in rat resistance arteries. Hypertension. 1999;34:659–665. doi: 10.1161/01.hyp.34.4.659. [DOI] [PubMed] [Google Scholar]
- 17.Matrougui K, Maclouf J, Levy BI, Henrion D. Impaired nitric oxide- and prostaglandin-mediated responses to flow in resistance arteries of hypertensive rats. Hypertension. 1997;30:942–947. doi: 10.1161/01.hyp.30.4.942. [DOI] [PubMed] [Google Scholar]
- 18.Widdop RE, Matrougui K, Levy BI, Henrion D. AT2 receptor-mediated relaxation is preserved after long-term AT1 receptor blockade. Hypertension. 2002;40:516–520. doi: 10.1161/01.hyp.0000033224.99806.8a. [DOI] [PubMed] [Google Scholar]
- 19.Bevan JA. Vascular myogenic or stretch-dependent tone. J Cardiovasc Pharmacol. 1985;7(Suppl 3):S129–136. doi: 10.1097/00005344-198500073-00015. [DOI] [PubMed] [Google Scholar]
- 20.Bevan JA, Garcia-Roldan JL, Joyce EH. Resistance artery tone is influenced independently by pressure and by flow. Blood Vessels. 1990;27:202–207. doi: 10.1159/000158811. [DOI] [PubMed] [Google Scholar]
- 21.Koller A, Huang A. Impaired nitric oxide-mediated flow-induced dilation in arterioles of spontaneously hypertensive rats. Circ Res. 1994;74:416–421. doi: 10.1161/01.res.74.3.416. [DOI] [PubMed] [Google Scholar]
- 22.Henrion D, Dechaux E, Dowell FJ, et al. Alteration of flow-induced dilatation in mesenteric resistance arteries of L-NAME treated rats and its partial association with induction of cyclo-oxygenase-2. Br J Pharmacol. 1997;121:83–90. doi: 10.1038/sj.bjp.0701109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schiffrin EL. Reactivity of small blood vessels in hypertension: relation with structural changes. State of the art lecture. Hypertension. 1992;19:II1–9. doi: 10.1161/01.hyp.19.2_suppl.ii1-a. [DOI] [PubMed] [Google Scholar]
- 24.Deng LY, Schiffrin EL. Effects of endothelin on resistance arteries of DOCA-salt hypertensive rats. Am J Physiol. 1992;262:H1782–1787. doi: 10.1152/ajpheart.1992.262.6.H1782. [DOI] [PubMed] [Google Scholar]
- 25.You D, Loufrani L, Baron C, Levy BI, Widdop RE, Henrion D. High blood pressure reduction reverses angiotensin II type 2 receptor-mediated vasoconstriction into vasodilation in spontaneously hypertensive rats. Circulation. 2005;111:1006–1011. doi: 10.1161/01.CIR.0000156503.62815.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li M, Liu K, Michalicek J, et al. Involvement of chymase-mediated angiotensin II generation in blood pressure regulation. J Clin Invest. 2004;114:112–120. doi: 10.1172/JCI20805. [DOI] [PMC free article] [PubMed] [Google Scholar]