

Abstract
BACKGROUND:
Generation of reactive oxygen species (ROS) is associated with cardioprotection imparted by ischemic preconditioning (IPC) and pharmacological PC (PPC). The authors have previously shown that IPC or PPC, using the mitochondrial ATP-sensitive K+ channel opener diazoxide (DZ), reduce mitochondrial Ca2+ ([Ca2+]m) during ischemia and reperfusion.
OBJECTIVES:
To test the hypothesis that both IPC and PPC (using DZ) lead to reduced [Ca2+]m and improved functional recovery via a ROS-dependent mechanism.
METHODS:
Intracellular Ca2+ ([Ca2+]i) and [Ca2+]m were measured in isolated perfused rat hearts loaded with the fluorescent indicator indo-1 acetoxymethyl ester. [Ca2+]m was determined by quenching the cytosolic indo-1 signal using manganese before ischemia (25 min). IPC and DZ (100 μM) group hearts were studied with and without the ROS scavenger N-2-mercaptopropionyl glycine (400 μM) (2-MPG).
RESULTS:
Both IPC and DZ significantly reduced [Ca2+]i and [Ca2+]m on reperfusion compared with the control. Administration of 2-MPG with washout before ischemia significantly attenuated the reduction in [Ca2+]m observed on reperfusion in both the IPC and DZ groups. Additionally, the myocardial functional protection imparted by IPC or DZ was lost with the administration of 2-MPG.
CONCLUSIONS:
The [Ca2+]m-reducing effect of IPC and DZ was attenuated with the administration of 2-MPG, resulting in decreased myocardial functional performance and increased release of creatine kinase, a marker of cellular injury. It can be concluded that IPC and DZ impart their protective effect via a mechanism involving ROS generation before the ischemic episode.
Keywords: Calcium, Ischemia, KATP channel, Oxygen radicals, Reperfusion
Ischemic preconditioning (IPC) and pharmacological treatment with mitochondrial ATP-sensitive K+ (mitoKATP) channel openers, such as diazoxide (DZ), appear to protect ischemic myocardium, in part via a reactive oxygen species (ROS)-dependent mechanism (1,2). Although clarification of this protective mechanism remains elusive, it potentially involves activation and translocation of protein kinase C (PKC) (1,3), diminution of ROS production during reperfusion (4) and maintenance of mitochondrial membrane potential (5). It has been demonstrated in selected species that IPC can only be blocked using a combination of PKC and tyrosine kinase blockers, demonstrating the nexus between ROS, kinase activity and IPC (6).
The association, and likely causal role, of mitochondrial Ca2+ ([Ca2+]m) overload with increased cellular injury during ischemia/reperfusion (I/R) has been established (7). Specifically, pharmacological inhibition of [Ca2+]m overload with ruthenium red has been shown to limit reperfusion injury (8), and the level of [Ca2+]m has been demonstrated to be inversely related to the degree of functional recovery following I/R (7). In these latter experiments, IPC and DZ were shown to attenuate the increase in [Ca2+]m during I/R, an effect eliminated by coadministration of the mitoKATP channel blocker 5-hydroxydecanoate (5-HD) (7). Given the data regarding the role of ROS in IPC and DZ administration, and the evidence that limitation of [Ca2+]m overload is associated with functional protection, we tested the hypothesis that lower [Ca2+]m on reperfusion and improved functional recovery are dependent on preischemic generation of ROS.
To test this hypothesis, an isolated perfused rat heart model of I/R was employed with beat-by-beat fluorescent measurements of intracellular Ca2+ ([Ca2+]i) and [Ca2+]m, and parallel measurements of hemodynamics and creatine kinase (CK), a marker of cellular injury. The ROS scavenger N-2-mercaptopropionyl glycine (2-MPG) was administered only before ischemia, bridging IPC episodes or during administration of DZ, and these results were compared with untreated hearts.
MATERIALS AND METHODS
Heart perfusion and measurement of function
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the United States National Institute of Health (NIH Publication No 85–23, revised 1996). As previously described (7), male Sprague-Dawley rat (250 g to 350 g) hearts were retrograde perfused with modified Krebs-Henseleit solution containing 11 mM glucose, 25 mM NaHCO3, 118 mM NaCl, 1 mM CaCl2, 4.6 mM KCl, 1.2 mM MgSO4 and 1.2 mM KH2PO4 at a perfusion pressure of 73.5 mmHg. Heart temperature was maintained at 37±0.5°C under all conditions and hearts were oxygenated with a gas mixture of 95% O2 and 5% CO2. Left ventricular end-diastolic pressure (LVEDP), systolic pressure (LVSP) and heart rate (HR) were continuously monitored, stored and analyzed using a computer-based data acquisition system (Biopac Systems, USA). Parallel experiments were performed for measurement of myocardial function and injury to exclude any effects of the Ca2+ indicator or MnCl2 loading on physiological measures.
Fluorescence instrumentation and measurements
Fluorescence measurements were performed as previously described in detail (9,10) using a modified spectrofluorometer (SLM Aminco 8100, Jobin Yvon, USA). Excitation light from a 450 W xenon arc lamp (Ushio Inc, Japan) was filtered through a 350 nm interference filter and focused onto the ingoing leg of a quartz bifurcated fibre bundle (Dolan-Jenner Industries Inc, USA). The common leg of this 1.57 mm diameter bundle fibre was girdled against the epicardial surface of the left ventricle, avoiding visible vessels. A shutter in front of the excitation light was opened for only seconds at a time during data acquisition to prevent bleaching of indo-1 acetoxymethyl (AM) ester fluorescence. The fluorescence signal was transferred via the outgoing leg of the bundle and separated by 385 nm and 456 nm interference filters before detection by photomultiplier tubes (Hamamatsu, Japan).
Indo-1 AM ester loading
After a 10 min equilibration period, baseline background fluorescence (primarily NADH [9,10]) was measured and subtracted from all subsequent fluorescence measurements upon calculation of Ca2+ values. Hearts were then loaded for 25 min to 30 min with modified Krebs-Henseleit buffer containing indo-1 AM ester (2.4 μM dissolved in DMSO and Pluronic F-127, 20% w/v, Molecular Probes, USA) and fetal bovine serum (1%). Probenecid (0.1 mM) was added to all perfusates to slow the extrusion of indo-1 AM ester from the myocytes (11). Residual indo-1 AM ester was washed out by perfusing with modified Krebs-Henseleit buffer for 10 min. An experiment was discarded if the fluorescence intensity at either 385 nm or 456 nm was less than twice the background fluorescence during any phase of the experimental protocol.
Manganese quenching of cytosolic fluorescence
To determine mitochondrial fluorescence, cytosolic fluorescence was quenched by adding MnCl2 to a final concentration of 17.5 μM to the perfusate 5 min to 10 min before I/R. Adequate quenching was verified by the loss of Ca2+ transients after perfusion with manganese. As in prior studies, the addition of manganese did not alter HR or developed pressure (7).
Experimental protocols
A schematic of the protocols is shown in Figure 1. All hearts had a 10 min equilibration period with baseline measurements of LVEDP, LVSP, HR and background fluorescence. Loading of indo-1 AM ester for 25 min to 30 min was followed by a 10 min washout period in each group. All hearts were subjected to 25 min of global ischemia followed by 30 min of reperfusion. Total Ca2+, [Ca2+]m, functional data and CK samples were obtained separately in parallel experiments. Hemodynamic and indo-1 fluorescence intensity measurements were performed every 5 min. Hearts were randomly assigned to one of six groups.
Figure 1).
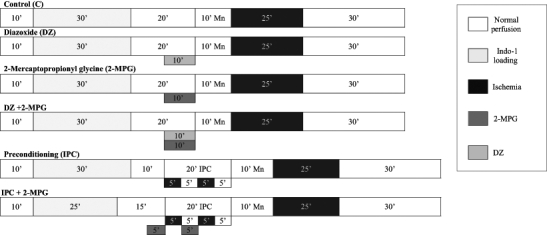
Protocol schematic illustrating the duration of each step for the groups in each protocol. Each group had either a 10 min (10’) manganese quench (with measurement of mitochondrial Ca2+) or an equivalent washout period using buffer. Indo-1 Indo-1 acetoxymethyl ester
Calculation of [Ca2+]i
[Ca2+]i was calculated using the standard equation for fluorescent Ca2+ indicators (12):
where R is the ratio of fluorescence at 385 nm and 456 nm, and Rmin and Rmax are the fluorescence ratios at zero and saturating [Ca2+]i, respectively, as determined in a previous study (9). A dissociation constant (Kd) of 594 nM was used to calculate [Ca2+]i (13,14). S456 is the ratio of fluorescence intensities during saturating and zero [Ca2+]i at 456 nm emission wavelength (13). Experiments in which the signal to noise ratio was less than two were not included in the data analysis. A sample set of fluorescent measurements is shown in Figure 2.
Figure 2).
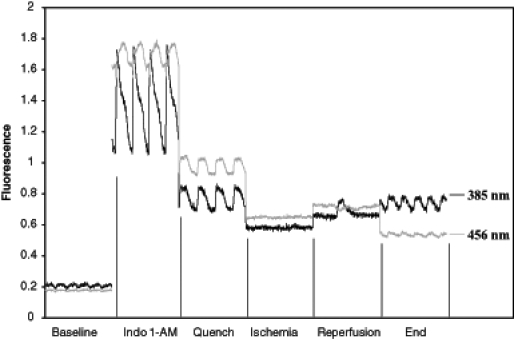
Representative fluorescent signals at 385 nm and 456 nm obtained over one second at specified time points in one experiment. Autofluorescence is measured under baseline conditions before loading with indo-1 acetoxymethyl ester. Following loading, Ca2+ transients are observed – these transients are abolished with a manganese quench. The resulting signals after quenching demonstrate concordance of the signals at the two wavelengths due to cardiac motion (ie, no Ca2+ transients are observed). Cardiac motion is absent during ischemia, resulting in flat traces. Reperfusion results in irregular contractions, which become regular by the end reperfusion period. AM acetoxymethyl
Calculation of [Ca2+]m
[Ca2+]m was calculated using the same equation employed to calculate [Ca2+]i from the fluorescent intensity after manganese quenching. On the basis of prior studies (15–17), the calibration parameters were determined to be the same in the cytosolic and mitochondrial compartments. Because of confounding factors such as changes in pH and NADH autofluoresence that modify the fluorescent signal or change the Kd of indo-1 AM ester during ischemia (10,13), Ca2+ was not determined during ischemia, but only under baseline and reperfusion conditions.
CK
A commercially available CK kit (Diagnostic Chemicals Limited, USA) was used to quantify the amount of cardiac damage sustained during I/R. An effluent sample was taken at the end of baseline perfusion, and total CK release was determined from the effluent collected on ice during the entire reperfusion period. Briefly, 40 μL of effluent was added to 1 mL of CK reagent and allowed to incubate for 3 min. Absorbance was measured at 340 nm every 30 s for 10 min following incubation. All reagents were stored and ran at 37 °C. The resultant slope was used to determine CK activity:
where ΔA/min is the change in absorbance/min, 1.040 mL is the total assay volume, 1000 converts U/mL to U/L, 6.22 M−1cm−1 is the absorbance coefficient of NADPH, 1 cm is the light path, and 0.040 mL is the sample volume. CK (U/L) was converted to a final reported value of U/g dry weight:
All materials were obtained from Sigma (USA) and were of the highest quality unless stated otherwise.
Statistical analysis
Data are presented as mean ± SEM. Differences in data between groups at each of the three time periods (baseline, 5 min reperfusion and end reperfusion) were analyzed using ANOVA. Bonferroni’s multiple comparison post-test was used if the ANOVA value was significant. A value of P<0.05 was considered statistically significant. A two-tailed unpaired Student’s t test was used when examining CK data. A value of P<0.05 was again considered statistically significant.
RESULTS
[Ca2+]i
Figures 3A and 3B show values of [Ca2+]i at baseline, 5 min of reperfusion and end reperfusion. No statistically significant differences in Ca2+ concentration between groups at baseline or at 5 min of reperfusion were observed. [Ca2+]i in the control group (685±110 nM) was significantly greater at end reperfusion than in all other groups (2-MPG [310±34 nM], IPC [310±21 nM], IPC plus 2-MPG [271±95 nM], DZ [306±29 nM], DZ plus 2-MPG [316±122 nM], P<0.01 versus control).
Figure 3).
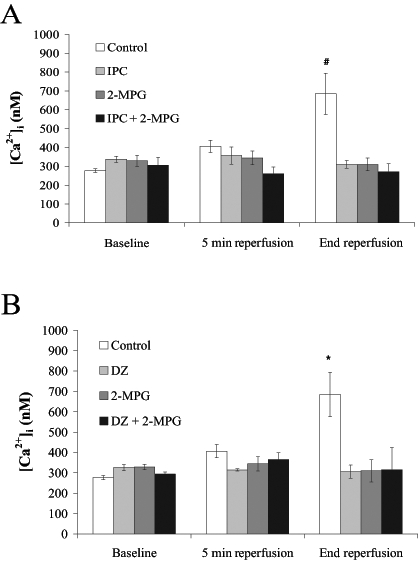
Intracellular Ca2+ concentration ([Ca2+]i) at baseline, 5 min reperfusion and end reperfusion. Rat hearts were subjected to A) control perfusion and ischemic preconditioning (IPC) ± N-2-mercaptopropionyl glycine (2-MPG) (400 μM); and B) control perfusion and pharmacological preconditioning with diazoxide (DZ) (100 μM) ± 2-MPG (400 μM). All values are expressed as mean ± SEM (n=5). Statistical significance was determined using one-way ANOVA with Bonferroni’s post-test. #IPC, 2-MPG and IPC plus 2-MPG were significantly different versus the control (P<0.01). *DZ, 2-MPG and 2-MPG plus DZ were significantly different versus the control (P<0.01)
[Ca2+]m
Figures 4A and 4B show [Ca2+]m at baseline, 5 min of reperfusion and end reperfusion. As in the measurement of [Ca2+]i, no statistically significant differences in Ca2+ were observed during baseline or after 5 min of reperfusion.
Figure 4).
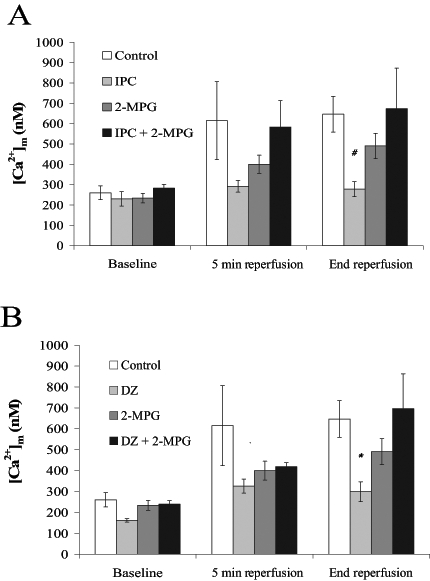
Mitochondrial Ca2+ concentration ([Ca2+]m) at baseline, 5 min reperfusion and end reperfusion. Rat hearts were subjected to A) control perfusion and ischemic preconditioning (IPC) ± N-2-mercaptopropionyl glycine (2-MPG) (400 μM); and B) control perfusion and pharmacological preconditioning with diazoxide (DZ) (100 μM) ± 2-MPG (400 μM). All values are expressed as mean ± SEM (n=5 to 12). Statistical significance was determined using one-way ANOVA with Bonferroni’s post-test. #IPC was significantly different from control and IPC plus 2-MPG (P<0.05). *DZ was significantly different from the control and from DZ plus 2-MPG (P<0.05)
Consistent with prior data (7), [Ca2+]m at end reperfusion was significantly lower in the IPC (306±34 nM) and DZ (299±47 nM) group hearts compared with control hearts (647±88 nM, P<0.05). Coadministration of 2-MPG resulted in increases in [Ca2+]m during reperfusion in both the DZ and IPC groups, resulting in significantly higher [Ca2+]m at end reperfusion (IPC plus 2-MPG [674±199 nM] versus IPC [P<0.05]; and DZ plus 2-MPG [697±166 nM] versus DZ [P<0.05]). The 2-MPG group had a [Ca2+]m concentration that was intermediate between control, DZ and IPC groups (491±62 nM).
HEMODYNAMICS
Baseline functional parameters
Baseline functional parameters for all six groups of animals are found in Table 1. No statistically significant differences were observed at baseline in any group or functional category.
TABLE 1.
Baseline measurements of myocardial functional characteristics
| Group | LVSP (mmHg) | LVEDP (mmHg) | DP (mmHg) | Heart rate (beats/min) |
|---|---|---|---|---|
| Control (n=9) | 72.8±3.7 | 6.1±0.77 | 66.7±4.1 | 310±9.0 |
| IPC (n=5) | 74.2±7.1 | 4.3±0.45 | 69.9±7.4 | 301±7.5 |
| DZ (n=5) | 62.9±5.0 | 4.7±0.77 | 63.2±7.4 | 303±22.0 |
| 2-MPG (n=5) | 63.3±5.9 | 4.4±0.52 | 58.9±5.7 | 307±13.6 |
| IPC plus 2-MPG (n=8) | 67.9±5.7 | 4.9±0.37 | 62.9±5.9 | 301±13.1 |
| DZ plus 2-MPG (n=6) | 73.9±2.3 | 4.4±0.33 | 69.4±2.8 | 286±13.4 |
Values are expressed as mean ± SEM. No statistical significance between any baseline measurements were observed. DP Developed pressure; DZ Diazoxide; IPC Ischemic preconditioning; LVEDP Left ventricular end-diastolic pressure; LVSP Left ventricular systolic pressure; 2-MPG N-2-mercaptopropionyl glycine
Left ventricular developed pressure
Figures 5A and 5B show left ventricular developed pressure (LVDP) as a percentage of baseline in the six groups at end reperfusion. IPC group hearts recovered 95±3% of their baseline LVDP compared with the IPC plus 2-MPG group which recovered only 26±12% (P<0.05 versus IPC). In a similar fashion, DZ group hearts recovered 89±15% of their baseline LVDP compared with the DZ plus 2-MPG group which recovered only 40±10% (P<0.05 versus DZ). When using ANOVA, the recovery of LVDP was not significantly greater in the IPC and DZ group hearts than either control or 2-MPG group hearts.
Figure 5).
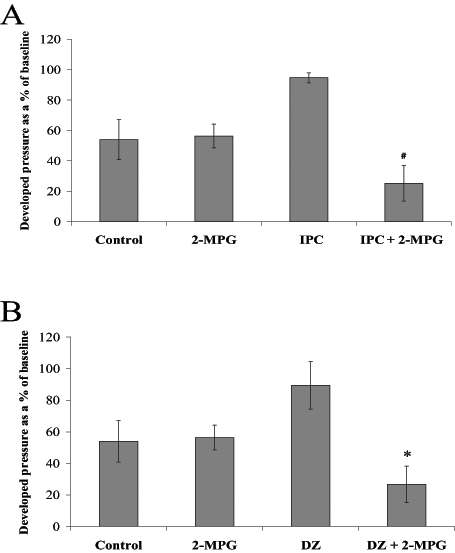
Left ventricular developed pressure at end reperfusion presented as a percentage of baseline. Rat hearts were subjected to A) control perfusion and ischemic preconditioning (IPC) ± N-2-mercaptopropionyl glycine (2-MPG) (400 μM); and B) control perfusion and pharmacological preconditioning with diazoxide (DZ) (100 μM) ± 2-MPG (400 μM). All values are expressed as mean ± SEM (n=5 to 9). Statistical significance was determined using one-way ANOVA with Bonferroni’s post-test. #IPC plus 2-MPG was significantly different from IPC (P<0.05). *DZ plus 2-MPG was significantly different from DZ (P<0.05)
LVEDP
LVEDP increased during I/R in all groups. DZ and IPC group hearts had the smallest increase in LVEDP (328±57% and 396±126% of baseline, respectively, versus 705±109% in control hearts). 2-MPG alone increased the LVEDP (719±98%), while coadministration of 2-MPG with IPC or DZ resulted in significant increases in LVEDP during I/R: IPC plus 2-MPG, 1004±145% and DZ plus 2-MPG, 1089±195%; IPC versus IPC plus 2-MPG (P<0.05), DZ versus DZ plus 2-MPG (P<0.01).
CK
CK release significantly increased from baseline to end reperfusion in all groups (Figures 6A and 6B). IPC and DZ reduced CK release on reperfusion by approximately 50%; however, this reduction, which is similar in magnitude to previous reports (7,18) did not reach statistical significance in the IPC group hearts. Coadministration of 2-MPG with DZ caused a significant increase in the amount of CK released compared with the intervention alone.
Figure 6).
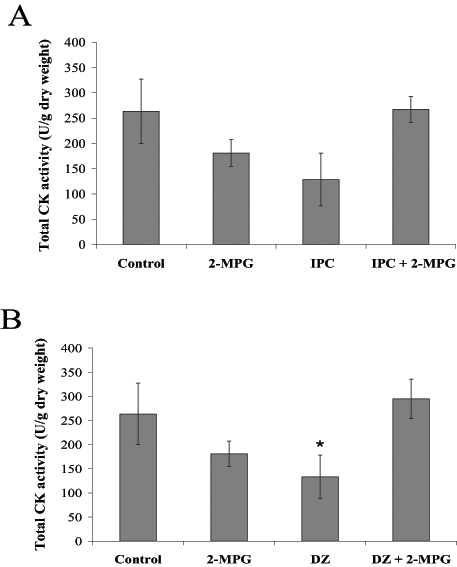
Total creatine kinase (CK) activity (U/g dry weight) for all groups measured throughout the reperfusion period. Rat hearts were subjected to A) control perfusion and ischemic preconditioning (IPC) ± N-2-mercaptopropionyl glycine (2-MPG) (400 μM); and B) control perfusion and pharmacological preconditioning with diazoxide (DZ) (100 μM) ± 2-MPG (400 μM). All values are expressed as mean ± SEM (n=5 to 6). A two-tailed unpaired Student’s t test was used for comparisons. *DZ was statistically different compared with the control and DZ plus 2-MPG (P<0.05)
Correlation among functional recovery, CK and [Ca2+]m
Previous studies have shown a significant correlation between [Ca2+]m at end reperfusion and myocardial functional recovery (7,8). Figure 7A shows the correlation between developed pressure and [Ca2+]m at end reperfusion (R=0.98), while Figure 7B shows the correlation between CK, a marker of cellular injury and [Ca2+]m at end reperfusion (R=0.97). These data indicate that minimal functional recovery does not occur above an average [Ca2+]m of approximately 600 nM. Although quantitatively different, these data parallel the findings of Wang et al (7), indicating that cellular impairment under these conditions is linearly related to [Ca2+]m during reperfusion.
Figure 7).
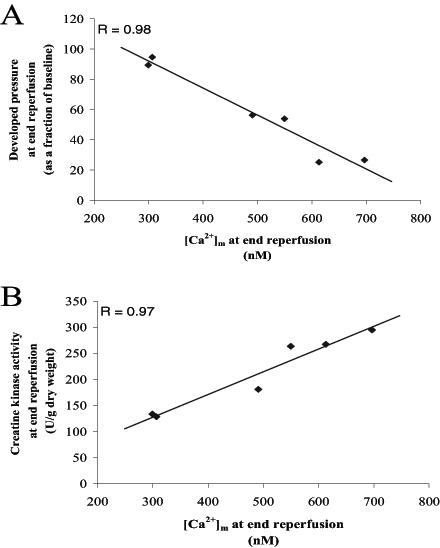
A) Least squares fit between developed pressure (as a fraction of baseline) and mitochondrial Ca2+ ([Ca2+]m) at end reperfusion. B) Least squares fit between creatine kinase release and [Ca2+]m at end reperfusion
DISCUSSION
These experiments add to the existing literature regarding the role of ROS in the protective effects of IPC and DZ. Importantly, scavenging of ROS before the 25 min ischemic episode, but not during ischemia or reperfusion, resulted in higher [Ca2+]m during reperfusion in both the DZ plus 2-MPG and IPC plus 2-MPG groups, and was associated with worse myocardial functional recovery and greater cellular injury. These findings suggest that ROS-mediated events lead to lower [Ca2+]m, the likely mechanism of myocardial protection. In addition, it is noteworthy that 2-MPG administration did not affect the accumulation of Ca2+ in the cytosol during reperfusion, and that lower [Ca2+]m and functional protection in IPC and DZ group hearts were independent of cytosolic Ca2+.
IPC and DZ lower [Ca2+]m during reperfusion
Consistent with prior experiments (7), both IPC and DZ resulted in functional protection and lower [Ca2+]m during reperfusion. The mechanism(s) of functional protection by IPC and DZ have been exhaustively investigated and, at present, include opening of mitoKATP channels (7), activation of kinases (such as PKC and tyrosine kinases) (6,19,20) and preischemic generation of ROS (21). These different mechanisms may be interdependent and can vary among species and experimental designs (6). For example, some studies have shown that the effect of IPC or DZ can be limited by 5-HD, a mitoKATP channel blocker (7,22), while in others, 5-HD has had no effect (23). Other studies have implicated translocation of prosurvival Bcl-2 family proteins into the mitochondria, resulting in relative closure of the mitochondrial transition pore (MTP), and maintenance of mitochondrial viability on reperfusion (20,24).
The mechanism by which IPC and DZ limit the accumulation of [Ca2+]m during reperfusion is unknown. This effect is clearly not dependent on a lower concentration of cytosolic Ca2+, because [Ca2+]i was similar in the IPC and DZ group hearts with or without concurrent exposure to 2-MPG. As indicated in Figures 4 and 5, hearts which had greater recovery were those with lower reperfusion [Ca2+]m, independent of cytosolic Ca2+. These findings are also inconsistent with the mitochondria acting solely in response to increases in cytosolic Ca2+, but rather suggest an active process by which mitochondria modulate matrix Ca2+ influx or efflux for a given concentration of cytosolic Ca2+. Possible processes include direct inhibition of influx pathways such as the mitochondrial uniporter, or changes in mitochondrial pH or membrane potential that result in lower Ca2+ uptake (25).
It should be noted that [Ca2+]i remained low in the DZ group hearts during reperfusion, in contrast to our prior study (7) in which cytosolic Ca2+, but not [Ca2+]m, increased during reperfusion. These different results may be due to further refinement of our procedures relating to regulation of cardiac temperature and buffer pH, and are more consistent with the functional protection afforded by DZ. Importantly, both sets of data demonstrate that these interventions result in lower [Ca2+]m, independent of [Ca2+]i.
Limiting [Ca2+]m is ROS-dependent
The ability of 2-MPG to counter the effects of IPC and DZ on [Ca2+]m and myocardial functional protection indicates that the mechanism of these effects involves ROS production before ischemia. The importance of ROS production in IPC was demonstrated by Vanden Hoek et al (21) who, using the probe 2′,7′-dichlorofluorescein diacetate (DCF) in embryonic ventricular cardiac myocytes, showed that an increase in DCF fluorescence (eg, ROS) occurred during hypoxic preconditioning. This increase in DCF fluorescence was associated with a significant reduction in ROS seen on reperfusion compared with myocytes without the preconditioning stimulus (26). Notably, ROS production by hypoxic preconditioning was limited by the concurrent administration of 2-MPG. Along with that study, numerous investigators have demonstrated the importance of ROS as a mechanism of IPC (2,27,28); however, there are no data before the present study showing that ROS production limits the increase in [Ca2+]m during I/R. It is currently unknown whether other protective mechanisms which are not ROS-dependent, such as adenosine (29), also limit [Ca2+]m.
How may ROS limit [Ca2+]m on reperfusion?
ROS production during hypoxic preconditioning (30), IPC (2) and DZ administration (2) generally originates in the mitochondrial electron transport chain, likely at complex III (31,32). This generation may depend on opening of mitoKATP channels, and involve translocation and phosphorylation of PKC and/or tyrosine kinases with secondary generation of ROS and nitric oxide (30). As in myocytes, there is evidence that the scavenging of ROS with either 2-MPG or Tiron (a superoxide scavenger) blocks the beneficial effect of these interventions in isolated rabbit hearts (30,31,33). How ROS generated by mitochondria result in lower [Ca2+]m during reperfusion is unclear. Possible mechanisms include ROS-mediated modification of Ca2+ transport proteins, such as the uniporter, to limit Ca2+ influx (20), interaction of ROS with the MTP (2), and/or ROS having an effect on mitochondrial pH.
Limiting [Ca2+]m improves function
Prior experiments (7,8) indicate that [Ca2+]m, rather than cytosolic Ca2+, is critical in defining myocardial functional recovery. The present results support that hypothesis, with a close correlation between [Ca2+]m at end reperfusion, and markers of cell injury or death such as CK release and LVDP (Figures 6A and 6B). This general finding may indicate that protected cells have mitochondria that are able to synthesize ATP, maintain their membrane potential and viability, and maintain physiological as opposed to lethal Ca2+ concentrations (20). However, these findings do not exclude the possibility that the limitation of [Ca2+]m by preischemic ROS generation is a marker of mitochondrial viability, rather than a causal factor.
Limitations
Brief administration of 2-MPG alone demonstrated some effects on both myocardial functional recovery and [Ca2+]m. Total [Ca2+]i at end reperfusion was similar to the IPC and DZ groups, while [Ca2+]m in the 2-MPG group hearts was intermediate between the control and IPC or DZ group hearts. Published data on the effects of 2-MPG alone are sparse and conflicting. While Baines et al (1) showed no alterations in infarct size when 2-MPG was administered and washed out before ischemia (1), Tanonaka et al (34) showed that the administration of 2-MPG without washout improved functional recovery after 30 min of ischemia. To test for possible residual 2-MPG after 10 min of exposure followed by 10 min of washout, 2-MPG concentration in the myocardium after washout (ie, immediately before ischemia) was measured in parallel experiments (n=4). Using a modified high-performance liquid chromatography protocol (35), samples of tissue were found to contain 14.3±0.6 pmol of 2-MPG/mg wet weight. Although this is an extremely low concentration, an effect of 2-MPG directly scavenging ROS on reperfusion cannot be excluded.
Further evidence against residual ROS protecting the 2-MPG group hearts and the IPC and DZ group hearts is the interaction of 2-MPG in the IPC and DZ group hearts. To the extent that IPC and DZ alone limited ROS production on reperfusion, the additional effect of 2-MPG should further reduce ROS content and facilitate recovery, the opposite of what was observed. One potential explanation for the 2-MPG results is based on the finding that 2-MPG alone can produce a moderate amount of ROS (36). As with IPC and DZ, this moderate ROS production could provide some protection in a manner similar to IPC and DZ; however, the data indicate that the ROS scavenging effects of 2-MPG in the presence of ROS production due to IPC or DZ overwhelm the modest protective effects of 2-MPG alone.
Technical issues affect the fluorescence measurements of Ca2+. Due to effects of changing pH and NADH concentration, measurements during ischemia were not considered. However, our previous work (7) showed that increases in NADH fluorescence during ischemia and reperfusion were similar in preconditioned and nonpreconditioned hearts. It is possible that manganese entry into mitochondria quenched some of the signal; however, our prior experiments have also shown that because the ratiometric technique is independent of quenching (as long as an adequate signal to noise ratio is present), the calculated [Ca2+]m is valid. Finally, differential quenching of mitochondria, and hence less contribution of these mitochondria to the overall signal, is a theoretical possibility that could bias the results.
CONCLUSIONS
The present results, and the results of others (7,37), confirm previous data showing that IPC and DZ lead to lower [Ca2+]m during reperfusion, and that this reduction in [Ca2+]m is directly associated with improved myocardial function (higher LVDP and lower LVEDP) and reduced CK release. The improved hemodynamics and reduced cell necrosis associated with lower [Ca2+]m on reperfusion are independent of total cellular Ca2+ concentration, a finding consistent with that of Wang et al (7) and Miyamae et al (8).
Furthermore, these data show that ROS generated by either IPC or DZ before the 25 min ischemic episode are necessary for the sparing effect on [Ca2+]m. Possible mechanisms for the effect of ROS on [Ca2+]m are either through an ROS-activated kinase cascade (20) or a direct effect of ROS on proteins such as the MTP or uniporter; however, the precise mechanism of this modulation (if present) remains unknown.
Acknowledgments
ACKNOWLEDGEMENTS AND SUPPORT: We would like to thank Tim Oroczo for his assistance in running high-performance liquid chromatography samples. This research was supported by a National American Heart Association Grant-In-Aid (0250236N) and a Philip Morris External Research program grant.
REFERENCES
- 1.Baines CP, Goto M, Downey JM. Oxygen radicals released during ischemic preconditioning contribute to cardioprotection in the rabbit myocardium. J Mol Cell Cardiol. 1997;29:207–16. doi: 10.1006/jmcc.1996.0265. [DOI] [PubMed] [Google Scholar]
- 2.Hausenloy D, Wynne A, Duchen M, Yellon D. Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation. 2004;109:1714–7. doi: 10.1161/01.CIR.0000126294.81407.7D. [DOI] [PubMed] [Google Scholar]
- 3.Lundmark JL, Ramasamy R, Vulliet PR, Schaefer S. Chelerythrine increases Na-K-ATPase activity and limits ischemic injury in isolated rat hearts. Am J Physiol. 1999;277:H999–H1006. doi: 10.1152/ajpheart.1999.277.3.H999. [DOI] [PubMed] [Google Scholar]
- 4.Vanden Hoek TL, Shao Z, Li C, Schumacker PT, Becker LB. Mitochondrial electron transport can become a significant source of oxidative injury in cardiomyocytes. J Mol Cell Cardiol. 1997;29:2441–50. doi: 10.1006/jmcc.1997.0481. [DOI] [PubMed] [Google Scholar]
- 5.Levraut J, Iwase H, Shao ZH, Vanden Hoek TL, Schumacker PT. Cell death during ischemia: Relationship to mitochondrial depolarization and ROS generation. Am J Physiol Heart Circ Physiol. 2003;284:H549–58. doi: 10.1152/ajpheart.00708.2002. [DOI] [PubMed] [Google Scholar]
- 6.Schulz R, Cohen MV, Behrends M, Downey JM, Heusch G. Signal transduction of ischemic preconditioning. Cardiovasc Res. 2001;52:181–98. doi: 10.1016/s0008-6363(01)00384-4. [DOI] [PubMed] [Google Scholar]
- 7.Wang L, Cherednichenko G, Hernandez L, et al. Preconditioning limits mitochondrial Ca(2+) during ischemia in rat hearts: Role of K(ATP) channels. Am J Physiol Heart Circ Physiol. 2001;280:H2321–8. doi: 10.1152/ajpheart.2001.280.5.H2321. [DOI] [PubMed] [Google Scholar]
- 8.Miyamae M, Camacho SA, Weiner MW, Figueredo VM. Attenuation of postischemic reperfusion injury is related to prevention of [Ca2+]m overload in rat hearts. Am J Physiol. 1996;271:H2145–53. doi: 10.1152/ajpheart.1996.271.5.H2145. [DOI] [PubMed] [Google Scholar]
- 9.Brandes R, Figueredo VM, Camacho SA, Baker AJ, Weiner MW. Quantitation of cytosolic [Ca2+] in whole perfused rat hearts using Indo-1 fluorometry. Biophys J. 1993;65:1973–82. doi: 10.1016/S0006-3495(93)81274-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brandes R, Figueredo VM, Camacho SA, Baker AJ, Weiner MW. Investigation of factors affecting fluorometric quantitation of cytosolic [Ca2+] in perfused hearts. Biophys J. 1993;65:1983–93. doi: 10.1016/S0006-3495(93)81275-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arkhammar P, Nilsson T, Berggren PO. Glucose-stimulated efflux of indo-1 from pancreatic beta-cells is reduced by probenecid. FEBS Lett. 1990;273:182–4. doi: 10.1016/0014-5793(90)81079-4. [DOI] [PubMed] [Google Scholar]
- 12.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–50. [PubMed] [Google Scholar]
- 13.Baker AJ, Brandes R, Schreur JH, Camacho SA, Weiner MW. Protein and acidosis alter calcium-binding and fluorescence spectra of the calcium indicator indo-1. Biophys J. 1994;67:1646–54. doi: 10.1016/S0006-3495(94)80637-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schreur JH, Figueredo VM, Miyamae M, Shames DM, Baker AJ, Camacho SA. Cytosolic and mitochondrial [Ca2+] in whole hearts using indo-1 acetoxymethyl ester: Effects of high extracellular Ca2+ Biophys J. 1996;70:2571–80. doi: 10.1016/S0006-3495(96)79828-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyata H, Lakatta EG, Stern MD, Silverman HS. Relation of mitochondrial and cytosolic free calcium to cardiac myocyte recovery after exposure to anoxia. Circ Res. 1992;71:605–13. doi: 10.1161/01.res.71.3.605. [DOI] [PubMed] [Google Scholar]
- 16.Miyata H, Silverman HS, Sollott SJ, Lakatta EG, Stern MD, Hansford RG. Measurement of mitochondrial free Ca2+ concentration in living single rat cardiac myocytes. Am J Physiol. 1991;261:H1123–34. doi: 10.1152/ajpheart.1991.261.4.H1123. [DOI] [PubMed] [Google Scholar]
- 17.Spurgeon HA, Stern MD, Baartz G, et al. Simultaneous measurement of Ca2+, contraction, and potential in cardiac myocytes. Am J Physiol. 1990;258:H574–86. doi: 10.1152/ajpheart.1990.258.2.H574. [DOI] [PubMed] [Google Scholar]
- 18.Zuurbier CJ, Eerbeek O, Goedhart PT, et al. Inhibition of the pentose phosphate pathway decreases ischemia-reperfusion-induced creatine kinase release in the heart. Cardiovasc Res. 2004;62:145–53. doi: 10.1016/j.cardiores.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 19.Uchiyama Y, Otani H, Wakeno M, et al. Role of mitochondrial KATP channels and protein kinase C in ischaemic preconditioning. Clin Exp Pharmacol Physiol. 2003;30:426–36. doi: 10.1046/j.1440-1681.2003.03853.x. [DOI] [PubMed] [Google Scholar]
- 20.Murphy E. Primary and secondary signaling pathways in early preconditioning that converge on the mitochondria to produce cardioprotection. Circ Res. 2004;94:7–16. doi: 10.1161/01.RES.0000108082.76667.F4. [DOI] [PubMed] [Google Scholar]
- 21.Vanden Hoek TL, Becker LB, Shao Z, Li C, Schumacker PT. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J Biol Chem. 1998;273:18092–8. doi: 10.1074/jbc.273.29.18092. [DOI] [PubMed] [Google Scholar]
- 22.Riess ML, Camara AK, Novalija E, Chen Q, Rhodes SS, Stowe DF. Anesthetic preconditioning attenuates mitochondrial Ca2+ overload during ischemia in Guinea pig intact hearts: Reversal by 5-hydroxydecanoic acid. Anesth Analg. 2002;95:1540–6. doi: 10.1097/00000539-200212000-00013. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz LM, Welch TS, Crago MS. Cardioprotection by multiple preconditioning cycles does not require mitochondrial K(ATP) channels in pigs. Am J Physiol Heart Circ Physiol. 2002;283:H1538–44. doi: 10.1152/ajpheart.00040.2002. [DOI] [PubMed] [Google Scholar]
- 24.Kowaltowski AJ, Vercesi AE, Fiskum G. Bcl-2 prevents mitochondrial permeability transition and cytochrome c release via maintenance of reduced pyridine nucleotides. Cell Death Differ. 2000;7:903–10. doi: 10.1038/sj.cdd.4400722. [DOI] [PubMed] [Google Scholar]
- 25.Gursahani H, Schaefer S. Acidification reduces mitochondrial calcium uptake in rat cardiac mitochondria. Am J Physiol Heart Circ Physiol. 2004;287:H2659–65. doi: 10.1152/ajpheart.00344.2004. [DOI] [PubMed] [Google Scholar]
- 26.Vanden Hoek T, Becker LB, Shao ZH, Li CQ, Schumacker PT. Preconditioning in cardiomyocytes protects by attenuating oxidant stress at reperfusion. Circ Res. 2000;86:541–8. doi: 10.1161/01.res.86.5.541. [DOI] [PubMed] [Google Scholar]
- 27.Stowe DF, Kevin LG. Cardiac preconditioning by volatile anesthetic agents: A defining role for altered mitochondrial bioenergetics. Antioxid Redox Signal. 2004;6:439–48. doi: 10.1089/152308604322899512. [DOI] [PubMed] [Google Scholar]
- 28.Otani H. Reactive oxygen species as mediators of signal transduction in ischemic preconditioning. Antioxid Redox Signal. 2004;6:449–69. doi: 10.1089/152308604322899521. [DOI] [PubMed] [Google Scholar]
- 29.Cohen MV, Baines CP, Downey JM. Ischemic preconditioning: From adenosine receptor of KATP channel. Annu Rev Physiol. 2000;62:79–109. doi: 10.1146/annurev.physiol.62.1.79. [DOI] [PubMed] [Google Scholar]
- 30.Lebuffe G, Schumacker PT, Shao ZH, Anderson T, Iwase H, Vanden Hoek TL. ROS and NO trigger early preconditioning: Relationship to mitochondrial KATP channel. Am J Physiol Heart Circ Physiol. 2003;284:H299–308. doi: 10.1152/ajpheart.00706.2002. [DOI] [PubMed] [Google Scholar]
- 31.Krenz M, Oldenburg O, Wimpee H, et al. Opening of ATP-sensitive potassium channels causes generation of free radicals in vascular smooth muscle cells. Basic Res Cardiol. 2002;97:365–73. doi: 10.1007/s003950200045. [DOI] [PubMed] [Google Scholar]
- 32.Oldenburg O, Critz SD, Cohen MV, Downey JM. Acetylcholine-induced production of reactive oxygen species in adult rabbit ventricular myocytes is dependent on phosphatidylinositol 3- and Src-kinase activation and mitochondrial K(ATP) channel opening. J Mol Cell Cardiol. 2003;35:653–60. doi: 10.1016/s0022-2828(03)00083-x. [DOI] [PubMed] [Google Scholar]
- 33.Pain T, Yang XM, Critz SD, et al. Opening of mitochondrial K(ATP) channels triggers the preconditioned state by generating free radicals. Circ Res. 2000;87:460–6. doi: 10.1161/01.res.87.6.460. [DOI] [PubMed] [Google Scholar]
- 34.Tanonaka K, Iwai T, Motegi K, Takeo S. Effects of N-(2-mercaptopropionyl)-glycine on mitochondrial function in ischemic-reperfused heart. Cardiovasc Res. 2003;57:416–25. doi: 10.1016/s0008-6363(02)00698-3. [DOI] [PubMed] [Google Scholar]
- 35.Lakritz J, Plopper CG, Buckpitt AR. Validated high-performance liquid chromatography-electrochemical method for determination of glutathione and glutathione disulfide in small tissue samples. Anal Biochem. 1997;247:63–8. doi: 10.1006/abio.1997.2032. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka K, Weihrauch D, Ludwig LM, Kersten JR, Pagel PS, Warltier DC. Mitochondrial adenosine triphosphate-regulated potassium channel opening acts as a trigger for isoflurane-induced preconditioning by generating reactive oxygen species. Anesthesiology. 2003;98:935–43. doi: 10.1097/00000542-200304000-00021. [DOI] [PubMed] [Google Scholar]
- 37.Ishida H, Hirota Y, Genka C, Nakazawa H, Nakaya H, Sato T. Opening of mitochondrial K(ATP) channels attenuates the ouabain-induced calcium overload in mitochondria. Circ Res. 2001;89:856–8. doi: 10.1161/hh2201.100341. [DOI] [PubMed] [Google Scholar]