
Figure 8).
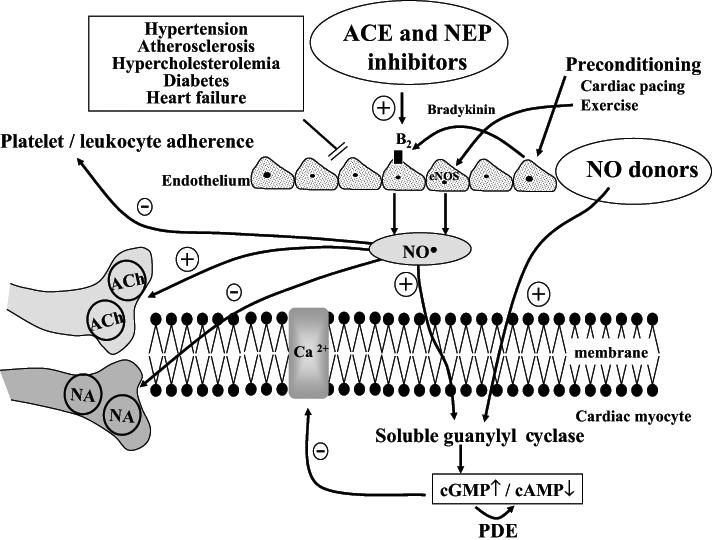
Hypothesis for the role of nitric oxide (NO) and other endothelium-derived endogenous protective mediators in the anti-arrhythmic effects of preconditioning. Ischemic preconditioning, cardiac pacing and exercise result in either a direct or a bradykinin-mediated increase in generation and subsequent release of NO from endothelial cells, and possibly also from cardiac myocytes. NO diffuses into the cardiac myocytes, stimulating soluble guanylyl cyclase and raising cyclic GMP (cGMP) levels. This both reduces cyclic AMP (cAMP) levels and Ca2+ entry through the L-type Ca2+ channels, as well as directly suppressing myocyte O2 consumption. NO can also inhibit noradrena-line (NA) release from sympathetic nerve endings and enhance, acting via a cGMP-dependent pathway, the effect of vagal nerve stimulation on presynaptic muscarinic receptors. It is hypothesized that under conditions where endothelial dysfunction is evident (hypertension, atherosclerosis, hypercholesterolemia, diabetes, etc), the ability to generate endogenous myocardial protective substances such as NO may become impaired. Thus, drugs that are able to replace the loss of these substaces (eg, NO donors), or drugs that enhance the production of these substances (eg, inhibition of kinin metabolism with angiotensin converting enzyme [ACE] and neutral endopeptidase [NEP] inhibitors which increase NO production) would be cardioprotective. Similarly, regular physical exercise, perhaps by increasing endothelial NO synthase (eNOS) gene expression, leads to increased NO production, reduced platelet and leukocyte adherence to the vascular endothelium, inhibition of cardiac sympathetic transmission and modulation of the cAMP/cGMP balance in cardiac myocytes. ACh Acetylcholine; B2 Bradykinin B2 receptor; PDE Phosphodiesterase