

Abstract
Sphingolipids are key lipid regulators of cell viability: ceramide is one of the key molecules in inducing programmed cell death (apoptosis), whereas other sphingolipids, such as ceramide 1-phosphate, are mitogenic. The thermotropic and structural behavior of binary systems of N-hexadecanoyl-D-erythro-ceramide (C16-ceramide) or N-hexadecanoyl-D-erythro-ceramide-1-phosphate (C16-ceramide-1-phosphate; C16-C1P) with 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) was studied with DSC and deuterium nuclear magnetic resonance (2H-NMR). Partial-phase diagrams (up to a mole fraction of sphingolipids X = 0.40) for both mixtures were constructed based on DSC and 2H-NMR observations. For C16-ceramide-containing bilayers DSC heating scans showed already at Xcer = 0.025 a complex structure of the main-phase transition peak suggestive of lateral-phase separation. The transition width increased significantly upon increasing Xcer, and the upper-phase boundary temperature of the mixture shifted to ∼65°C at Xcer = 0.40. The temperature range over which 2H-NMR spectra of C16-ceramide/DPPC-d62 mixtures displayed coexistence of gel and liquid crystalline domains increased from ∼10° for Xcer = 0.1 to ∼21° for Xcer = 0.4. For C16-C1P/DPPC mixtures, DSC and 2H-NMR observations indicated that two-phase coexistence was limited to significantly narrower temperature ranges for corresponding C1P concentrations. To complement these findings, C16-ceramide/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and C16-C1P/POPC mixtures were also studied by 2H-NMR and fluorescence techniques. These observations indicate that DPPC and POPC bilayers are significantly less perturbed by C16-C1P than by C16-ceramide and that C16-C1P is miscible within DPPC bilayers at least up to XC1P = 0.30.
Introduction
Ceramide (see Fig. 1) functions as a second messenger in several cellular processes including apoptosis, growth suppression, differentiation, and cell senescence, see, e.g., (1). Notwithstanding the emergence of detailed protein mediated pathways executing the actions of ceramides, these molecules are highly hydrophobic; thus all actions that these lipids perform have to take place within the lipid bilayer of cellular membranes. Several studies have previously demonstrated that ceramides are incorporated within the lipid bilayers and that they alter significantly the physical state of the membrane at a two- and three-dimensional level (2–10). Bilayers containing ceramides are highly ordered and show an increased main-phase transition temperature probably due to minimal hydration and strong hydrogen bonding capacity between ceramide headgroups (11,12). Furthermore, ceramide even at low contents (mole fraction of ceramide Xcer < 0.10), is laterally segregated into microdomains (e.g., 3–7). Compared to traditional lipid second messengers, such as the platelet-activating factor, found only at very low molar concentrations in cells, the levels of ceramides are not low. Actually, they may in some circumstances represent several mol % of lipids in activated cells. Furthermore, compartmentalized production of ceramide from sphingomyelin by the action of sphingomyelinase may elevate these levels severalfold at the site of production. These high concentrations challenge the view of ligand-receptor interaction. We have earlier proposed (3–7) that universal changes in the state of the cellular membranes may mediate the biological responses of this lipid.
Figure 1.

Structures of DPPC, N-hexadecanoyl-D-erythro-ceramide (C16-ceramide), and N-hexadecanoyl-D-erythro-ceramide-1-phosphate (C16-C1P) used in this study.
Interestingly, it appears that it is not just the presence of lipid second messengers in cellular membranes that determine the fate of the cell; it is more the balance of the relative levels of lipid second messengers. A good example is given by the levels of chemically similar diacylglycerols and ceramides. Whereas diacylglycerols have been shown to be highly mitogenic in several cell lines (13,14), ceramides seem to drive cells toward death (e.g., 1,15,16). The precursor molecules such as phosphatidylcholine and sphingomyelin, which serve as the major phospholipid species in the plasma membrane, seem not to have any profound role in inducing cells to undergo major cellular changes. Yet, the breakdown products of these lipids, diacylglycerol and ceramide then prompt the cell to flourish and die, respectively. The hydrolysis of ceramide by ceramidase yields sphingosine, which is a well-known inhibitor of protein kinase C (17). Alternatively phosphorylation of ceramide by ceramide kinase (CERK) produces ceramide 1-phosphate (C1P, Fig. 1), which curiously enough is mitogenic (18,19). Thus, to summarize, it is not the emergence of lipid second messengers that seem to be important in driving the cell fate; rather it is the relative levels of these molecules that is the determining factor. In keeping with this view, it was recently shown that cells undergoing apoptosis with concomitant appearance of increasing levels of ceramides downregulate also CERK (20) and thus prevent formation of C1P. We hypothesize that changes in the physical state of the membrane would play a role in determining the fate of the cell. Accordingly, the objective of this study is to compare the changes in the physical state of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayers with increasing content of ceramide or C1P.
Materials and Methods
Materials
EDTA, HEPES, and NaCl were from Sigma-Aldrich (St. Louis, MO). DPPC, chain-perdeuterated DPPC (DPPC-d62), POPC, chain-perdeuterated POPC (POPC-d31), and N-hexadecanoyl-D-erythro-ceramide (C16-ceramide) were obtained from Avanti Polar Lipids (Alabaster, AL). N-hexadecanoyl-D-erythro-ceramide-1-phosphate (C16-ceramide-1-phosphate; C16-C1P) was custom synthesized by Matreya (Matreya LLC, Pleasant Gap, PA). 1-Palmitoyl-2-[(pyren-1-yl)]decanoyl-sn-glycero-3-phosphocholine (PPDPC) and 1,6-diphenyl-1,3,5-hexatriene (DPH) were from Sigma-Aldrich. Concentrations of the lipids were determined gravimetrically and those of fluorescent probes spectrophotometrically using appropriate molar exctinction coefficients. The purity of the above lipids was verified using thin-layer chromatography. Water was freshly deionized in a Milli RO/Milli Q (Millipore, Bedford, MA) filtering system.
Liposome preparation
For DSC experiments, appropriate amounts of the lipid stock were weighed and dissolved into chloroform (DPPC = 10 mg/ml, C16-ceramide 5 mg/ml, and C16-C1P 1 mg/ml). These solutions were mixed in chloroform to obtain the desired compositions. The resulting mixtures were then evaporated to dryness under a stream of nitrogen, and traces of solvent were removed by evacuating under reduced pressure for 6–24 h. The lipid residues were hydrated at 80°C in a buffer of 20 mM HEPES, 0.1 mM EDTA, 150 mM NaCl, with a pH of 7.4 to yield multilamellar vesicles (MLVs) with a lipid concentration of 1 mM and maintained at this temperature for 30 min. At Xcer = 0.40 (Xcer standing for the mole fraction of C16-ceramide), the obtained solution was visibly aggregated. All other solutions were opaque in line with formation of MLVs.
The preparation of samples for 2H-NMR experiments differed only slightly. Amounts of dry lipid appropriate to the composition of a given sample and sufficient to ensure at least 15 mg of deuterated lipid per sample were dissolved in chloroform/methanol 2:1 (v/v) and mixed together. Solvent was removed by rotary evaporation at 45°C, and samples were then further dried under vacuum for 8–10 h. Samples for 2H-NMR experiments were then hydrated, also in a buffer of 20 mM HEPES, 0.1 mM EDTA, 150 mM NaCl, with a pH of 7.4, by using a small amount of buffer to wash lipid from the walls of the flask while rotating the flask for ∼1 h at 35–45°C. Samples were further hydrated by three cycles of alternate vortexing and heating to 85°C. Because of the large difference between the main transition temperatures of POPC and C16-ceramide bilayers, dispersions containing POPC-d31 underwent an additional hydration step consisting of five freeze-thaw cycles as described by Hsueh et al. (10). After hydration, MLV suspensions were transferred to 400 μl NMR tubes with a diameter of 8 mm and allowed to settle for 30 min after which excess buffer, if any, was removed. Sample tubes were then sealed and vortexed lightly for no more than 10 s.
DSC
Differential heat capacity scans were recorded at a lipid concentration of 1 mM (MLVs) and at a heating rate of 0.5°C/min. Before their loading into precooled DSC cuvettes, the samples were equilibrated on ice for ∼24 h and thereafter degassed at low pressure. The calorimeter (VP-DSC, MicroCal, Northampton, MA) was interfaced to a PC, and data were analyzed using the routines of the software provided with the instrument. All samples were scanned by heating from 10°C to 80°C followed by cooling from 80°C to 10°C. All experiments were done in duplicates.
2H-NMR
Spectra were obtained using a locally constructed wideline NMR spectrometer in conjunction with a 3.5 T superconducting magnet (Nalorac Cryogenics, Martinez, CA) in which the deuteron Larmor frequency is 23.215 MHz. Spectra were obtained using a quadrupole echo sequence (21) consisting of two π/2 pulses of 3.1–3.7 μs duration, differing in phase by 90°, and separated by 35 μs. Depending on the amount of deuterated material in a given sample, 4000–12,000 transients were averaged with a repetition time of 0.7 s. Oversampling (22) was used to give effective sampling times of 4 μs for liquid crystalline samples and 2 μs for gel-phase samples. The spectrometer was tuned and phased to minimize signal in the imaginary channel. A five-point smoothing algorithm was used to interpolate the echo signal and locate a time-domain point at the echo maximum (23). Quadrupole echo signals were then Fourier transforming by discarding points to the left of the echo peak and zero filling to 4096 points. Each quadrupole echo signal was Fourier transformed a second time with the imaginary channel zeroed to give a symmetric spectrum that was used for calculation of the first spectral moment.
Within the NMR probe, the 400 μl tube containing a given sample was inserted into a radio frequency coil enclosed within a copper oven. Temperature in the oven was controlled to ± 0.1°C using a CYC3200 temperature controller (Omega Engineering, Stamford, CT) coupled to a copper-constantan thermocouple mounted close to the sample. Spectra were collected from high to low temperature with steps of 2°C except near the main-phase transition where steps of 1°C were used. After each step, temperature was allowed to stabilize after which the sample was allowed to equilibrate for an additional 30 min before initiation of signal acquisition.
Measurement of Ie/Im for PPDPC and fluorescence anisotropy for DPH
A monomeric excited-state pyrene may relax to ground state by emitting photons with a maximum wavelength at ∼380 nm (Im). During its lifetime, the excited-state pyrene may form a complex with a ground-state pyrene producing a characteristic excimer or excited dimer. This complex relaxes back to two ground-state pyrenes by emitting a broad band centered at ∼480 nm (Ie). Essentially the excimer/monomer fluorescence intensity ratio (Ie/Im) is proportional to the rate of collisions between the pyrenes. Consequently, for PPDPC the value for Ie/Im reflects the lateral mobility as well as the local concentration of this lipid analog in the membrane. Fluorescence emission spectra for POPC/C16-ceramide and POPC/C16-C1P MLVs labeled with PPDPC (X = 0.01) were recorded with a Perkin-Elmer (Waltham, MA) LS55 spectrofluorometer equipped with a magnetically stirred, thermostated cuvette compartment. The excitation wavelength was 344 nm, and the excitation and emission bandwidths were 4 nm. Two milliliters of liposome solution (45 nmol of lipid) in a four-window quartz cuvette were used in each measurement with temperature maintained at 25°C. Each sample was equilibrated for 2 min before recording the spectrum. Three scans were averaged, and the emission intensities at ∼380 nm and 480 nm were taken for Im and Ie, respectively. DPH was included into POPC/C16-ceramide and POPC/C16-C1P MLVs to yield a lipid/DPH molar ratio of 500:1. Polarized emission was measured in the L-format using Polaroid-film-type prisms in the Perkin-Elmer LS55 spectrofluorometer. Excitation at 360 nm and emission at 450 nm were selected with monochromators and using 5 nm bandwidths. The samples were maintained in the cuvette for 2 min before the measurement of anisotropy, averaging the signal over a 10 s interval. Values of steady-state fluorescence anisotropy (r) were calculated using software routines provided by Perkin-Elmer. All experiments were done in duplicate.
Results
DSC
Representative DSC up- and downscans for DPPC/C16-ceramide and DPPC/C16-C1P MLVs are illustrated in Fig. 2 a–d. Pure DPPC shows three transitions: a subtransition (Ts) at 17.0°C, a pretransition (Tp) at 34.5°C, and a main transition (Tm) at 41.5°C, in agreement with published data (24). Increasing Xcer (the mole fraction of C16-ceramide) from 0 to 0.15 decreased the subtransition temperature from 17.0°C to 16.6°C with continuous widening of the endotherm peak and decrease in the enthalpy of this transition. The pretransition is observed up to Xcer = 0.10 at slightly higher temperatures. Increasing Xcer from 0 to 0.025 broadened the main transition endothermic peak and also shifted the Tm to 42.0–42.1°C. Furthermore, an endothermic shoulder centered at 45.2°C appeared suggestive of lateral-phase separation. At Xcer = 0.05, the main endotherm was observed at 42.0°C, yet the peak became significantly broader, and the clearly visible endothermic shoulder shifted to 47.6°C. Whereas the endotherm at 42–43°C remained at approximately the same temperature (for Xcer = 0.025–0.40), a new endotherm appeared at 48°C (at Xcer = 0.05–0.30). The clearly visible third endotherm was progressively shifted to higher temperatures reaching 62.3°C at Xcer = 0.30. At Xcer = 0.40, the sample became clearly aggregated, which prevented a comprehensive DSC analysis. Yet, the broad and asymmetric main-phase transition became even more clearly segregated to at least three main peaks. A new endotherm appeared at temperatures >80°C suggestive of crystallized, pure C16-ceramide (25).
Figure 2.

(a) High-sensitivity DSC heating scans for multilamellar DPPC/C16-ceramide vesicles (MLVs). (b) Data for the same MLVs recorded upon cooling. Also shown are the (c) heating and (d) cooling scans for DPPC/C16-C1P MLVs. The mole fraction of ceramide, Xcer, or ceramide-1-phosphate, XC1P, is indicated in the figure. The total lipid concentration was 1 mM in 20 mM HEPES, 0.1 mM EDTA, and 150 mM NaCl (pH 7.4).
In cooling scans for DPPC, the subtransition was not observed, the pretransition was shifted to lower temperature (Tp = 34.3°C), and the main transition was observed at 41.0°C. The sub- or pretransition was not observed in any sample having C16-ceramide. The peak at 41.0°C remained at 40.9–41.0°C up to Xcer = 0.15. Yet, another clearly visible endotherm appeared at 44.2°C already at Xcer = 0.025. This endotherm shifted monotonously to higher temperatures reaching 67°C at Xcer = 0.40. At Xcer = 0.30, a third endotherm was observed at 57.3°C, and at Xcer = 0.40, a shoulder on the high temperature side appeared at 70°C.
In heating scans for DPPC/C16-C1P mixtures, the subtransition was observed up to XC1P = 0.075 at approximately the same temperature of 17.0–17.2°C. At higher XC1P this endotherm was no longer discernible. The pretransition was observed up to XC1P = 0.15 and was shifted continuously to higher temperatures reaching 38.5°C at XC1P = 0.15. The main-phase transition temperature observed for DPPC at 41.5°C was progressively shifted to higher temperatures with increasing XC1P and reaching 43.2°C for XC1P = 0.40 with concomitant broadening of this peak. The main-phase transition peak remained rather symmetric up to XC1P = 0.40 in heating scans.
In cooling scans for DPPC mixtures with C16-C1P the subtransition could not be resolved from the DSC traces. The pretransition was observed up to XC1P = 0.15 and the pretransition temperature increased monotonously from 29.2°C (XC1P = 0) to 36.3°C (XC1P = 0.15). The main transition observed at 41.0°C (XC1P = 0) also shifted to slightly higher temperatures reaching 42.2°C at XC1P = 0.40 and increasing XC1P induced broadening of the endotherm. At XC1P = 0.30, a shoulder appeared in the endotherm at the higher temperature side, and at XC1P = 0.40, two overlapping endotherms were observed centered at 42.2°C and 43.9°C.
2H-Nuclear magnetic resonance
Fig. 3 shows 2H-NMR spectra at selected temperatures for DPPC-d62 (panel a), DPPC-d62/C16-ceramide at Xcer = 0.2 (panel b), DPPC-d62/C16-C1P at XC1P = 0.2 (panel c), DPPC-d62/C16-ceramide at Xcer = 0.4 (panel d), and DPPC-d62/C16-C1P at XC1P = 0.4 (panel e). All samples were in the form of vesicles suspended in a buffer of 20 mM HEPES, 0.1 mM EDTA, and 150 mM NaCl at pH = 7.4.
Figure 3.
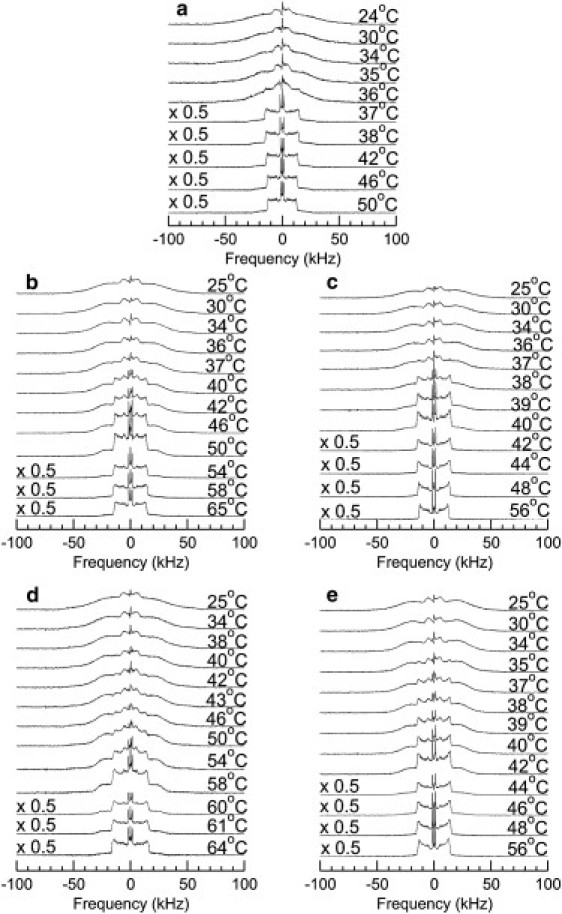
2H-NMR spectra at selected temperatures for (a) DPPC-d62, (b) DPPC-d62 plus C16-ceramide at Xcer = 0.2, (c) DPPC-d62 plus C16-C1P at XC1P = 0.2, (d) DPPC-d62 plus C16-ceramide at Xcer = 0.4, and (e) DPPC-d62 plus C16-C1P at XC1P = 0.4. All samples were in the form of vesicles suspended in a buffer of 20 mM HEPES, 0.1 mM EDTA, and 150 mM NaCl at pH = 7.4.
The spectra in Fig. 3 a for DPPC-d62 reflect a sharp transition from fluid- to gel-phase bilayers. In the fluid phase, the spectrum is a superposition of Pake doublets characteristic of fast, axially symmetric reorientation about the bilayer normal. For deuterons on a given segment of a DPPC-d62 acyl chain, the splitting between the prominent edges of the doublet spectrum is given by , where 167 kHz is the quadrupole coupling constant for carbon-deuterium bonds and is the orientation order parameter for a given carbon-deuterium bond. In the expression for , the angle is the instantaneous angle between the carbon-deuterium bond and the local bilayer normal, and the average is over chain conformations accessed during the characteristic time, ∼10−5 s, of the 2H-NMR experiment. In the spectrum for the fluid phase, doublets with the largest splittings are contributed by deuterons closest to the headgroup end of the DPPC-d62 where motions are most constrained. The prominent edges with splittings that increase from ∼12 kHz to 15 kHz as temperature is lowered from 50°C to 37°C reflect the clustering of orientation order parameter values, referred to as the orientation order parameter profile plateau, for methylene groups at the headgroup end of the DPPC acyl chains (23,26,27). The sharp doublet with the smallest splitting is contributed by the acyl chain methyl groups whose motions are only slightly constrained and which can also reorient rapidly about the methyl axis. The DPPC-d62 spectra in the gel phase reflect orientationally ordered chains reorienting about the molecular axis with a correlation time that is not short relative to the characteristic time of the experiment. The central feature of the gel-phase spectrum, with a width of ∼14 kHz, is contributed by the acyl chain methyl groups, which continue to reorient about the methyl axis in the gel phase.
Fig. 3, b and d, show spectra at selected temperatures for DPPC- d62 containing C16-ceramide at Xcer = 0.2 and 0.4, respectively. The most significant difference between these two spectral series and the DPPC-d62 spectra in Fig. 3 a is the range of temperatures over which the DPPC-d62/C16-ceramide spectra are characteristic of coexisting fluid- and gel-phase domains. For Xcer = 0.2, the spectra between 54°C and 37°C contain both fluid-phase spectral components. The gel-phase spectral component, characterized by enhanced intensity beyond ±30 kHz and the growth of the gel-phase methyl feature with a width of ∼14 kHz, begins to emerge between 56°C and 54°C and grows in prominence as the temperature is lowered. The fluid-phase spectral component, indicated by the presence of prominent edges characteristic of fast, axially symmetric reorientation and gel spectral components, persist down to 37°C and is absent for lower temperatures. For Xcer = 0.4, a gel-phase spectral component begins to emerge at 60°C, and fluid-phase spectral features persist to between 40°C and 38°C.
Fig. 3, c and e, show spectra at selected temperatures for DPPC-d62 containing C16-C1P at XC1P = 0.2 and 0.4, respectively. These spectral series also contain spectra that simultaneously display both fluid and gel spectral components, but the temperature ranges over which of the two phases that coexist are significantly narrower than for the corresponding DPPC-d62/C16-ceramide samples. For XC1P = 0.2 (Fig. 3 c), a gel-phase spectral component begins to emerge between 41°C and 40°C, and the fluid-phase spectral component is absent below 37°C. For XC1P = 0.4 (Fig. 3 e), the gel-phase spectral component begins to emerge between 46°C and 45°C, and the fluid-phase spectral component disappears below 34°C.
Inspection of the spectra in Fig. 3 suggests that for a given concentration, the temperature range over which gel and fluid phases coexist for DPPC-d62/C16-ceramide is approximately twice that for DPPC-d62/C16-C1P. The mixing behaviors of DPPC-d62/C16-ceramide and DPPC-d62/C16-C1P can also be distinguished by comparing temperature dependences of the first spectral moments (M1) for both mixtures at a series of concentrations. The first spectral moment is proportional to the intensity-weighted splitting for a given spectrum. In fluid-phase spectra, which are characteristic of axially symmetric reorientation, M1 is proportional to the orientation order parameter averaged over all chain deuterons. In the gel phase, where spectra cannot be described in terms of doublet splittings, M1 provides an indication of chain order but must be interpreted carefully.
Fig. 4 shows the temperature dependence of M1 for spectra of both lipid mixtures at a series of concentrations. For DPPC-d62, the fluid to gel transition is marked by a sharp increase in M1. For each of the mixtures, the onset of two-phase coexistence on cooling is marked by an increase in the slope of M1 versus temperature. For each concentration, the onset of two-phase coexistence for DPPC-d62/C16-ceramide occurs at a significantly higher temperature than for DPPC-d62/C16-C1P. On the M1 versus temperature plots for the lipid mixtures, the lower limit of two-phase coexistence is not as sharply defined as the upper limit, but there is clearly less variation among samples in the lower temperature limit of two-phase coexistence.
Figure 4.

Temperature dependence of 2H-NMR first spectral moments (M1) for DPPC-d62 plus (a) C16-ceramide or (b) C16-C1P. Both panels show results for (○) DPPC-d62 alone and DPPC-d62 plus C16-ceramide or C16-C1P at mole fractions of (∇) X = 0.1, (Δ) X = 0.2, (◊) X = 0.3, and (□) X = 0.4.
The plots of first moment versus temperature provide an indication of how the range, in temperature, of two-phase coexistence varies with composition for the DPPC-d62/C16-ceramide and DPPC-d62/C16-C1P mixtures. For a given concentration, the two-phase region for DPPC-d62/C16-ceramide covers a significantly larger temperature range than for DPPC-d62/C16-C1P. It should also be noted that the M1 versus temperature plot for Xcer = 0.4 is only slightly displaced from that for Xcer = 0.3. As discussed later, this may reflect the existence of a threshold concentration beyond which ceramide cannot be accommodated in the DPPC/ceramide bilayers.
The M1 versus temperature plots in Fig. 4 also indicate how DPPC-d62 chain order is perturbed by the other lipid component above and below the two-phase region. C16-ceramide significantly increases DPPC-d62 chain order at any given temperature in the fluid phase. It also raises the average chain order just above the onset of two phase coexistence. Below the range of two-phase coexistence, DPPC-d62 chain order in the gel phase is also higher in the presence of C16-ceramide than for DPPC-d62 alone. C16-C1P has a weaker ordering effect on DPPC-d62 chain order in the fluid phase, and chain order just above the onset of two-phase coexistence is roughly independent of C16-C1P concentration. In the gel phase, C16-C1P does not perturb DPPC-d62 chain order significantly.
To determine whether C16-ceramide and C16-C1P mix differently with more fluid phosphatidylcholine bilayers, mixtures of these sphingolipids with POPC-d31 were also observed with 2H-NMR. Fig. 5, panels a–c, shows 2H-NMR spectra at selected temperatures for POPC-d31 (panel a), POPC-d31/C16-ceramide at Xcer = 0.2 (panel b), and POPC-d31/C16-C1P at XC1P = 0.2 (panel c). All samples were in the form of vesicles suspended in a buffer of 20 mM HEPES, 0.1 mM EDTA, and 150 mM NaCl at pH = 7.4, and hydrated by a series of freeze-thaw cycles (10). Fig. 5, panel d, shows the temperature dependence of M1 for these samples.
Figure 5.

Panels a–c show 2H-NMR spectra at selected temperatures for (a) POPC-d31, (b) POPC-d32 plus C16-ceramide at Xcer = 0.2, and (c) POPC-d31 plus C16-C1P at XC1P = 0.2. Panel d shows the temperature dependence of 2H-NMR first spectral moments (M1) for (○) POPC-d31, (□) POPC-d31 plus C16-ceramide at Xcer = 0.2, and (Δ) POPC-d31 plus C16-C1P at XC1P = 0.2. All samples were in the form of vesicles suspended in a buffer of 20 mM HEPES, 0.1 mM EDTA, and 150 mM NaCl at pH = 7.4 using a freeze-thaw hydration protocol.
The spectra of POPC-d31 (Fig. 5 a) and the temperature dependence of the corresponding first spectral moments (circles in Fig. 5 d) indicate that the POPC-d31 dispersion went through a sharp transition from liquid crystal to gel as it was cooled to below −6°C. In contrast, the spectra of POPC-d31/C16-ceramide at Xcer = 0.2 (Fig. 5 b) contain a small but distinct gel-phase component for all temperatures between 30°C and the completion of the transition, for this mixture, just below −6°C. The gel-phase spectral component is shown, in the magnified baselines of the 15°C and 5°C spectra, by the presence of intensity in the −60 kHz to −30 kHz range of frequency. These frequencies are beyond the ±30 kHz range of the liquid crystalline spectral component at those temperatures. The limited intensity of the gel-phase spectral component above 0°C for POPC-d31/C16-ceramide at Xcer = 0.2 is consistent with the expectation that gel domains in this mixture should be highly enriched in C16-ceramide except for temperatures close to the solidus boundary of the two-phase coexistence region in the POPC/C16-ceramide binary phase diagram (10). Nevertheless, the strong perturbation of POPC chain orientational order by C16-ceramide between 30°C and −6°C is apparent from the differences between POPC-d31 and POPC-d31/C16-ceramide M1 values over this range.
Spectra for POPC-d31/C16-C1P at XC1P = 0.2 (Fig. 5 c) show no evidence of a gel-phase component above 10°C. The spectra and first moments indicate that, in this POPC-d31/C16-C1P mixture, most of the ordering of POPC-d31from its more fluid, liquid crystalline state to its more ordered state, characteristic of the gel phase, occurs between ∼5°C and −3°C. The spectrum at −1°C contains liquid crystal and gel-phase components, but the spectra above 1°C and below −3°C are not indicative of phase separation. The narrow temperature range over which the transition occurs indicates that POPC-d31 bilayers are perturbed more weakly by C16-C1P than by C16-ceramide. The difference between the mixing properties of the two sphingolipids with POPC is thus consistent with the difference seen between their mixing properties with DPPC.
Fluorescence and anisotropy measurements
Finally, a complementary technique, fluorescence spectroscopy, was used to characterize the perturbations of POPC bilayers by C16-ceramide and C16-C1P. The use of POPC as the matrix lipid allowed the comparison of ceramide and C1P effects on bilayers to be extended to a more fluid bilayer environment that better approximates biological membranes. First, a pyrene labeled lipid analog, PPDPC, was used to estimate lateral segregation, and then DPH anisotropy was used to assess changes in lipid acyl-chain ordering. The value for Ie/Im measured for PPDPC in fluid POPC matrix is low, indicating the absence of lateral segregation of PPDPC in POPC (Fig. 6, panel a). Upon increasing Xcer, a profound increase in Ie/Im is seen. This could result either from an increased rate of lipid lateral diffusion or lateral segregation of the probe. To resolve between these two mechanisms, we measured DPH fluorescence anisotropy (r, Fig. 6, panel b). A rapid increase in DPH anisotropy was seen upon increasing Xcer. Based on our previous findings (3,4), this is caused by increased lateral packing density in the bilayer causing decrement in membrane lateral diffusion. Accordingly, microdomains are formed that are enriched in C16-ceramide. For C16-C1P up to XC1P = 0.30 in POPC bilayers, no evident changes are seen in Ie/Im for PPDPC after which increasing XC1P to 0.40 causes an increase in Ie/Im. Likewise, DPH anisotropy remains practically unaltered up XC1P = 0.20, whereafter a small gradual increase in DPH anisotropy is seen. Yet, these changes for POPC/C16-C1P bilayers in both Ie/Im for PPDPC and DPH anisotropy are subtle compared to POPC/C16-ceramide bilayers.
Figure 6.

(a) Intermolecular Ie/Im for PPDPC (X =0.01) measured for MLVs composed of POPC and the indicated contents of C16-ceramide (■) or C16-C1P (○). (b) Fluorescence anisotropy (r) for DPH (X = 0.002) residing in binary MLVs composed of POPC and C16-ceramide (■) or C16-C1P (○). The total lipid concentration was 22.5 μM in 20 mM HEPES, 0.1 mM EDTA, and 150 mM NaCl (pH 7.4). The temperature was maintained at 25°C with a circulating waterbath.
Discussion
In this article, we have addressed properties of DPPC/C16-ceramide and DPPC/C16-C1P mixtures using DSC and NMR. To better mimic natural membranes, these results were complemented with studies using fluid POPC as the matrix lipid. Both sphingolipids were composed of a sphingosine backbone having a saturated palmitoyl hydrocarbon chain and a palmitoyl N-acyl chain esterified at the NH2 group. For C16-C1P, an additional phosphate group is attached to the C1 position. The first three carbons (C1–C3) of the sphingosine backbone of ceramide or C16-C1P are intuitively equivalent to the three glycerol carbons of glycerophospholipids such as phosphatidylcholine, whereas C4 is more or less equivalent to the sn-1 ester-bond oxygen of phosphatidylcholine. Intuitively C16-ceramide or C16-C1P, having two palmitoyl chains, are sterically close to DPPC, which has saturated C16 chains, and should therefore match a DPPC bilayer with regard to its hydrophobic length. The structurally closely related sphingomyelin, having the phosphocholine group esterified to ceramide, is miscible in phosphatidylcholine membranes as long as there is no hydrophobic mismatch and the Tms do not differ too much (28,29). Maulik and Shipley (30) studied the mixing behavior of N-stearoyl-sphingomyelin (C18-SM) with DPPC using DSC and x-ray diffraction. Their data showed complete miscibility of these lipids in the gel and the fluid phase. Likewise, N-palmitoyl-SM (C16-SM) was miscible in dimyristoyl-phosphatidylcholine (DMPC) bilayers (28), and recent atomistic simulations have also indicated sphingomyelin to mix in fluid and raft-like membranes (31). In line with these data, we showed that C16-SM was miscible in fluid palmitoyl-oleoyl-phosphatidylcholine (POPC) bilayers (4). Yet, C16-ceramide was laterally segregated in fluid POPC bilayers, and biologically more relevantly, enzymatic formation of C16-ceramide from C16-SM by the action of sphingomyelinase induced also immiscibility. These results are also confirmed and show that ceramide exhibits a strong propensity to induce phase separation. This behavior is consistent with recent atom-scale simulations that showed ceramide to act like cholesterol, meaning that they both order lipid acyl chains in palmitoyl-oleoyl-phosphatidylcholine bilayers (32). In contrast, C16-C1P is miscible (up to XC1P ∼ 0.2–0.30) in DPPC and POPC bilayers, and compared to C16-ceramide induces significantly less phase separation near the transition.
Partial phase diagrams of mixtures of DPPC with either C16-ceramide or C16-C1P were constructed from the DSC data recorded during upscans and from 2H-NMR observations (Fig. 7). Because chain perdeuteration lowers the DPPC main-phase transition temperature by ∼4° and because the 2H-NMR spectra may not reliably distinguish the Pβ′ and Lβ′ gel phases, the phase diagrams derived from DSC and 2H-NMR observations are presented and discussed separately.
Figure 7.

Panels a and b show partial-phase diagrams obtained from DSC observations for DPPC with C16-ceramide up to Xcer = 0.40 (a) and DPPC with C16-C1P up to XC1P = 0.40 (b). See text for details. The lines connecting measured data points represent guides to the eye. Panels c and d show partial phase diagrams for DPPC-d62 with C16-ceramide determined from NMR studies up to Xcer = 0.40 (c) and DPPC-d62 with C16-C1P up to XC1P = 0.40 (d).
The phase diagrams derived from DSC observations are presented in Fig. 7, a and b. Some approximation was necessary because some of the phase boundaries could not be determined accurately. The DPPC/C16-ceramide phase diagram is constructed based on our previous partial-phase diagram for DMPC/C16-ceramide (6). It should be emphasized that the phase behavior of the DPPC/C16-ceramide mixture is complex, and therefore, the phase diagram shown represents the simplest one consistent with our data. In DSC scans at Xcer = 0.025, a significantly broader peak with a shoulder in the high temperature side is evident, with an additional subtransition peak and a pretransition peak. These indicate a narrow phase coexistence region between the Pβ′ phase and the Lα phase. As Xcer is increased to 0.05, a clear second peak is observed, and at still higher ceramide contents, at least three peaks are observed in the DSC scans at high temperatures. The upper phase boundary of the gel/Lα phase coexistence region can be determined independently and accurately from the DSC scans and from changes in 2H NMR lineshape. Because the Pβ′ phase is only observed up to Xcer = 0.10, there must be a point at Xcer > 0.10, whereby the lower gel (probably Lβ′)/Lα coexistence line starts to ascend from ∼40°C. Because Tm for pure C16-ceramide is ∼80–90°C (25), a possible scenario for the remaining part of the phase diagram could be that the horizontal phase boundary at T ∼40°C starts to bend upward for higher ceramide contents, so that the gel/Lα phase coexistence envelope will close up at T = 80–90°C.
The partial-phase diagram for DPPC/C16-C1P is simpler than the one for DPPC/C16-ceramide (Fig. 7 b). The slight increase in the pretransition temperature of the DPPC/C16-C1P bilayers with increasing C16-C1P content indicates that C16-C1P prefers the Lβ′ gel phase to the Pβ′ gel phase. In the gel phase, the glycerophospholipid counterpart, phosphatidic acid, adopts an ordered phase with a small tilt angle of 5–10° (33). It is thus feasible to suggest that C16-C1P would adopt a similar configuration in the gel phase. This could lead to gel-phase immiscibility at high C16-C1P concentrations. Yet, at XC1P ≤ 0.40, we did not find evidence for such behavior. At higher temperatures for DPPC/C16-C1P mixtures, the perturbation of DPPC packing is only modestly affected by the presence of C16-C1P, and therefore the shape of the endotherm remains symmetric and only the peak width is widened indicative of coexistence of Lα with a gel-phase (possibly Lβ) phase. The coexistence region is narrow even at XC1P = 0.30. DSC cooling scans do not suggest significant DPPC/C16-C1P phase separation at XC1P < 0.30. At XC1P = 0.40, the end of the main phase transition is difficult to determine precisely and Lα-gel (Lβ) coexistence region is widened. The solidus fluid line is constantly shifted to higher temperatures as XC1P is increased.
The 2H-NMR observations are consistent with these interpretations of the DPPC/C16-ceramide and DPPC/C16-C1P phase diagrams. It should be noted that chain perdeuteration lowers the main DPPC-d62 phase transition by ∼4° from that of normal DPPC. Fig. 7, c and d, show the range of two-phase coexistence deduced from inspection of the spectral series obtained while cooling each sample. As it is difficult to reliably distinguish different gel phases from 2H-NMR spectra of DPPC-d62, Fig. 7, c and d, do not show a boundary corresponding to the pretransition. Spectra characteristic of fast, axially symmetric reorientation in the fluid phase are superpositions of Pake doublets. The prominent edges, typically with splittings of 30–40 kHz, correspond to the most ordered portion of the chains on molecules in regions of the sample where the bilayer normal is perpendicular to the applied magnetic field. For such spectra, intensity is limited to a frequency range equal to twice the splitting of the prominent 90° edges. Upon cooling a sample from the Lα phase, the onset of two-phase coexistence is indicated by the first appearance of intensity just beyond this theoretical limit. Within the two-phase region, spectra are superpositions of the fluid-phase spectral component and the wider gel-phase spectrum. The lower limit of two-phase coexistence is identified by the disappearance of the sharp doublet features characteristic of the fluid spectral component.
In a study of deuterium-labeled ceramide in POPC, Hsueh et al. (10) observed spectra indicative of solid or crystalline phase ceramide for Xcer > 0.2. In this study, the label in the sphingolipid/DPPC mixtures is applied to DPPC, and it is not possible to detect separation of solid-phase ceramide directly from the observed spectra. However, by making use of spectral differences (10,34–37), it was possible to infer that a fraction of the ceramide in the Xcer = 0.4 sample of DPPC-d62/C16-ceramide was not participating in the fluid/gel-phase equilibrium.
The relative intensities of the fluid and gel spectral components within the two-phase region are determined by the lever rule. This has been exploited to obtain tie-line endpoint concentrations, and thus two-phase boundaries, in a variety of model membrane systems using differences between 2H-NMR spectra, at a given temperature, for samples with different concentrations within the two-phase region (10,34–37). For a given temperature, within the two-phase region, two samples having different overall compositions will have different relative amounts of the gel and fluid spectra corresponding to the tie-line endpoints at that temperature. The amount of one spectrum that must be subtracted from the other to obtain either the fluid or gel endpoint spectrum can be used to calculate the corresponding endpoint composition. Spectral subtractions were carried out between 40°C and 55°C using the DPPC-d62/C16-ceramide spectra obtained from the Xcer = 0.2 and Xcer = 0.4 samples. These subtractions consistently yielded fluid-phase endpoints that fell significantly below the endpoint concentrations that could be inferred by interpolation of the boundary points obtained from direct inspection of spectra for the range of C16-ceramide concentrations studied. In particular, the sample with Xcer = 0.1 effectively located the fluid-phase endpoint concentration for the tie line at 46°C. It was found that spectral subtraction of the 46°C spectra for Xcer = 0.2 and Xcer = 0.4 could only be consistent with this identified endpoint if the actual C16-ceramide concentration in the higher concentration sample was assumed to be in the range 0.27 < Xcer< 0.3. This implies that some of the C16-ceramide in the Xcer = 0.4 did not participate in the phase equilibrium and was presumably separated into a solid or crystalline phase. This would be consistent with the observation, from Fig. 4 a, that the first spectral moments from the Xcer = 0.4 sample of DPPC-d62/C16-ceramide differed only slightly from those of the Xcer = 0.3. In light of this evidence for separation of C16-ceramide from the Xcer = 0.4 sample, the partial-phase diagrams in Fig. 7, a and c, for DPPC-d62/C16-ceramide reflect DSC observations and 2H-NMR spectra from samples limited to Xcer ≤ 0.3.
Because DPPC is not a major component of cellular membranes, POPC was used as the matrix lipid in an additional set of experiments. 2H-NMR comparisons of POPC-d31/C16-ceramide and POPC-d31/C16-C1P, both at X = 0.2, were consistent with the finding, from 2H-NMR observations of DPPC-d62/C16-ceramide and DPPC-d62/C16-C1P, that C16-ceramide perturbs phosphatidylcholine lipid bilayers more strongly and is more conducive to phase separation in phosphatidylcholine bilayers than C16-C1P.
Using fluorescent probes PPDPC and DPH, we confirmed our previous findings (4) that C16-ceramide is laterally segregated into microdomains already at Xcer < 0.1. In keeping with the slightly increased 2H-NMR first spectral moments for binary mixtures of POPC and C16-C1P, a small increase in DPH anisotropy was seen at XC1P ≥ 0.30. Keeping with the NMR data, this increase is minor compared to the effect of C16-ceramide. PPDPC data suggest that C16-C1P is miscible with POPC at least up to XC1P = 0.20. At XC1P = 0.30, C16-C1P may show some immiscibility, and at XC1P = 0.40 this tendency is more clear. It should be emphasized that compared to C16-ceramidem C16-C1P shows much weaker propensity for lateral segregation in POPC bilayers.
The mechanism causing increase in Ie/Im for PPDPC upon increasing Xcer is of interest. If PPDPC would reside within the C16-ceramide-enriched domains, an increase in Xcer would dilute PPDPC and thus decrease Ie/Im. Yet, the opposite is observed. On the other hand, preferential partitioning of PPDPC into POPC domains would increase Ie/Im as XPOPC decreases. However, increments of Ie/Im can be estimated to be negligible. Packing of the saturated ceramide in microdomains can be anticipated to be tight, and accommodation of bulky PPDPC into these domains is not likely. Accordingly, the perhaps most feasible explanation is that PPDPC becomes enriched into the C16-ceramide-rich domain boundaries. We have previously shown that DPH induces large local perturbations in phosphatidylcholine bilayers (38), and therefore it is expected that DPH would also be excluded from the tightly packed C16-ceramide-enriched microdomains. Anisotropy depends on the average angular motion of the fluorophore. A decrease in the membrane-free volume allows for more hindered wobbling of the fluorophore and thus increases anisotropy. Accordingly, we suggest that DPH is reporting the dynamics of the domain interface as well as that of POPC. Similar reasoning can be applied to POPC/C16-C1P membranes, although the segregation of C16-C1P is much weaker compared to ceramide.
The main implication of the findings presented here is that phosphorylation of ceramide to yield C16-C1P should inhibit or reverse the formation of laterally segregated gel-like ceramide-enriched domains. Biologically, the rapid emergence of ceramides, in events such as apoptosis or inflammation, is catastrophic as the biophysical properties of the membranes where ceramide is produced are rapidly changed. To alleviate this, the cells strive to activate enzymes to process ceramides into lipids that are more miscible into the surrounding lipid matrix. In line with this, Luberto and Hannun (39) showed that in SV40-transformed lung fibroblasts, exogenous addition of ceramide induced a rapid increase in sphingomyelin synthase activity. A similar increase in levels of sphingomyelin (and also ceramide) was recently observed in cells lacking CERK (20), suggesting that sphingomyelin synthase might be enhanced. It is tempting to suggest that in normal cells rapid emergence of ceramides would also activate CERK to reduce the levels of ceramides. Importantly in contrast to ceramides, sphingomyelin, and C1P are easily accommodated into cellular membranes, and thus the physical state of the membranes would be sustained.
Acknowledgments
The authors acknowledge Douglas Grzetic and Bretta Russell-Schulz for their assistance with the acquisition and analysis of NMR results reported here.
This work was supported by funding from the Sigrid Juselius Foundation (J.M.H.), the Finnish Cultural Foundation (J.M.H.), Evald and Hilda Nissi Foundation (J.M.H.), The Finnish Eye Foundation (J.M.H.), the Academy of Finland (A.H., I.V., S.W.), and from the Natural Sciences and Engineering Research Council of Canada (M.R.M.).
Footnotes
This article is dedicated to deceased colleague Anne Helle.
References
- 1.Hannun Y.A. Functions of ceramide in coordinating cellular responses to stress. Science. 1996;274:1855–1859. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- 2.Huang H.W., Goldberg E.M., Zidovetski R. Ceramide induces structural defects into phosphatidylcholine bilayers and activates phospholipase A2. Biochem. Biophys. Res. Commun. 1996;220:834–838. doi: 10.1006/bbrc.1996.0490. [DOI] [PubMed] [Google Scholar]
- 3.Holopainen J.M., Lehtonen J.Y.A., Kinnunen P.K.J. Lipid microdomains in dimyristoylphosphatidylcholine-ceramide liposomes. Chem. Phys. Lipids. 1997;88:1–13. doi: 10.1016/s0009-3084(97)00040-6. [DOI] [PubMed] [Google Scholar]
- 4.Holopainen J.M., Subramanian M., Kinnunen P.K.J. Sphingomyelinase induces lipid microdomain formation in a fluid phosphatidylcholine/sphingomyelin membrane. Biochemistry. 1998;37:17562–17570. doi: 10.1021/bi980915e. [DOI] [PubMed] [Google Scholar]
- 5.Holopainen J.M., Angelova M.I., Kinnunen P.K.J. Vectorial budding of vesicles by asymmetrical enzymatic formation of ceramide in giant liposomes. Biophys. J. 2000;78:830–838. doi: 10.1016/S0006-3495(00)76640-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holopainen J.M., Lemmich J., Richter F., Mouritsen O.G., Rapp G. Dimyristoylphosphatidylcholine/C16:0-ceramide binary liposomes studied by differential scanning calorimetry and wide- and small-angle x-ray scattering. Biophys. J. 2000;78:2459–2469. doi: 10.1016/S0006-3495(00)76790-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holopainen J.M., Brockman H.L., Brown R.E., Kinnunen P.K.J. Interfacial interactions of ceramide with dimyristoylphosphatidylcholine: impact of the N-acyl chain. Biophys. J. 2001;80:765–775. doi: 10.1016/S0006-3495(01)76056-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrer D.C., Maggio B. Phase behavior and molecular interactions in mixtures of ceramide with dipalmitoylphosphatidylcholine. J. Lipid Res. 1999;40:1978–1989. [PubMed] [Google Scholar]
- 9.Veiga M.P., Arrondo J.L., Goni F.M., Alonso A. Ceramides in phospholipid membranes: effects on bilayer stability and transition to nonlamellar phases. Biophys. J. 1999;76:342–350. doi: 10.1016/S0006-3495(99)77201-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsueh Y.-W., Giles R., Kitson N., Thewalt J. The effect of ceramide on phosphatidylcholine membranes: a deuterium NMR study. Biophys. J. 2002;82:3089–3095. doi: 10.1016/S0006-3495(02)75650-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore D.J., Rerek R.E., Mendelsohn R. Lipid domains and orthorhombic phases in model stratum corneum: evidence from Fourier transform infrared spectroscopy studies. Biochem. Biophys. Res. Commun. 1997;231:797–801. doi: 10.1006/bbrc.1997.6189. [DOI] [PubMed] [Google Scholar]
- 12.Li L., Tang X., Taylor K.G., DuPre D.B., Yappert M.C. Conformational characterization of ceramides by nuclear magnetic resonance spectroscopy. Biophys. J. 2002;82:2067–2080. doi: 10.1016/S0006-3495(02)75554-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jarvis W.D., Fornari F.A., Jr., Browning J.L., Gewirtz D.A., Kolesnick R.N. Attenuation of ceramide-induced apoptosis by diglyceride in human myeloid leukemia cells. J. Biol. Chem. 1994;269:31685–31692. [PubMed] [Google Scholar]
- 14.Flores I., Jones D.R., Merida I. Changes in the balance between mitogenic and antimitogenic lipid second messengers during proliferation, cell arrest, and apoptosis in T-lymphocytes. FASEB J. 2000;14:1873–1875. doi: 10.1096/fj.99-1066fje. [DOI] [PubMed] [Google Scholar]
- 15.Kolesnick R.N., Krönke M. Regulation of ceramide production and apoptosis. Annu. Rev. Physiol. 1998;60:643–665. doi: 10.1146/annurev.physiol.60.1.643. [DOI] [PubMed] [Google Scholar]
- 16.Pettus B.J., Chalfant C.E., Hannun Y.A. Ceramide in apoptosis: an overview and current perspectives. Biochim. Biophys. Acta. 2002;1585:114–125. doi: 10.1016/s1388-1981(02)00331-1. [DOI] [PubMed] [Google Scholar]
- 17.Hannun Y.A., Loomis C.R., Merrill A.H., Jr., Bell R.M. Sphingosine inhibition of protein kinase C activity and of phorbol dibutyrate binding in vitro and in human platelets. J. Biol. Chem. 1986;261:12604–12609. [PubMed] [Google Scholar]
- 18.Chalfant C.E., Spiegel S. Sphingosine 1-phosphate and ceramide 1-phosphate: expanding roles in cell signaling. J. Cell Sci. 2005;118:4605–4612. doi: 10.1242/jcs.02637. [DOI] [PubMed] [Google Scholar]
- 19.Gomez-Munoz A. Ceramide 1-phosphate/ceramide, a switch between life and death. Biochim. Biophys. Acta. 2006;1758:2049–2056. doi: 10.1016/j.bbamem.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 20.Mitra P., Maceyka M., Payne S.G., Lamour N., Milstien S. Ceramide kinase regulates growth and survival of A549 human lung adenocarcinoma cells. FEBS Lett. 2007;581:735–740. doi: 10.1016/j.febslet.2007.01.041. [DOI] [PubMed] [Google Scholar]
- 21.Davis J.H., Jeffrey K.R., Bloom M., Valic M.I., Higgs T.P. Quadrupole echo deuteron magnetic resonance spectroscopy in ordered hydrocarbon chains. Chem. Phys. Lett. 1976;42:390–394. [Google Scholar]
- 22.Prosser R.S., Davis J.H., Dahlquist F.W., Lindorfer M.A. 2H nuclear magnetic resonance of the gramicidin A backbone in a phospholipids bilayer. Biochemistry. 1991;30:4687–4696. doi: 10.1021/bi00233a008. [DOI] [PubMed] [Google Scholar]
- 23.Davis J.H. The description of membrane lipid conformation, order and dynamics by 2H-NMR. Biochim. Biophys. Acta. 1983;737:117–171. doi: 10.1016/0304-4157(83)90015-1. [DOI] [PubMed] [Google Scholar]
- 24.Mabrey S., Sturtevant J.M. Investigation of phase transitions of lipids and lipid mixtures by high sensitivity differential scanning calorimetry. Proc. Natl. Acad. Sci. USA. 1976;73:3862–3866. doi: 10.1073/pnas.73.11.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shah J., Atienza J.M., Duclos R.I., Jr., Rawlings A.V., Dong Z. Structural and thermotropic properties of synthetic C16:0 (palmitoyl) ceramide: effect of hydration. J. Lipid Res. 1995;36:1936–1944. [PubMed] [Google Scholar]
- 26.Seelig J. Deuterium magnetic resonance: theory and application to lipid membranes. Q. Rev. Biophys. 1977;10:353–418. doi: 10.1017/s0033583500002948. [DOI] [PubMed] [Google Scholar]
- 27.Davis J.H. Deuterium magnetic resonance study of the gel and liquid crystalline phases of dipalmitoyl phosphatidylcholine. Biophys. J. 1979;27:339–358. doi: 10.1016/S0006-3495(79)85222-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calhoun W.I., Shipley G.G. Sphingomyelin-lecithin bilayers and their interaction with cholesterol. Biochemistry. 1979;18:1717–1722. doi: 10.1021/bi00576a013. [DOI] [PubMed] [Google Scholar]
- 29.Bar L.K., Barenholz Y., Thompson T.E. Effect of sphingomyelin composition on the phase structure of phosphatidylcholine-sphingomyelin bilayers. Biochemistry. 1997;36:2507–2516. doi: 10.1021/bi9625004. [DOI] [PubMed] [Google Scholar]
- 30.Maulik P.R., Shipley G.G. Interactions of N-stearoyl sphingomyelin with cholesterol and dipalmitoylphosphatidylcholine in bilayer membranes. Biophys. J. 1996;70:2256–2265. doi: 10.1016/S0006-3495(96)79791-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niemelä P., Ollila S., Hyvönen M.T., Karttunen M., Vattulainen I. Assessing the nature of lipid raft membranes. PLoS Comput. Biol. 2007;3:304–312. doi: 10.1371/journal.pcbi.0030034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pandit S., Chiu S.-W., Jakobsson E., Grama A., Scott H.L. Cholesterol surrogates: a comparison of cholesterol and 16:0 ceramide in POPC bilayers. Biophys. J. 2007;92:920–927. doi: 10.1529/biophysj.106.095034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jähnig F., Harlos K., Vogel H., Eibl H. Electrostatic interactions at charged lipid membranes. Electrostatically induced tilt. Biochemistry. 1979;18:1459–1468. doi: 10.1021/bi00575a012. [DOI] [PubMed] [Google Scholar]
- 34.Huschilt J.C., Hodges R.S., Davis J.H. Phase equilibria in an amphiphilic peptide-phospholipid model membrane by deuterium nuclear magnetic resonance difference spectroscopy. Biochemistry. 1985;24:1377–1386. [Google Scholar]
- 35.Vist M.R., Davis J.H. Phase equilibria of cholesterol /dipalmitoylphosphatidylcholine mixtures: 2H nuclear magnetic resonance and differential scanning calorimetry. Biochemistry. 1990;29:451–464. doi: 10.1021/bi00454a021. [DOI] [PubMed] [Google Scholar]
- 36.Morrow M.R., Srinivasan R., Grandal N. The phase diagram of dimyristoyl phosphatidylcholine and chain-perdeuterated distearoyl phosphatidylcholine: a deuterium NMR spectral difference study. Chem. Phys. Lipids. 1991;58:63–72. [Google Scholar]
- 37.Morrow M.R., Singh D., Lu D., Grant C.W.M. Glycolipid phase behaviour in unsaturated phosphatidylcholine bilayers: a 2H-NMR study. Biochim. Biophys. Acta. 1992;1106:85–93. doi: 10.1016/0005-2736(92)90225-b. [DOI] [PubMed] [Google Scholar]
- 38.Repakova J., Holopainen J.M., Morrow M.R., MacDonald M.C., Capkova P. Influence of DPH on the structure and dynamics of a DPPC bilayer. Biophys. J. 2005;88:3398–3410. doi: 10.1529/biophysj.104.055533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luberto C., Hannun Y.A. Sphingomyelin synthase, a potential regulator of intracellular levels of ceramide and diacylglycerol during SV40 transformation. Does sphingomyelin synthase account for the putative phosphatidylcholine-specific phospholipase C? J. Biol. Chem. 1998;273:14550–14559. doi: 10.1074/jbc.273.23.14550. [DOI] [PubMed] [Google Scholar]