

Abstract
Glycogen synthase kinase -3 (GSK-3) is a key enzyme involved in numerous physiological events and in major diseases, such as Alzheimer's disease, diabetes, cardiac hypertrophy. Indirubins are bis-indoles which can be generated from various natural sources or chemically synthesized. While highly potent and selective as GSK-3 inhibitors, most indirubins exhibit low water solubility. To address the issue of solubility we have designed novel analogues of 6-bromo-indirubin-3′-oxime with increased hydrophilicity, based on the GSK-3/indirubins co-crystal structures. The new derivatives with an extended amino side chain attached at position 3′ showed potent GSK-3 inhibitory activity, enhanced selectivity and dramatically increased water solubility. Furthermore, some of them displayed little or no cytotoxicity. The new indirubins inhibit GSK-3 in a cellular reporter model. They alter the circadian period measured in rhythmically expressing cell cultures, suggesting that they might constitute tools to investigate circadian rhythm regulation.
INTRODUCTION
Among the 518 protein kinases which constitute the human kinome, glycogen synthase kinase 3 (GSK-3) stand out as a particularly interesting and well-studied family of serine/threonine kinases review in 1-5. There are only two GSK-3 forms (GSK-3α and GSK-3ß), which share extensive similarity (84% overall identity, 98% within the catalytic domain), the main difference coming from an extra Gly-rich stretch in the N terminal domain of GSK-3α. GSK-3 are highly conserved protein kinases present from unicellular parasites 6,7 to yeast 8 up to mammals. These kinases are involved in numerous critical physiological events such as Wnt and Hedgehog signaling, embryonic development (pattern specification and axial orientation), transcription, insulin action, cell division cycle, cell death, cell survival, differentiation, multiple neuronal functions, circadian rhythm regulation, stem cell differentiation, etc… In addition GSK-3s are implicated in a large diversity of human diseases, including nervous system disorders such as Alzheimer's disease 9-11, schizophrenia 12, bipolar disorder 13, diabetes 14, heart hypertrophy 15,16, renal diseases 17, shock and inflammation 18, cancers 19, etc… There is thus a strong rationale supporting the search for potent and selective GSK-3 inhibitors for their use as pharmacological tools in basic research, as potential drugs for the treatment of specific diseases and for the maintenance of pluripotent stem cells in the absence of feeder cells 20. Numerous GSK-3 inhibitory scaffolds have been described review in 21-24. Interestingly many of these inhibitors also interact with cyclin-dependent kinases (CDKs), another family of well-studied key regulatory enzymes 25.
Among GSK-3 inhibitors, derivatives of the bis-indole indirubin (collectively referred to as indirubins) appear as a class of original and promising tools and agents review in 26. Their moderate selectivity might be an inconvenient when used as a research reagent, but their combined effects on several disease-relevant targets (in particular CDKs and GSK-3) may constitute an advantage for potential therapeutic applications 24. Among many indirubins, 6-bromo-indirubin-3′-oxime (6BIO) 27-29 has been widely used to investigate the physiological role of GSK-3 in various cellular settings and to alter the fate of embryonic stem cells 20. While highly potent and relatively selective kinase inhibitory indirubins have been developed, they usually exhibit low water solubility. To address the solubility problem of these promising compounds we have designed novel analogues of 6BIO with increased hydrophilicity. Improvement of the hydrophilic character of a molecule may be approached by several ways. The decrease of the aromatic character of indirubin scaffold by changing the hybridization state of an aromatic carbon atom to sp3 has been proposed as a way to enhance solubility 30. An alternative method is the introduction of hydrophilic groups on the molecule 31. Obviously, it is essential that the optimization of hydrophilicity does not negatively impact on either the potency or on the selectivity of the molecule towards the target kinase. The choice of the substitution position is thus highly significant since there are two important areas of the molecule that cannot be altered without dramatic decrease of efficacy on kinases. The first one is the pharmacophore consisting of the lactam nitrogen and carbonyl and the heterocyclic nitrogen of the bis-indole core that form the key hydrogen bonding interaction pattern with the active site of the kinase targets 27,28,32-36. The second is the bromine substitution at position 6 which is the selectivity determinant of 6BIO towards GSK-3ß 27,28,37. A detailed analysis of the crystal structure of GSK-3ß in complex with 6BIO 27,28 provided critical information suggesting that the 3′ position might be ideal for carrying out chemical modifications on the indirubin scaffold. We thus designed and synthesized a series of 6-bromo-indirubins with various substitutions on position 3′. Some of these molecules displayed high potency towards GSK-3, enhanced selectivity and much increased water-solubility. These molecules were evaluated for their GSK-3 inhibitory actions in several cellular systems.
RESULTS AND DISCUSSION
Rationale - Binding cavity mapping
The biochemical and potential therapeutic importance of 6BIO being a highly potent and selective GSK-3β ATP-competitive inhibitor is undermined by its poor water solubility (<5mg/l). Chemical synthesis was chosen to address the question of improving this physicochemical property. Modifications of the 6BIO molecule had to be done with care, as the aim was to introduce substitutions that might improve solubility and simultaneously maintain affinity and selectivity.
Both affinity and selectivity are attributed to particular sites of the 6BIO molecule that, as a consequence, should not be altered. The pharmacophore is an essential part of the molecule which needs to be kept intact. It accommodates the main stabilizing interactions of the ligand which is anchored at the kinase active site through three hydrogen bonds. Furthermore, substitution at position 6 is crucial for selectivity. A bulky group at that position takes advantage of the difference in the active site “gatekeeper” residue between GSK-3β (leu132) and the CDKs (phe80CDK2). Selectivity results from the steric clash between the substituent at position 6- and the phenynalanine residue of the CDKs. Positions 4 and 7 are not suitable for substitution. An atom larger than hydrogen at position 4 distorts the planarity of the molecule. Substitution on position 7 causes unfavorable contacts with the protein backbone 37,38. An introduction of hydrophilic groups at position 5 has already been performed 39,40. Nevertheless, an unfavorable interaction with the catalytic residues lys85 and asp200 of the active site is possible. Substituents at position 5′ and 6′ would be oriented towards the side chain of arg141 forming π-π face-to-edge stacking interactions, but analogues carrying bulky substitutions at these positions have demonstrated a diminished potency 25,27,28,40,41.
The study of crystal structures of indirubin 3′-oxime bound to CDK5/p25 (1UNH) 34 and 6BIO bound to GSK3β (1UV5) 27,28 indicated the oxime moiety as an ideal site for introducing groups that will increase the hydrophilic character of 6BIO. The oxime of the bound ligand lies at the periphery of the binding cleft being partly exposed to the solvent (Figure 1). As a consequence, the introduction of bulky groups or chains at this position seems to be sterically acceptable. Most importantly, the rich polar environment provided by the ribose sub-site where the oxime is positioned could facilitate novel favorable interactions. The binding mode of ATP to CDK2 as it appears in crystal structures (1B38, 1HCK), demonstrates the significance of interactions formed at the ribose sub-site on stabilizing the inhibitor-receptor complex (Figure 2). The ribose moiety of ATP is anchored by the side chain carboxyl of asp86 and by the backbone carbonyl of gln131 through hydrogen bonds (Figure 1). In the structure of ATP bound to CDK2/Cyclin A (1FIN), a conformational change is induced by cyclin binding which results in a reorientation of ATP in the cavity. However, ribose retains a strong interaction with the cavity environment, in this case with the backbone carbonyl of ile10 of the gly-loop. The ribose sub-site of the GSK-3β binding cavity is similar to that of CDK2. Ile10CDK2 corresponds to ile62GSK3β, gln131CDK2 to gln185GSK3β and asp86CDK2 to thr138GSK3β. The ribose sub-site thus, provides a rich environment of polar groups that could be targeted as interaction partners of improved 6BIO analogues.
Figure 1. Mapping of the hydrophobic (green), positive (blue) and negative (red) regions of the binding pocket of GSK-3.

Three distinct regions are visible, the hydrophobic active site where 6BIO molecule is bound, the ribose sub-site where the sugar of ATP forms stabilizing interactions with the receptor and the phosphate sub-site where the three phosphate groups interact with catalytic residues of the kinase. The ribose and phosphate sub-sites provide a polar environment which could accommodate favourable electrostatic or hydrogen bonding interactions with the inhibitor molecule.
Figure 2. Superposition of crystal structures of ATP bound to monomeric CDK2 (orange) and CDK2/cyclin A (blue).
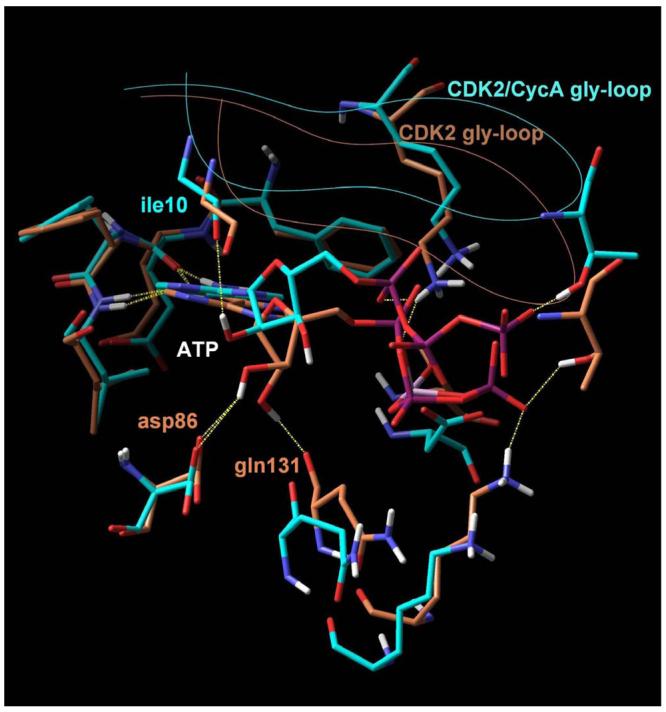

In both cases, two hydrogen bonds formed between the adenine and the receptor (yellow dashed lines) stabilize the complex. An additional set of favourable interactions exists at the ribose sub-site of the binding pocket where the sugar moiety of ATP interacts through hydrogen bonds with residues that constitute the periphery of the binding pocket, namely asp86 and gln131 in the monomeric kinase and ile10 in the CDK2/cyclin A dimer. Visible is also the Cα trace of the glycine loop at the top of the binding pocket.
By examining crystal structures of ATP or ATP-competitive inhibitors bound to various kinases, an additional part of the cavity was located where favorable interactions could be facilitated. It is the place where the phosphate groups of ATP interact with the receptor, called phosphate sub-site. Key interactions between the ATP phosphates and the kinase are formed there and more specifically between the α-phosphate and the side chain of lys33, between the β-phosphate and the side chains of lys129 and gln131 and between the γ-phosphate and the side chains of lys129 and thr14. These residues along with asn132 and the catalytic asp145 (the D of the DFG conserved motif) form the phosphate sub-site in CDK2. In GSK-3β the homologous residues are ser66 (thr14CDK2), lys85 (lys33CDK2), lys183 (lys129CDK2), gln185 (gln131CDK2), asn186 (asn132CDK2) and asp200 (asp145CDK2). A grid-based mapping of the hydrophilic and hydrophobic sites of the binding cavity of GSK-3β was performed by the Sitemap module of Maestro. Finally, two water molecules (pdb 1UV5, waters 49 and 74) that are proposed to bridge the interaction between thr138 and the oxime were considered as part of the cavity and possible mediators of favorable interactions 28.
Rationale - Energetics
On the basis of the GSK-3ß active site inspection, the substitution of the 3′oxime by a hydrophilic chain containing appropriate chemical groups that could interact with residues of the ribose and the phosphate sites of the receptor cavity was considered as suitable (Figure 1). Such a substitution is expected to resolve the solubility problem of indirubins without affecting the selectivity towards GSK-3ß as the 6Br substitution remains unaltered. Introducing hydrophilic chemical groups on 6BIO could have undesirable consequences on binding affinity as an increase of the polar character of a molecule results in a larger desolvation energy penalty upon binding. In addition, conformational entropy cost proportional to molecular flexibility could further decrease binding affinity. Energetics of binding imply that the finest way to overcome the impact of introducing flexible hydrophilic groups on the affinity of 6BIO for GSK-3β is the formation of novel stabilizing interactions between these groups and the kinase. Maximizing the attractive interactions in an otherwise highly flexible moiety could equilibrate the expected entropic loss in binding affinity due to the degrees of freedom inherent in such a system as well as the desolvation energy loss and optimize the affinity of the inhibitors.
Synthesis of 3′-substituted, 6-bromo-indirubins
In an effort to prepare derivatives consistent with the above rationale we synthesized 3′-substituted oximes of 6BIO bearing side chains with amine groups 5-15 and the corresponding salts 16-25. The synthesis was based on the reaction of the 3′-[O-(2-bromoethyl)oxime] intermediate 1 with the appropriate amine: pyrrolidine, morpholine, piperazine, N-methylpiperazine, hydroxyethylpiperazine, methoxyethylpiperazine, dimethylamine and diethylamine, N,N-bis-2-hydroxyethylamine, N-2,3-dihydroxypropyl-N-methyl amine, and N-2-hydroxyethoxyethyl piperazine. The intermediate 1 was prepared by the reaction of 6BIO with 1,2-dibromoethane in DMF and Et3N at room temperature. In addition, the carbamate 4 was prepared by the reaction of 6BIO with N,N-diethylcarbamyl chloride. The alcohols 2 and 3 were prepared by the reaction of 6BIO with the appropriate bromo alcohol in DMF and Et3N at room temperature. Indirubin and 6BIO were synthesized as previously reported 28.
Building analogues - kinase inhibitory properties
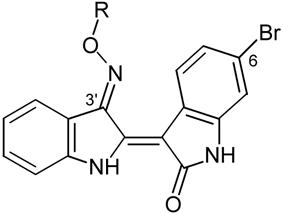
Several chemical groups were introduced on the oxime oxygen of 6BIO and designed analogues along with their IC50 values for three different kinases (GSK-3α/β, CDK1/cyclin B, CDK5/p25) are summarized in Table 1. As the initial objective was to increase solubility, these groups were chosen according to their polarity and their potential to ionize. The biological results of the new 6BIO analogues have provided interesting information about structure-activity relations concerning GSK-3. In addition, docking calculations have provided insight in the binding mode of the novel analogues. The physicochemical properties of representative compounds such as calculated pKa, clogD and water solubility are summarized in Table 2.
Table 1. Effects of indirubins 1-26 on three protein kinases and on the survival of human neuroblastoma SH-SY5Y cells.
Indirubins were tested at various concentrations on GSK-3α/ß, CDK1/cyclin B, CDK5/p25, as described in Experimental Section. IC50 values, calculated from the dose-response curves, are reported in μM. The compounds were tested at various concentrations for their effects on SH-SY5Y cells survival after 48 h incubation as estimated using the MTS reduction assay. IC50 values, calculated from the dose-response curves, are reported in μM.
 | |||||
|---|---|---|---|---|---|
| Cpd # | R | GSK3 | CDK1 | CDK5 | SH-SY5Y |
| 6BIO | H | 0.005 | 0.320 | 0.083 | 9.0 |
| 1 | —CH2CH2Br | 0.14 | > 10 | > 10 | > 100 |
| 2 | —CH2CH2OH | 0.003 | > 10 | > 10 | > 100 |
| 3 | —CH2CH(OH)CH2OH | 0.034 | 0.110 | 0.025 | 0.94 |
| 4 | 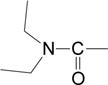 |
0.03 | > 10 | 10 | > 100 |
| 5 | 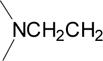 |
0.033 | 0.490 | 0.100 | 5.4 |
| 16 | 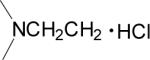 |
0.029 | 0.19 | 0.053 | 5.8 |
| 6 | 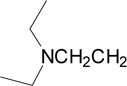 |
0.035 | 0.09 | 0.02 | 78.0 |
| 17 | 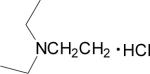 |
0.027 | 0.19 | 0.04 | > 100 |
| 9 | 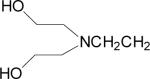 |
0.040 | 0.60 | 0.21 | 6.6 |
| 20 | 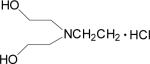 |
0.041 | 1.00 | 0.10 | 9.0 |
| 10 | 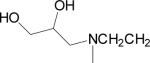 |
0.067 | 0.24 | 0.24 | 5.8 |
| 21 | 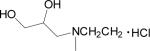 |
0.023 | 0.15 | 0.10 | 6.0 |
| 7 | 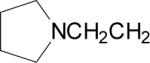 |
0.026 | 0.50 | 0.13 | 5.5 |
| 18 | 0.054 | 0.45 | 0.10 | 4.2 | |
| 8 | 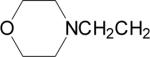 |
0.060 | 1.10 | 0.60 | 72 |
| 19 | 0.110 | 1.80 | 0.90 | 74 | |
| 11 | 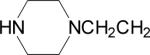 |
0.0033 | 0.3 | 0.2 | 13.2 |
| 22 | 0.0013 | 0.2 | 0.18 | 5.9 | |
| 12 | 0.0070 | 0.4 | 0.4 | 5.4 | |
| 23 | 0.0050 | 0.3 | 0.3 | 5.4 | |
| 13 | 0.0050 | 0.6 | 0.2 | 28.0 | |
| 24 | 0.0042 | 0.4 | 0.2 | 16.7 | |
| 14 | 0.0110 | 2.8 | 0.5 | > 100 | |
| 25 | 0.0200 | 1.0 | 0.4 | > 100 | |
| 15 | 0.014 | 0.90 | 0.31 | 94.4 | |
| 26 | 0.033 | 0.50 | 0.33 | 97.6 | |
Table 2.
Water solubility of indirubin salts and calculated physicochemical properties pKa and logD at pH 7.4 of corresponding bases.
| No. | Solubility (g/l) | pKa | LogD |
|---|---|---|---|
| 6BIO | < 0.005 | - | 2.59 |
| 16 | 0.141 | 8.59 | 1.69 |
| 17 | 0.192 | 9.38 | 1.19 |
| 18 | 0.195 | 9.18 | 1.16 |
| 21 | 1.45 | 7.48 | 1.79 |
| 22 | 1.61 | 8.85 | −0.87 |
| 23 | 1.50 | 7.65 | 1.74 |
| 24 | 1.14 | 7.65 | 1.41 |
| 25 | 0.57 | 7.47 | 1.90 |
| 26 | 4.253 | 7.58 | 1.46 |
Analogue 3 carries a side chain with two aliphatic hydroxyls at a distance of two carbon atoms from the oxime oxygen, which were expected to mimic the 2′ and 3′ hydroxyl groups of ATP ribose. It demonstrated a 6-fold decrease of activity compared to 6BIO. This decrease was accompanied by a mediocre improvement of solubility. This result was indicative of the unfavorable influence of chain flexibility on affinity. As flexibility was implicit for groups that would be used to improve solubility, the amplification of their stabilizing interactions was considered as a way to equalize the entropic loss. A series of analogues carrying a tertiary amine two carbons from the oxime was prepared in order to enhance the strength of hydrogen bonding interactions that are expected to form at the ribose sub-site. Analogue 5 carries a dimethylamino group two carbon atoms from the oxime oxygen. Analogue 6 carrying a bulkier diethylamino group was designed to scan the extent by which the ATP channel could accommodate larger hydrophobic interactions. The ethyl carbons of 6 were fused to a pyrrolidine ring in analogue 7 to reduce conformational entropy. Their activity was only slightly improved. Docking calculations demonstrated that the main interaction formed by those analogues was a charge-assisted hydrogen bond between the protonated secondary amine nitrogen and the side chain of the catalytic asp200. However, the geometry of this bond was not optimal. As a result, alternative binding modes were observed where residues asn64 (gly-loop), asn186 (ribose-site) and water 47 participated with polar interactions. Keeping the tertiary amine unaltered was encouraged by this observed binding mode. As a result, further scanning of the cavity towards the phosphate sub-site was performed by the extended chains of 9, 10 and 8. The first two carry a short aliphatic extension bearing two hydroxyl groups. The free rotation of the extended chain was restricted in 8, where the two hydroxyethyl groups were fused to a morpholine ring. Compounds 8 and 9 demonstrated a noteworthy decrease of affinity (8-fold and 12-fold). According to these results and modeling it was deduced that affinity could not be optimized by introduction of hydroxyl groups. Interactions formed by hydroxyls at the ribose sub-site included hydrogen bonds with gln185 and a water-mediated bridge with thr138. Docking calculations suggested that these bonds could not compensate for the hydroxy-carrying group inherent flexibility. The conversion of the heterocyclic oxygen of the rigid morpholine to nitrogen would thus add a tertiary positively charged amine group closer to asp200. Analogue 11/22 carrying a piperazine ring demonstrated the highest affinity (3.3 nM/ 1.3 nM). The proposed binding mode of 11 is depicted in Figure 3a and shows an optimal pattern of interactions between the ligand and the active site of GSK-3β accounting for the excellent affinity of this compound. The 4-nitrogen of the piperazine ring is protonated and donates a high-quality charge-assisted hydrogen bond to the carboxylate oxygen of asp200. The piperazine ring adopts the lowest energy chair conformation which permits an optimal approach and orientation between donor and acceptor atoms (1.8Å). In addition, the oxime oxygen forms a hydrogen bond with water49 in a similar to the other compounds mode. However, the high ionizing potential of 11 has a negative impact on its physicochemical profile which is reflected on the calculated logD value (−0.87). A conversion of the secondary 4-nitrogen to tertiary was utilized as a way to limit its basicity. This conversion was combined with a gradual extension of the molecule that assisted on mapping the full extent by which the cavity can accommodate bulky ligands. The piperazine ring was substituted by a methyl, a hydroxyethyl, a methoxyethyl and a hydroxyethoxyethyl group in analogues 12, 13, 14 and 15, respectively (and corresponding salts: 23, 24, 25, 26). Within this last group of analogues, 12 and 13 share the best consensus pharmacological and physicochemical profile. They demonstrate affinity similar to that of 6BIO, improved selectivity of one order of magnitude for GSK-3, increased water solubility and acceptable lipophilicity. The proposed binding mode of 13 is depicted in Figure 3b.
Figure 3. Binding mode of analogues 11 (A) and 13 (B) to the binding pocket of GSK-3β.
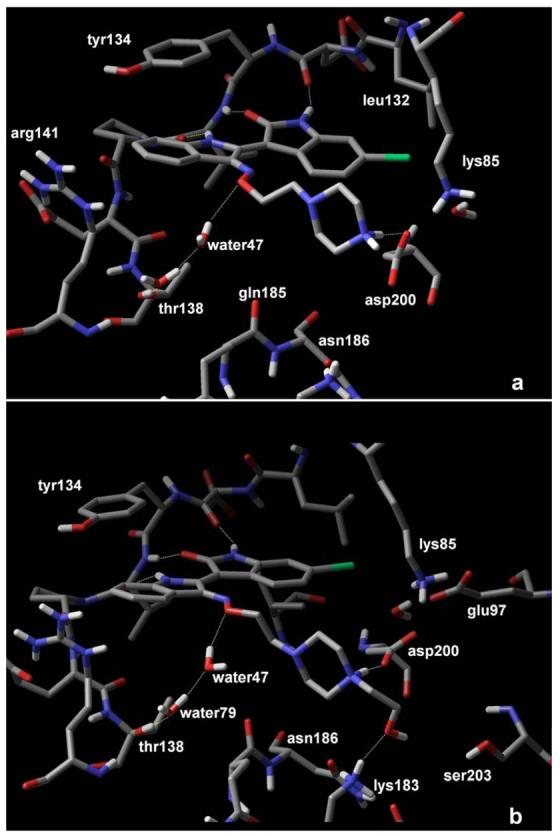
The piperazine substitution of both analogues interacts with asp200 and residues located at the phosphate sub-site of the binding pocket in addition to the hydrogen bonds (yellow dashed lines) formed between the indirubin scaffold and the receptor backbone
Cytotoxicity of the 6BIO analogues
All compounds were next tested for their effects on survival of SH-SY5Y neuroblastoma cells using an MTS reduction assay. These assays revealed that increased potency on GSK-3 was not associated with enhanced cell death (Table 1). Analogues 13, 14 and 15 (and their corresponding salts, 24, 25, 26) had little cell death inducing activities. The IC50 values were respectively 28 μM, >100 μM, 94 μM (salts: 17 μM, >100 μM, 98 μM) to compare with the IC50 of 6BIO, 9 μM). Therefore the substituted piperazine ring extension, not only favours selectivity and efficacy towards GSK-3, allows better solubility, but also reduces their cytotoxicity. These features are particularly favorable for the use of these compounds in the study of GSK-3 in cellular systems, and also as potential therapeutic leads in the context of neurodegenerative diseases and diabetes.
Confirmation of intracellular inhibition of GSK-3 by indirubins
To investigate whether the new indirubins were effective at inhibiting GSK-3 in a cellular context we measured their effects on the phosphorylation of β-catenin at GSK-3 specific sites in SH-SY5Y neuroblastoma cells. Cells were exposed to various concentrations of 10 μM of each indirubin in the presence of a constant level of MG132 (an inhibitor of the proteasome which prevented the rapid degradation of β-catenin once phosphorylated by GSK-3). The level of GSK-3 -phosphorylated was estimated either by Western blotting (with an antibody that specifically cross-reacts with β-catenin when phosphorylated at a GSK-3 site) following SDS-PAGE (Figure 4a) or by an ELISA assay (Figure 4b). Results revealed a dose-dependent inhibition of GSK-3 selective phosphorylation sites on β-catenin, demonstrating that these compounds are actually able to inhibit GSK-3 in cells. The most efficient compounds were 6BIO, 3, 5, 9, 11, 12 and 13 (and their salts when available, i.e. 16, 20, 22, 23, 24). Dose-response curves were obtained with ELISA assay (Figure 4b). The kinase inactive derivative 1-methyl-6BIO was ineffective in the cellular assay.
Figure 4. β-catenin phosphorylation at GSK-3 phosphorylation sites is inhibited by the indirubin derivatives.

A. SH-SY5Y neuroblastoma cells were exposed for 6 hours to 10 μM of each indirubin, in the presence of a constant 2 μM level of the proteasome inhibitor MG132. The level of GSK-3 -phosphorylated β-catenin was estimated by Western blotting following SDS-PAGE, using an antibody that specifically cross-reacts with GSK-3 phosphorylated β-catenin. Lack or reduction of the signal indicates that the indirubin has been able to inhibit GSK-3 within the neuroblastoma cells. C, control (DMSO); 6B, 6BIO; M6B, Methyl-6BIO (a control inactive analog of 6BIO 27); K, kenpaullone, a structurally unrelated GSK-3 inhibitor 49. B. Dose-response curves for a selection of indirubins were run in an ELISA assay using the same antibodies directed against GSK-3 phosphorylated β-catenin. SH-SY5Y cells were exposed for 6 hours to a range of concentrations of each indirubin, in the presence of MG132, and extracts were assessed in the ELISA assay. Activity was expressed as percentage of phosphorylated β-catenin in untreated control cells.
Effects on circadian rhythm in mammalian cell cultures
GSK-3 is a key regulator of the circadian rhythm (aka the daily biological clock) 44,45. The circadian rhythm can be partially reproduced in a cellular system which is an excellent model system for circadian clocks in non-neural, peripheral tissues46. We therefore used this system to explore the possibility that GSK-3 inhibition could affect the circadian rhythm. Rat-1 fibroblasts stably transfected with a PPer2::Fluc reporter construct show a robust circadian rhythm of luminescence as a gauge of clock-controlled Per2 promoter (PPer2). Cells were cultured and treated first with 10 μM indirubin 15 as described in the Experimental section and their circadian rhythm of Per expression dependent luminescence was monitored during 4 days. A gradual shortening of the period length was clearly observed (Figure 5a). Similar experiments were next performed with a small selection of indirubins and the period length was calculated as in Figure 5a. The most efficient compounds in shortening the period length (Figure 5b) were also the most efficient at inhibiting β-catenin phosphorylation in the cellular assay (Figure 4), which supports the hypothesis that the action of the indirubins in shortening the circadian period is upon GSK-3. Previous studies have suggested a key action of GSK-3 in regulating the circadian rhythms of mammalian cells using lithium as a pharmacological tool 45. Lithium lengthens the period of the circadian rhythm, whereas indirubins shorten the period (Figure 5). However, the concentrations of lithium used in the previous investigations of circadian rhythms and GSK-3 were 10-20 mM 46, whereas our results with indirubins were obtained with concentrations 1000X lower (10 μM). These comparisons suggest that the circadian period-lengthening effects of lithium may be due to side effects of lithium. Therefore, indirubins might constitute a useful pharmacological tool to investigate the role of GSK-3 in the regulation of the circadian rhythm.
Figure 5. Indirubins alter the circadian period in mammalian fibroblasts.

Rat-1 fibroblasts stably transfected with a PPer2::Fluc reporter construct show a robust circadian rhythm of luminescence as a gauge of clock-controlled Per2 promoter (PPer2) activity. Cells were cultured up to 100% confluence and treated for 2 h with 0.1 μM dexamethasone to synchronize the oscillators. The medium was then replaced with assay medium supplemented with 0.1 mM luciferin and the luminescence rhythm was monitored for 4 days or more. Compounds were added to the culture dishes at 10 μM and left continuously. DMSO was used as a solvent control. Regression analyses were used to determine period and phase of the luminescence rhythms. A. Time-course of a typical luminescence rhythm recorded in control cells (○) or cells treated with indirubin 15 (●). White arrows indicate the peaks of luminescence in control cells, black arrows indicate the peaks of luminescence in indirubin 15 treated cells. B. Period lengths calculated in cells exposed to various compounds. C, control; K, kenpaullone.
Conclusions
Through a rationale analysis of the key interactions of indirubins at the ATP-binding site of the disease-relevant glycogen synthase kinase -3, and the synthesis and biological evaluation of analogues exploring various modifications at the 3′ site, we were able to uncover new interaction sites offering further stabilization to the inhibitor/GSK-3 complexes. Consequently, extensions at this site provide enhanced activity and selectivity towards GSK-3 and also provided the opportunity to introduce substitutions favoring enhanced water-solubility. These molecules were less cytotoxic than the parent 6BIO compound, and demonstrated potent GSK-3 inhibition in two cellular models. These results open new directions towards the design of pharmacologically favorable indirubins with potential development against Alzheimer's disease, diabetes, heart hypertrophy or in the field of embryonic stem cell pluripotency maintenance.
EXPERIMENTAL SECTION
Chemistry
General chemistry experimental procedures
All chemicals were purchased from Aldrich Chemical Co. NMR spectra were recorded on Bruker DRX 400 and Bruker AC 200 spectrometers [1H (400 and 200 MHz) and 13C (50 MHz)]; chemical shifts are expressed in ppm downfield from TMS. The 1H-1H and the 1H-13C NMR experiments were performed using standard Bruker microprograms. Melting points were determined with a Sanyo Gallencamp apparatus. MS spectra were determined on a MSQ Thermofinnigan spectrometer. All UV/vis spectra were recorded on a Shimadzu UV-160A spectrophotometer.
Solubility Measurements
Equilibrium solubilities were determined by adding an excess amount of solid to the medium (water, double distilled) followed by 5 min of sonification and overnight equilibration by stirring at ambient temperature (25±0.1 °C). The samples were centrifuged and aliquots were removed. Standard solutions were prepared for each compound in order to quantify the aforementioned saturated solutions, and reference curves were plotted for each compound. The absorbance of each saturated and standard solution was measured with a UV/vis spectrophotometer at wavelength that varied between 515 and 518 nm.
General Procedure for the Preparation of the Ethers 1-3
To a solution of 6-Bromoindirubin-3′-oxime (1.0 g, 2.83 mmol) in DMF (90 ml) were added triethylamine (0.5 ml) and the appropriate bromide (2 equiv.) under Ar, and the mixture was stirred for 17 h. at room temperature. Then water (300 ml) was added and the precipitate formed was collected by filtration and washed with water.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2-bromoethyl)-oxime] (1)
Yield: 95%. Mp 252 °C. 1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 11.67 (1H, s, H-1′), 10.93 (1h, s, H-1), 8.49 (1H, d, J = 8.3 Hz, H-4), 8.21 (1H, d, J = 7.8 Hz, H-4′), 7.45 (2H, brs, H-6′, H-7′), 7.16 (1H, dd, J = 8.3 / 2.0 Hz, H-5), 7.07 (2H, m, H-5′, H-7), 4.93 (2H, t, J = 5.6 Hz, H-1″), 3.97 (2H, t, J = 5.6 Hz, H-2″). APCI-MS m/z 462, 464, 466 (M+H)+. Anal. (C18H13N3O2Br2) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2-hydroxyethyl)-oxime] (2)
Yield: 96%. Mp >300 °C. 1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 11.70 (1H, brs, H-1′), 10.90 (1H, brs, H-1), 8.54 (1H, d, J = 8.4 Hz, H-4), 8.18 (1H, d, J = 7.6 Hz, H-4′), 7.44 (2H, m, H-7′, H-6′), 7.16 (1H, dd, J = 8.4 / 1.9 Hz, H-5), 7.05 (2H, m, H-5′, H-7), 5.02 (1H, t, J = 5.4 Hz, -OH), 4.62 (2H, t, J = 4.8 Hz, H-1″), 3.89 (2H, m, H-2″). CI-MS m/z 400, 402 (M+H)+. Anal. (C18H14N3O3Br) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2,3-dihydroxypropyl)-oxime](3)
Yield: 95%. Mp >300 °C. 1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 11.70 (1H, s, H-1′), 10.90 (1H, s, H-1), 8.56 (1H, d, J = 8.5 Hz, H-4), 8.17 (1H, d, J = 7.7 Hz, H-4′), 7.44 (2H, m, H-6′, H-7′), 7.15 (1H, dd, J = 8.5, 1.9 Hz, H-5), 7.06 (1H, m, H-5′), 7.03 (1H, d, J = 1.9 Hz, H-7), 5.14 (1H, d, J = 5.0 Hz, −CHOH), 4.84 (1H, t, J = 5.6 Hz, −CH2OH), 4.65 (1H, dd, J = 10.9, 3.7 Hz, H-1″a), 4.50 (1H, dd, J = 10.9, 6.6 Hz, H-1″b), 3.99 (1H, m, H-2″), 3.50 (2H, m, H-3″). APCI-MS (+) m/z 430, 432 (M+H)+. Anal. (C19H16N3O4Br2) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(N,N-diethylcarbamyl)-oxime] (4)
To a solution of 6-Bromoindirubin-3′-oxime (36 mg, 0.11 mmol) in DMF (10 ml) were added triethylamine (0.5 ml) and 0.3 ml (3.66 mmol) N,N-diethylcarbamylchloride under Ar, and the mixture was stirred for 12 h. at room temperature. Then water (40 ml) was added and the precipitate formed was collected by filtration and washed with water to give 4 quantitatively. Yield: 90%. Mp 237 °C. 1H-NMR (400 MHz, pyridine d-5, δ ppm, J in Hz) 12.36 (1H, s, H-1′), 12.15 (1H, s, H-1), 9.93 (1H, d, J = 8.6 Hz, H-4), 8.13 (1H, d, J = 7.8 Hz, H-4′), 7.61 (1H, dd, J = 8.6 / 1.9 Hz, H-5), 7.43 (1H, m, H-6′), 7.35 (1H, d, J = 1.9 Hz, H-7′), 7.14 - 7.06 (2H, m, H-5′, H-7), 3.44 (4H, brs, −N(CH2CH3)2), 1.18 (6H, t, J = 7.0 Hz, −N(CH2CH3)2). APCI-MS (+) m/z 455, 457 (M+H)+. Anal. (C21H19N4O3Br) C, H, N.
General Procedure for the Preparation of the Amines 5-15
100 mg of 6-bromoindirubin-3′-[O-(2″-bromoethyl)-oxime] (1) was dissolved in 5 ml of anhydrous DMF. An excess of the appropriate amine was added under magnetic stirring and the mixture was then heated at 50 °C. After the completion of the reaction, the mixture was poured into water (30 ml) and the precipitate was filtered and washed with water and cyclohexane. Dimethylamine, diethylamine, pyrrolidine, morpholine, diethanolamine, 3-methylamine-1,2-propanediole, piperazine, 1-methylpiperazine, 1-(2-methoxyethyl) piperazine, 1-(2-hydroxyethyl) piperazine and 1-[2-(2-hydroxyethoxy)-ethyl] piperazine afforded products (5) - (15), correspondingly, in qualitative yields.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2-dimethylaminoethyl)-oxime] (5)
Mp 230 °C. 1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 11.71 (1H, brs, H-1′), 10.92 (1H, brs, H-1), 8.55 (1H, d, J = 8.5 Hz, H-4), 8.14 (1H, d, J = 7.5 Hz, H-4′), 7.44 (2H, d, J = 4.1 Hz, H-6′, H-7′), 7.15 (1H, dd, J = 8.5 / 2.0 Hz, H-5), 7.05 (2H, m, H-5′, H-7), 4.69 (2H, t, J = 5.8 Hz, H-1″), 2.80 (2H, t, J = 5.8 Hz, H-2″), 2.27 (6H, s, –N(CH3)2 ). APCI-MS m/z 427, 429 (M+H)+. Anal. (C20H19N4O2Br) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2-diethylaminoethyl)-oxime] (6)
Mp 232 °C. 1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 11.71 (1H, s, H-1′), 10.92 (1H, s, H-1), 8.55 (1H, d, J = 8.2 Hz, H-4), 8.16 (1H, d, J = 7.8 Hz, H-4′), 7.44 (2H, d, J = 3.4 Hz, H-6′, H-7′), 7.13 (1H, dd, J = 8.2 / 2.1 Hz, H-5), 7.09 - 7.02 (2H, m, H-7, H-5′), 4.65 (2H, t, J =6.0 Hz, H-1″), 2.94 (2H, t, J = 6.0 Hz, H-2″), 2.58 (2H, q, J = 7.2 Hz, –N(CH2CH3)2 ), 0.98 (6H, t, J = 7.2 Hz, –N(CH2CH3)2 ). APCI-MS (+) m/z 455, 457 (M+H)+. Anal. (C22H23N4O2Br) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2-pyrrolidin-1-ylethyl)oxime] (7)
Mp 208 °C. 1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 11.70 (1H, s, H-1′), 10.93 (1H, s, H-1), 8.54 (1H, d, J = 8.5 Hz, H-4), 8.14 (1H, d, J = 7.7 Hz, H-4′), 7.45 (2H, m, H-6′, H-7′), 7.14 (1H, d, J = 8.5 / 1.9 Hz, H-5), 7.05 (2H, m, H-5′, H-7), 4.70 (2H, t, J = 5.8 Hz, H-1″), 2.98 (2H, brt, J = 5.8 Hz, H-2″), 2.57 (4H, brs, H-3″, H-6″), 1.69 (4H, m, H-4″, H-5″). APCI-MS (+) m/z 453, 455 (M+H)+. Anal. (C22H21N4O2Br) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2-morpholin-1-ylethyl)oxime] (8)
Mp 235 °C. 1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 11.70 (1H, s, H-1′), 10.90 (1H, s, H-1), 8.52 (1H, d, J = 8.5 Hz, H-4), 8.15 (1H, d, J = 7.6 Hz, H-4′), 7.43 (2H, m, H-6′, H-7′), 7.14 (1H, dd, J = 8.5 / 1.9 Hz, H-5), 7.05 (1H, m, H-5′), 7.02 (1H, d, J = 1.9 Hz, H-7), 4.70 (2H, t, J = 5.8 Hz, H-1″), 3.57 (4H, t, J = 4.5 Hz, H-4″, H-5″), 2.86 (2H, t, J = 5.8 Hz, H-2″), 2.50 (4H, m, H-3″, H-6″, overlapped with DMSO). APCI-MS (+) m/z 469, 471 (M+H)+. Anal. (C22H21N4O3Br) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2-(N,N-(2-hydroxyethyl)aminoethyl) oxime] (9)
Mp 201 °C. 1H NMR (400 MHz, pyridine d-5, δ ppm, J in Hz) 12.31 (1H, brs, H-1′), 12.25 (1H, brs, H-1), 8.93 (1H, d, J = 8.2 Hz, H-4), 8.42 (1H, d, J = 7.8 Hz, H-4′), 7.47 (1H, dd, J = 8.2, 1.8 Hz, H-5), 7.39 (1H, d, J = 1.8 Hz, H-7), 7.34 (1H, t, J = 7.2 Hz, H-6′), 7.04 (2H, m, H-5′, H-7′), 5.89 (1H, brs, –OH), 4.86 (2H, t, J = 6.3 Hz, H-1″), 4.00 (4H, m, —N(CH2CH2OH)2 ), 3.38 (2H, t, J = 6.3 Hz, H-2″), 3.08 (4H, t, J = 5.9 Hz, —N(CH2CH2OH)2 ). APCI-MS (+) m/z 487, 489 (M+H)+. Anal. (C22H23N4O4Br) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-(O-{2-[N-methyl, N-(2,3-dihydroxypropyl) amino]ethyl}oxime] (10)
Mp 195 °C. 1H NMR (400 MHz, pyridine d-5, δ ppm, J in Hz) 12.27 (2H, m, H-1, H-1′), 8.90 (1H, d, J = 8.8 Hz, H-4), 8.41 (1H, d, J = 7.5 Hz, H-4′), 7.46 (1H, dd, J = 8.8,1.8 Hz, H-5), 7.38 (1H, d, J = 1.8 Hz, H-7′), 7.36 (1H, t, J = 7.5 Hz, H-6′), 7.05 (2H, m, H-5′, H-7), 4.80 (2H, t, J = 6.1 Hz, H-1″), 4.29 (1H, m, H-4″), 4.11 (1H, dd, J = 11.0, 4.6 Hz, H-5″a), 4.04 (1H, dd, J = 11.0, 5.5 Hz, H-5″b), 3.14 (2H, t, J = 6.1 Hz, H-2″), 2.93 (2H, m, H-3″), 2.49 (3H, s, –NCH3). CI-MS m/z 487, 489 (M+H)+. Anal. (C22H23N4O4Br) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2-piperazine-1-ylethyl)oxime] (11)
Mp 255 °C (dec.). 1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 11.69 (1H, s, H-1′), 10.92 (1H, s, H-1), 8.53 (1H, d, J = 8.5 Hz, H-4), 8.15 (1H, d, J = 7.4 Hz, H-4′), 7.43 (2H, m, H-6′, H-7′), 7.14 (1H, d, J = 8.5 Hz, H-5), 7.03 (2H, m, H-5′, H-7), 4.69 (2H, br t, H-1″), 2.83 (2H, br t, H-2″), 2.71 (4H, brs, H-4″, H-5″), 2.46 (4H, brs, H-3″, H-6″, partially overlapped with DMSO). APCI-MS (+) m/z 468, 470 (M+H)+. Anal. (C22H22N5O2Br) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-{O-[2-(4-methyl-piperazin-1-yl)ethyl]oxime} (12)
Mp 222 °C. 1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 11.68 (1H, s, H-1′), 10.90 (1H, s, H-1), 8.40 (1H, d, J = 8.5 Hz, H-4), 8.14 (1H, d, J = 7.7 Hz, H-4′), 7.42 (2H, m, H-6′, H-7′), 7.13 (1H, dd, J = 8.5 / 1.9 Hz, H-5), 7.04 (1H, m, H-5′), 7.02 (1H, d, J = 1.9 Hz, H-7), 4.68 (2H, t, J = 5.9 Hz, H-1″), 2.85 (2H, t, J = 5.9 Hz, H-2″), 2.50 (4H, brs, H-3″, H-6″, overlapped with DMSO), 2.31 (4H, brs, H-4″, H-5″), 2.13 (3H, s, –NCH3). APCI-MS (+) m/z 482, 484 (M+H)+. Anal. (C23H24N5O2Br) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-(O-{2-[4-(2-hydroxyethyl)piperazin-1-yl]ethyl}oxime) (13)
Mp 187 °C. 1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 8.52 (1H, d, J = 8.5 Hz, H-4), 8.16 (1H, d, J = 7.6 Hz, H-4′), 7.43 (2H, m, H-6′, H-7′), 7.13 (1H, dd, J = 8.5 / 1.8 Hz, H-5), 7.05 (1H, m, H-5′), 7.02 (1H, d, J = 1.8 Hz, H-7), 4.69 (2H, t, J = 5.7 Hz, H-1″), 3.45 (2H, t, J = 6.3 Hz, H-8″), 2.85 (2H, t, J = 5.7 Hz, H-2″), 2.50 (4H, H-3″, H-6″, overlapped with DMSO), 2.42 (4H, H-4″, H-5″), 2.34 (2H, t, J = 6.3 Hz, H-7″). APCI-MS (+) m/z 512, 514 (M+H)+. Anal. (C24H26N5O3Br) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-(O-{2-[4-(2-methoxyethyl)piperazin-1-yl]ethyl}oxime) (14)
Mp 184 °C. 1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 11.70 (1H, s, H-1′), 10.90 (1H, s, H-1), 8.50 (1H, d, J = 8.5 Hz, H-4), 8.16 (1H, d, J = 7.6 Hz, H-4′), 7.44 (2H, m, H-6′, H-7′), 7.15 (1H, dd, J = 8.5, 1.7 Hz, H-5), 7.07 (2H, m, H-5′, H-7), 4.70 (2H, t, J = 5.6 Hz, H-1″), 3.40 (2H, H-8″, overlapped with water), 3.21 (3H, s, −OCH3), 2.87 (2H, brt, H-2″), 2.66-2.40 (H-4″, H-5″, H-3″, H-6″, H-7″, overlapped with DMSO). APCI-MS (+) m/z 526, 528 (M+H)+. Anal. (C25H28N5O3Br) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2-{4-[2-(2-hydroxyethoxy)-ethyl]piperazin-1-yl}ethyl)oxime] (15)
Mp 183 °C. 1H NMR (400 MHz, pyridine d-5, δ ppm, J in Hz) 12.31 (1H, s, H-1′), 12.25 (1H, s, H-1), 8.89 (1H, d, J = 8.3 Hz, H-4), 8.39 (1H, d, J = 7.9 Hz, H-4′), 7.45 - 7.33 (3H, m, H-5, H-7′, H-6′), 7.09 (2H, m, H-5′, H-7), 4.78 (2H, t, J = 5.8 Hz, H-1″), 3.96 (2H, t, J = 5.0 Hz, H-10″), 3.70 (2H, t, J = 5.0 Hz, H-9″), 3.66 (2H, t, J = 5.8 Hz, H-8″), 2.94 (2H, t, J = 5.8 Hz, H-2″), 2.68 (2H, brs, H-3″, H-6″), 2.57 (8H, t, J = 5.8 Hz, H-4″, H-5″, H-7″). APCI-MS (+) m/z 556, 558 (M+H)+. Anal. (C26H30N5O4Br) C, H, N.
General Procedure for the Preparation of the Amine Salts 16-26
The appropriate indirubin derivative 5-15 (0.10 mmol) was dissolved in anhydrous THF (50 ml) and 0.2 ml of a saturated solution of hydrochloric acid in ether was added dropwise. The reaction mixture was left to cool in an ice bath and the precipitate formed was collected by filtration.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2-dimethylaminoethyl)oxime] Hydrochloride (16)
Sw (g/l) 0.141. 1H-NMR (400 MHz, DMSO d-6, δ ppm J in Hz) 11.70 (1H, s, H-1′), 10.97 (1H, s, H-1), 8.49 (1H, d, J = 8.3 Hz, H-4), 8.22 (1H, J = 7.4 Hz, H-4′), 7.46 (2H, m, H-7, H-6′), 7.20 (1H, dd, J = 8.3 / 1.7 Hz, H-5), 7.05 (2H, m, H-5′, H-7′), 4.95 (2H, brs, H-1″), 3.58 (2H, m, H-2″), 2.81 (6H, brs, −N(CH3)2). Anal. (C20H20N4O2BrCl) C, H, N.
Data for (2Z-3′E)-6-Bromoindirubin-3′-[O-(2-diethylaminoethyl)oxime] Hydrochloride (17)
Sw (g/l) 0.192. 1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 11.70 (1H, s, H-1′), 10.98 (1H, s, H-1), 8.49 (1H, d, J = 8.6 Hz, H-4), 8.21 (1H, d, J = 7.4 Hz, H-4′), 7.47 (2H, m, H-7, H-6′), 7.21 (1H, dd, J = 8.6 / 1.9 Hz, H-5), 7.09 - 7.04 (2H, m, H-5′, H-7′), 5.00 (2H, brs, H-1″), 3.58 (2H, brs, H-2″), 3.24 (4H, brs, −N(CH2CH3)2 ), 1.21 (6H, t, J = 7.0 Hz, −N(CH2CH3)2 ). Anal. (C22H24N4O2BrCl) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2-pyrrolidin-1-ylethyl)oxime] Hydrochloride (18)
Sw (g/l) 0.195. 1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 11.71 (1H, s, H-1′), 10.97 (1H, s, H-1), 8.48 (1H, d, J = 8.6 Hz, H-4), 8.22 (1H, d, J = 7.4 Hz, H-4′), 7.44 - 7.52 (2H, m, H-7, H-6′), 7.20 (1H, dd, J = 8.6 / 1.9 Hz, H-5), 7.07 (2H, m, H-5′, H-7′), 4.94 (2H, brs, H-1″), 3.64 (2H, brs, H-2″), 3.13 (4H, m, H-3″, H-6″), 2.02 (4H, m, H-4″, H-5″). Anal. (C22H22N4O2BrCl) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2-morpholin-1-ylethyl)oxime] Hydrochloride (19)
1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 11.69 (1H, s, H-1′), 10.98 (1H, s, H-1), 8.47 (1H, d, J = 8.5 Hz, H-4), 8.22 (1H, d, J = 7.8 Hz, H-4′), 7.45 (2H, m, H-7, H-6′), 7.21 (1H, dd, J = 8.3, 1.8 Hz, H-5), 7.06 (2H, m, H-5′, H-7′), 5.05 (2H, brs, H-1″), 3.95 (2H, m, H-2″), 3.75 (4H, m, H-4″, H-5″), 3.27 (4H, H-3″, H-6″, overlapped with water). Anal. (C22H22N4O3BrCl) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2-(N,N-(2-hydroxyethyl)aminoethyl)oxime] (20)
1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 11.71 (1H, s, H-1′), 10.98 (1H, s, H-1), 8.48 (1H, d, J = 8.3 Hz, H-4), 8.22 (1H, d, J = 7.9 Hz, H-4′), 7.46 (2H, m, H-7, H-6′), 7.21 (1H, dd, J = 8.3 / 1.8 Hz, H-5), 7.06 (2H, m, H-5′, H-7′), 5.35 (2H, brs, OH), 5.03 (2H, brs, H-1″), 3.84 (2H, brs, H-2″), 3.78 (4H, brs, −N(CH2CH2OH)2 ), 3.38 (4H, m, −N(CH2CH2OH)2, overlapped with water). Anal. (C22H24N4O4BrCl) C, H, N.
Data for (2Z-3′E)-6-Bromoindirubin-3′-(O-{2-[N-methyl, N-(2,3-dihydroxypropyl)amino]ethyl}oxime] Hydrochloride (21)
Sw (g/l) 1.45. 1H NMR (400 MHz, DMSO d-6, δ ppm, J in Hz) 11.69 (1H, s, H-1′), 10.96 (1H, s, H-1), 8.48 (1H, d, J = 8.5 Hz, H-4), 8.20 (1H, d, J = 7.9 Hz, H-4′), 7.4 (2H, m, H-7, H-6′), 7.19 (1H, dd, J = 8.5 / 1.8 Hz, H-5), 7.05 (2H, m, H-5′, H-7′), 4.95 (2H, brs, H-1″), 3.89 (1H, brs, H-4″), 3.38 (4H, H-3″, H-5″, overlapped with water), 2.83 (2H, brs, H-3″), 2.50 (3H,−N(CH3), overlapped with DMSO).Anal. (C22H24N4O4BrCl) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2-piperazine-1-ylethyl)oxime] Dihydrochloride (22)
Sw (g/l) 1.61. 1H NMR (400 MHz, D2O, δ ppm, J in Hz) 7.65 (1H, d, J = 8.5 Hz, H-4), 7.55 (1H, d, J = 7.5, H-4′), 7.26 (1H, t, J = 7.5 Hz, H-6′), 6.85 (1H, t, J = 7.2 Hz, H-5′), 6.76 (1H, d, J = 8.5 Hz, H-5), 6.72 (1H, d, J = 7.5 Hz, H-7′), 6.54 (1H, s, H-7), 4.42 (2H, brt, H-1″), 3.38 (4H, brt, H-4″, H-5″), 3.11 (6H, brs, H-3″, H-6″, H-2″). Anal. (C22H24N5O2BrCl2) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-{O-[2-(4-methylpiperazin-1-yl)ethyl]oxime} Dihydrochloride (23)
Sw (g/l) 1.50. 1H NMR (400 MHz, D2O, δ ppm, J in Hz) 7.63 (1H, d, J = 8.2 Hz, H-4), 7.53 (1H, d, J = 7.5, H-4′), 7.25 (1H, t, J = 7.6 Hz, H-6′), 6.84 (1H, t, J = 7.6 Hz, H-5′), 6.75 (1H, d, J = 8.2 Hz, H-5), 6.70 (1H, d, J = 7.5 Hz, H-7′), 6.53 (1H, s, H-7), 4.39 (2H, brs, H-1″), 3.39 (4H, brs, H-3″, H-6″), 3.11 (6H, brs, H-2″, H-4″, H-5″), 2.90 (3H, s, −NCH3). Anal. (C23H26N5O2BrCl2) C, H, N.
Data for (2′Z-3E)-6-Bromoindirubin-3′-(O-{2-[4-(2-hydroxyethyl)piperazin-1-yl]ethyl}oxime) Dihydrochloride (24)
Sw (g/l) 1.14. 1H NMR (400 MHz, D2O, δ ppm, J in Hz) 7.68 (1H, d, J = 8.5 Hz, H-4), 7.56 (1H, d, J = 7.4, H-4′), 7.27 (1H, brt, J = 7.2 Hz, H-6′), 6.85 (1H, brt, J = 7.2 Hz, H-5′), 6.78 (1H, d, J = 8.5 Hz, H-5), 6.73 (1H, d, J = 7.4 Hz, H-7′), 6.57 (1H, s, H-7), 4.42 (2H, brs, H-1″), 3.89 (2H, brs, H-8″), 3.39 (4H, brs, H-3″, H-6″), 3.26 (2H, brs, H-7″), 3.12 (6H, brs, H-2″, H-4″, H-5″). Anal. (C24H28N5O3BrCl2) C, H, N.
Data for (2′Z-3E)-6-Bromoindirubin-3′-(O-{2-[4-(2-methoxyethyl)piperazin-1-yl]ethyl}oxime) Dihydrochloride (25)
Sw (g/l) 0.57. 1H NMR (400 MHz, D2O, δ ppm, J in Hz) 7.70 (1H, brs, H-4), 7.61 (1H, brs, H-4′), 7.28 (1H, brt, J = 7.2 Hz, H-6′), 6.88 (1H, brt, J = 7.5 Hz, H-5′), 6.77 (2H, brs, H-5, H-7′), 6.58 (1H, s, H-7), 4.52 (2H, brs, H-1″), 3.74 (2H, brs, H-8″), 3.55-3.22 (12H, H-2″, H-3″, H-4″, H-5″, H-6″, H-7″), 3.35 (3H, s, −OCH3). Anal. (C25H30N5O3BrCl2) C, H, N.
Data for (2′Z-3′E)-6-Bromoindirubin-3′-[O-(2-{4-[2-(2-hydroxyethoxy)-ethyl]piperazin-1-yl}ethyl)oxime] Dihydrochloride (26)
Sw (g/l) 4.253. 1H NMR (400 MHz, D2O, δ ppm, J in Hz) 7.54 (1H, d, J = 8.1 Hz, H-4), 7.44 (1H, d, J = 7.2 Hz, H-4′), 7.21 (1H, brt, J = 7.6 Hz, H-6′), 6.78 (1H, brt, J = 7.2 Hz, H-5′), 6.69 (1H, d, J = 7.2 Hz, H-7′), 6.61 (1H, d, J = 8.1 Hz, H-5), 6.34 (1H, s, H-7), 4.28 (2H, brs, H-1″), 3.82, 3.72, 3.63, (6H, H-8″, H-9″, H-10″), 3.46-2.72 (12H, H-2″, H-3″, H-4″, H-5″, H-6″, H-7″). Anal. (C26H32N5O4BrCl2) C, H, N.
Molecular modeling
All compounds were designed and minimized. Compounds carrying nitrogen in their side chain were designed in their free and protonated states as well. Ligand partial charges were calculated in a semi-empirical level with the AM1 hamiltonian using MOPAC6 42.The receptor was prepared from the pdb entry of 6BIO bound to GSK-3β (1UV5). Protons were added and all crystallographic water molecules were deleted except waters 47 and 79 that mediate the interaction of the oxime with thr138 and water 1 that is buried in the cavity 28. The kinase was minimized in order to relieve the crystallographic strain in the absence of the ligand and then all molecules were manually docked in the active site by superimposition on crystallographic 6BIO coordinates. Docking was performed with the Monte Carlo Multiple Minimum search algorithm as implemented in Macromodel 9 software 43. The three key hydrogen bonds of each complex were restrained at their crystallographic values by application of harmonic distance and angle constraints. Force constraints were applied on waters 49 and 79. The remaining atoms within 6Å around the ligand were free to move. All calculations were performed with a truncated Newton conjugate gradient minimizer, a convergence criterion of 0.05kJ/Ǻ-mol, the AMBER* forcefield including protein partial charges and the GB/SA implicit solvent model. Values of LogD and pKa were calculated by Pallas 3.1 software.
Biology
Kinase preparation and assays
Kinase activities were assayed in buffer A (10 mM MgCl2, 1 mM EGTA, 1 mM DTT, 25 mM Tris-HCl pH 7.5, 50 μg heparin/ml) or C (homogenization buffer but 5 mM EGTA, no NaF and no protease inhibitors), at 30 °C, at a final ATP concentration of 15 μM. Blank values were subtracted and activities calculated as pmoles of phosphate incorporated during a 30 min. incubation. The activities were expressed in % of the maximal activity, i.e. in the absence of inhibitors. Controls were performed with appropriate dilutions of dimethylsulfoxide. Phosphorylation of the substrate was assessed by the P81 phosphocellulose assay.
CDK1/cyclin B was extracted in homogenization buffer (60 mM ß-glycerophosphate, 15 mM p-nitrophenylphosphate, 25 mM Mops (pH 7.2), 15 mM EGTA, 15 mM MgCl2, 1 mM DTT, 1 mM sodium vanadate, 1 mM NaF, 1 mM phenylphosphate, 10 μg leupeptin/ml, 10 μg aprotinin/ml, 10 μg soybean trypsin inhibitor/ml and 100 μM benzamidine) from M phase starfish (Marthasterias glacialis) oocytes and purified by affinity chromatography on p9CKShs1-sepharose beads, from which it was eluted by free p9CKShs1 as previously described 25. The kinase activity was assayed in buffer C, with 1 mg histone H1 /ml, in the presence of 15 μM [γ-33P] ATP (3,000 Ci/mmol; 10 mCi/ml) in a final volume of 30 μl. After 30 min. incubation at 30 °C, 25 μl aliquots of supernatant were spotted onto 2.5 × 3 cm pieces of Whatman P81 phosphocellulose paper, and, 20 sec. later, the filters were washed five times (for at least 5 min. each time) in a solution of 10 ml phosphoric acid/liter of water. The wet filters were counted in the presence of 1 ml ACS (Amersham) scintillation fluid.
CDK5/p25 was reconstituted by mixing equal amounts of recombinant human CDK5 and p25 expressed in E. coli as GST (Glutathione-S-transferase) fusion proteins and purified by affinity chromatography on glutathione-agarose (vectors kindly provided by Dr. L.H. Tsai) (p25 is a truncated version of p35, the 35 kDa CDK5 activator). Its activity was assayed with histone H1 in buffer C as described for CDK1/cyclin B.
GSK-3α/β was purified from porcine brain by affinity chromatography on immobilized axin 47. It was assayed, following a 1/100 dilution in 1 mg BSA/ml 10 mM DTT, with 4 μM GS-1 (YRRAAVPPSPSLSRHSSPHQSpEDEEE), a GSK-3 specific substrate obtained from Millegen (Labege, France), in buffer A, in the presence of 15 μM [γ-33P] ATP (3,000 Ci/mmol; 10 mCi/ml) in a final volume of 30 μl. After 30 min. incubation at 30 °C, 25 μl aliquots of supernatant were processed as described above.
Cellular assays
Cell culture conditions and cell survival assessment
SH-SY5Y human neuroblastoma cell line (kindly provided by Dr. Jacint Boix, Lleida) was grown at 37 °C with 5% CO2 in DMEM supplemented with 2mM L-glutamine (Invitrogen, Cergy Pontoise, France), plus antibiotics (penicillin-streptomycin) from Lonza, and a 10% volume of fetal calf serum (FCS) (Invitrogen). Drug treatments were performed on exponentially growing cultures at the indicated time and concentrations. Control experiments were carried also using appropriate dilutions of DMSO. Cell viability was determined by means of the MTS (3- (4,5-dimethylthiazol-2-yl)-5- (3-carboxymethoxyphenyl)-2- (4-sulfophenyl)-2H -tetrazolium) method after 48 hr of treatment as previously described 48.
β-catenin phosphorylation in SH-SY5Y human neuroblastoma cells
Nearly confluent SH-SY5Y human neuroblastoma cells were grown in 96 plates in DMEM (supplemented with 10% FCS and antibiotics). Cells were co-treated with tested compounds and 2 μM MG132 (to allow accumulation of phosphorylated β-catenin) for 6 hours. Final DMSO concentration did not exceed 1%. Cells were then subjected to an ELISA assay using antibodies directed against Ser33/Ser37/Thr41-phosphorylated (1:1000) β-catenin obtained from Cell Signaling Technology. Results are expressed in percentage of maximal β-Catenin phosphorylation, i.e. in untreated cells exposed to MG132 only as positive control (100% phosphorylation).
Cell culture and luminescence assay of circadian rhythmicity
These experiments used Rat-1 fibroblasts that have been stably transfected with a PPer2::Fluc reporter construct that shows a robust circadian rhythm of luminescence as a gauge of clock-controlled Per2 promoter (PPer2) activity 46. The cells were cultured in DMEM (11965-092, GIBCO/Invitrogen) supplemented with 5% FBS, 50 units/ml penicillin, and 50 μg/ml streptomycin in a 5% CO2 incubator at 37°C. Approximately 5 × 105 cells were seeded in a 35 mm dish at least 5 days before the experiment. Three days after the cells reached 100% confluence, the cells were treated with 0.1 μM dexamethasone (Sigma) for 1h to synchronize the oscillators among the cells in the population. At the end of the treatment, the medium was replaced with assay medium [DMEM without phenol red, supplemented with bicarbonate (350 mg/L), 5% FBS, 10mM HEPES (pH 7.2), antibiotics (25 units/ml penicillin, 25 μg/ml streptomycin), and 0.1 mM luciferin (Promega)]. Culture dishes were sealed with a 40-mm microscope glass cover slip and high-vacuum grease to prevent the evaporation of culture medium. The luminescence rhythm was monitored in a LumiCycle (Actimetrics Inc., Evanston, IL, USA). Before being sealed, drugs were added to the culture dishes to different final concentrations and left continuously with the cells thereafter while the luminescence patterns were recorded for 5 days or more. DMSO was used as a solvent control. Regression analyses to determine period and phase of the luminescence rhythms were performed with the Chrono II program (courtesy of Dr. Till Roenneberg).
Electrophoresis and Western blotting
Cells were resuspended, lysed for 30 min at 4 °C in Homogenization Buffer and sonicated. After centrifugation (14000 r.p.m. for 15 min at 4 °C), the protein concentration was determined in the supernatants by the Bradford protein assay (Bio-Rad). Following heat denaturation for 5 min, proteins were separated by 10% NuPAGE pre-cast Bis-Tris Acetate polyacrylamide mini gel (Invitrogen) with MOPS SDS running buffer. Proteins were transferred to 0.45 μm nitrocellulose filters (Schleicher and Schuell). These were blocked with 5% low fat milk in Tris-Buffered Saline - Tween-20, incubated overnight at 4 °C with antibodies directed against Ser33/Ser37/Thr41-phosphorylated β-catenin (1:1000) (Cell Signaling Technology) and analyzed by Enhanced Chemiluminescence (ECL, Amersham).
Scheme 1.

Reagents: (i) dibromoethane, triethylamine, DMF anh., 25°C; (ii) 2-bromoethanol, triethylamine, DMF anh., 25°C; (iii) 3-bromo-1,2-propanediol, triethylamine, DMF anh., 25°C; (iv)N,N-diethylcarbamyl chloride, triethylamine, DMF anh., 25°C. (v) DMF anh, 25°C, amine a-k
ACKNOWLEDGMENTS
We are grateful to Dr. J. Boix for SH-SY5Y cell line. This research was supported by grants from the EEC (FP6-2002-Life Sciences & Health, PRO-KINASE Research Project) (L.M.), and TEMPO (FP6 program, LSHG-CT-2006-037543) (L.M.), PENED program (V.M.), the Canceropole Grand-Ouest (L.M.), the “Association France-Alzheimer” (Finistère) and the ACCAMBA project (CT500485)). KB was supported by a fellowship from the “Ministère de la Recherche”. Funded in C.H. Johnson's lab by a grant from the National Institutes of Health (grant number R21 NS054991) and a Vanderbilt Institute for Chemical Biology Pilot Project Grant.
Abbreviations
- 6BIO
6-bromoindirubin-3′-oxime
- CDK
cyclin-dependent kinase
- FCS
fetal calf serum
- GSK-3
glycogen synthase kinase-3
- IO
indirubin-3′-oxime
- MTS
(3- (4,5-dimethylthiazol-2-yl)-5- (3-carboxymethoxyphenyl)-2- (4-sulfophenyl)-2H -tetrazolium)
Footnotes
SUPPORTING INFORMATION AVAILABLE: Elemental Analysis. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- 1.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jope RS, Johnson GVW. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Dajani R, et al. Crystal structure of glycogen synthase kinase 3β: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell. 2001;105:721–732. doi: 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 4.ter Haar E, et al. Structure of GSK3β reveals a primed phosphorylation mechanism. Nat. Struct. Biol. 2001;8:593–596. doi: 10.1038/89624. [DOI] [PubMed] [Google Scholar]
- 5.Kockeritz L, Doble B, Patel S, Woodgett JR. Glycogen synthase kinase-3--an overview of an over-achieving protein kinase. Curr. Drug Targets. 2006;7:1377–88. doi: 10.2174/1389450110607011377. [DOI] [PubMed] [Google Scholar]
- 6.Droucheau E, Primot A, Thomas V, Mattei D, Knockaert M, Richardson C, Sallicandro P, Alano P, Jafarshad A, Baratte B, Kunick C, Parzy D, Pearl L, Doerig C, Meijer L. Plasmodium falciparum glycogen synthase kinase -3, molecular model, expression, intracellular localisation and selective inhibitors. Biochim. Biophys. Acta (Proteins and Proteomics) 2004;1697:181–196. doi: 10.1016/j.bbapap.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 7.Xingi E, Smirlis D, Bisti S, Myrianthopoulos V, Magiatis P, Meijer L, Mikros E, Alexios- Skaltsounis AL, Soteriadou K. Role of Leishmania donovani glycogen synthase kinase-3β in parasite cellular processes: a potential drug target. Mol. Microbiol. 2008 submitted. [Google Scholar]
- 8.Kassir Y, Rubin-Bejerano I, Mandel-Gutfreund Y. The Saccharomyces cerevisiae GSK-3 beta homologs. Curr Drug Targets. 2006;7:1455–1465. doi: 10.2174/1389450110607011455. [DOI] [PubMed] [Google Scholar]
- 9.Muyllaert D, Kremer A, Jaworski T, Borghgraef P, Devijver H, Croes S, Dewachter I, Van Leuven F. Glycogen synthase kinase-3beta, or a link between amyloid and tau pathology? Genes Brain Behav. 2008;7(Suppl 1):57–66. doi: 10.1111/j.1601-183X.2007.00376.x. [DOI] [PubMed] [Google Scholar]
- 10.Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem. 2008;104:1433–9. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Avila J, Hernández F. GSK-3 inhibitors for Alzheimer's disease. Expert Rev Neurother. 2007;7:1527–33. doi: 10.1586/14737175.7.11.1527. [DOI] [PubMed] [Google Scholar]
- 12.Lovestone S, Killick R, Di Forti M, Murray R. Schizophrenia as a GSK-3 dysregulation disorder. Trends Neurosci. 2007;30:142–9. doi: 10.1016/j.tins.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Rowe MK, Wiest C, Chuang DM. GSK-3 is a viable potential target for therapeutic intervention in bipolar disorder. Neurosci. Biobehav. Rev. 2007;31:920–31. doi: 10.1016/j.neubiorev.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanabe K, Liu Z, Patel S, Doble BW, Li L, Cras-Méneur C, Martinez SC, Welling CM, White MF, Bernal-Mizrachi E, Woodgett JR, Permutt MA. Genetic deficiency of glycogen synthase kinase-3beta corrects diabetes in mouse models of insulin resistance. PLoS Biol. 2008;6:e37. doi: 10.1371/journal.pbio.0060037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugden PH, Fuller SJ, Weiss SC, Clerk A. Glycogen synthase kinase 3 (GSK3) in the heart: a point of integration in hypertrophic signalling and a therapeutic target? A critical analysis. Br. J. Pharmacol. 2008;153(Suppl 1):S137–53. doi: 10.1038/sj.bjp.0707659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kerkelä R, Woulfe K, Force T. Glycogen synthase kinase-3beta - actively inhibiting hypertrophy. Trends Cardiovasc. Med. 2007;17:91–96. doi: 10.1016/j.tcm.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Obligado SH, Ibraghimov-Beskrovnaya O, Zuk A, Meijer L, Nelson PJ. CDK/GSK-3 inhibitors as therapeutic agents for parenchymal renal diseases. Kidney Int. 2008;73:684–90. doi: 10.1038/sj.ki.5002731. [DOI] [PubMed] [Google Scholar]
- 18.Dugo L, Collin M, Thiemermann C. Glycogen synthase kinase 3beta as a target for the therapy of shock and inflammation. Shock. 2007;27:113–123. doi: 10.1097/01.shk.0000238059.23837.68. Erratum in: Shock2007, 27, 709. [DOI] [PubMed] [Google Scholar]
- 19.Ougolkov AV, Billadeau DD. Targeting GSK-3: a promising approach for cancer therapy? Future Oncol. 2006;2:91–100. doi: 10.2217/14796694.2.1.91. [DOI] [PubMed] [Google Scholar]
- 20.Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou A. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3 specific inhibitor. Nature Med. 2004;10:55–63. doi: 10.1038/nm979. [DOI] [PubMed] [Google Scholar]
- 21.Martinez A. Preclinical efficacy on GSK-3 inhibitors: Towards a future generation of powerfuldrugs. Med. Res. Rev. 2008 Feb 12; doi: 10.1002/med.20119. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 22.Patel DS, Dessalew N, Iqbal P, Bharatam PV. Structure-based approaches in the design of GSK-3 selective inhibitors. Curr. Protein Pept. Sci. 2007;8:352–64. doi: 10.2174/138920307781369409. [DOI] [PubMed] [Google Scholar]
- 23.Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat. Rev. Drug Discov. 2004;3:479–87. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- 24.Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol. Sci. 2004;25:471–80. doi: 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 25.Leclerc S, Garnier M, Hoessel R, Marko D, Bibb JA, Snyder GL, Greengard P, Biernat J, Mandelkow E-M, Eisenbrand G, Meijer L. Indirubins inhibit glycogen synthase kinase - 3β and CDK5/p25, two kinases involved in abnormal tau phosphorylation in Alzheimer's disease - A property common to most CDK inhibitors ? J. Biol. Chem. 2001;276:251–260. doi: 10.1074/jbc.M002466200. [DOI] [PubMed] [Google Scholar]
- 26.Meijer L, Guyard N, Skaltsounis LA, Eisenbrand G, editors. Indirubin; the red shade of indigo. Editions «Life in Progress». Station Biologique; Roscoff: 2006. p. 297. 27 chapters. [Google Scholar]
- 27.Meijer L, Skaltsounis AL, Magiatis P, Polychonopoulos P, Knockaert M, Leost M, Ryan XP, Vonica CD, Brivanlou A, Dajani R, Tarricone A, Musacchio A, Roe SM, Pearl L, Greengard P. GSK-3 selective inhibitors derived from Tyrian purple indirubins. Chem. & Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 28.Polychronopoulos P, Magiatis P, Skaltsounis L, Myrianthopoulos V, Mikros E, Tarricone A, Musacchio A, Roe SM, Pearl L, Leost M, Greengard P, Meijer L. Structural basis for the synthesis of indirubins as potent and selective inhibitors of glycogen synthase kinase -3 and cyclin-dependent kinases. J. Med. Chem. 2004;47:935–946. doi: 10.1021/jm031016d. [DOI] [PubMed] [Google Scholar]
- 29.Ribas J, Bettayeb K, Ferandin Y, Garrofé-Ochoa X, Knockaert M, Totzke F, Schächtele C, Mester J, Polychronopoulos P, Magiatis P, Skaltsounis AL, Boix J, Meijer L. 7-bromoindirubin-3′-oxime induces caspase-independent cell death. Oncogene. 2006;25:6304–6318. doi: 10.1038/sj.onc.1209648. [DOI] [PubMed] [Google Scholar]
- 30.Jautelat R, Brumby T, Schafer M, Briem H, Eisenbrand G, Schwahn S, Kruger M, Lucking U, Prien O, Siemeister G. From the insoluble dye indirubin towards highly active, soluble CDK2-inhibitors. Chem. Biochem. 2005;6:531–540. doi: 10.1002/cbic.200400108. [DOI] [PubMed] [Google Scholar]
- 31.Merz KH, Schwahn S, Hippe F, Mühlbeyer S, Jakobs S, Eisenbrand G. Novel indirubin derivatives, promising anti-tumor agents inhibiting cyclin-dependent kinases. Int. J. Clin. Pharmacol. Ther. 2004;42:656–658. doi: 10.5414/cpp42656. [DOI] [PubMed] [Google Scholar]
- 32.Hoessel R, Leclerc S, Endicott J, Noble M, Lawrie A, Tunnah P, Leost M, Damiens E, Marie D, Marko D, Niederberger E, Tang W, Eisenbrand G, Meijer L. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nature Cell Biology. 1999;1:60–67. doi: 10.1038/9035. [DOI] [PubMed] [Google Scholar]
- 33.Davies TG, Tunnah P, Meijer L, Marko D, Eisenbrand G, Endicott JA, Noble MEM. Inhibitor binding to active and inactive CDK2. The crystal structure of a CDK2-cyclin A/indirubin-5-sulphonate. Structure. 2001;9:389–397. doi: 10.1016/s0969-2126(01)00598-6. [DOI] [PubMed] [Google Scholar]
- 34.Mapelli M, Massimiliano L, Crovace C, Seeliger M, Tsai LH, Meijer L, Musacchio A. Mechanism of CDK5/p25 binding by CDK inhibitors. J. Med. Chem. 2005;48:671–679. doi: 10.1021/jm049323m. [DOI] [PubMed] [Google Scholar]
- 35.Holton S, Merckx A, Burgess D, Doerig C, Noble M, Endicott J. Structures of P. falciparum PfPK5 test the CDK regulation paradigm and suggest mechanisms of small molecule inhibition. Structure. 2003;11:1329–1337. doi: 10.1016/j.str.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 36.Bertrand JA, Thieffine S, Vulpetti A, Cristiani C, Valsasina B, Knapp S, Kalisz HM, Flocco M. Structural characterization of the GSK-3β active site using selective and non-selective ATP-mimetic inhibitors. J. Mol. Biol. 2003;33:393–407. doi: 10.1016/j.jmb.2003.08.031. [DOI] [PubMed] [Google Scholar]
- 37.Myrianthopoulos V, Magiatis P, Ferandin Y, Skaltsounis AL, Meijer L, Mikros E. An integrated computational approach to the phenomenon of potent and selective inhibition of Aurora kinases B and C by a series of 7- substituted indirubins. J. Med. Chem. 2007;50:4027–4037. doi: 10.1021/jm070077z. [DOI] [PubMed] [Google Scholar]
- 38.Ferandin Y, Bettayeb K, Kritsanida M, Lozach O, Polychronopoulos P, Magiatis P, Skaltsounis AL, Meijer L. 3′-Substituted, 7-halogenoindirubins, a new class of cell death inducing agents. J. Med. Chem. 2006;49:4638–4649. doi: 10.1021/jm060314i. [DOI] [PubMed] [Google Scholar]
- 39.Beauchard A, Ferandin Y, Frère S, Lozach O, Blairvacq M, Meijer L, Thiéry V, Besson T. Synthesis of novel 5- substituted indirubins as potential inhibitor of protein kinases. Bioorg. Med. Chem. 2006;14:6434–6443. doi: 10.1016/j.bmc.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 40.Moon MJ, Lee SK, Lee JW, Song WK, Kim SW, Kim JI, Cho C, Choi SJ, Kim YC. Synthesis and structure-activity relationships of novel indirubin derivatives as potent anti-proliferative agents with CDK2 inhibitory activities. Bioorg. Med. Chem. 2006;14:237–246. doi: 10.1016/j.bmc.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 41.Knockaert M, Blondel M, Bach S, Leost M, Elbi C, Hager G, Naggy SR, Han D, Denison M, Ffrench M, Ryan XP, Magiatis P, Polychronopoulos P, Greengard P, Skaltsounis L, Meijer L. Independent actions on cyclin-dependent kinases and aryl hydrocarbon receptor mediate the anti-proliferative effects of indirubins. Oncogene. 2004;23:4400–4412. doi: 10.1038/sj.onc.1207535. [DOI] [PubMed] [Google Scholar]
- 42.Stewart JJP. MOPAC-A semiempirical molecular orbital program. J. Comput. Aided Mol. Des. 1990;4:1–105. doi: 10.1007/BF00128336. [DOI] [PubMed] [Google Scholar]
- 43.Mohamadi F, Richards NG, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. Macromodel - an intergrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990;11:440–467. [Google Scholar]
- 44.Martinek S, Inonog S, Manoukian AS, Young MW. A role for the segment polarity gene shaggy/GSK-3 in the Drosophila circadian clock. Cell. 2001;105:769–779. doi: 10.1016/s0092-8674(01)00383-x. [DOI] [PubMed] [Google Scholar]
- 45.Iitaka C, Miyazaki K, Akaike T, Ishida N. A role for glycogen synthase kinase-3beta in the mammalian circadian clock. J. Biol. Chem. 2005;280:29397–29402. doi: 10.1074/jbc.M503526200. [DOI] [PubMed] [Google Scholar]
- 46.Izumo M, Sato TR, Straume M, Johnson CH. Quantitative analyses of circadian gene expression in mammalian cell cultures. PLoS Computational Biology. 2006;2:e136. doi: 10.1371/journal.pcbi.0020136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Primot A, Baratte B, Gompel M, Borgne A, Liabeuf S, Romette JL, Costantini F, Meijer L. Purification of GSK-3 by affinity chromatography on immobilised axin. Protein Expr. & Purif. 2000;20:394–404. doi: 10.1006/prep.2000.1321. [DOI] [PubMed] [Google Scholar]
- 48.Ribas J, Boix J. Cell differentiation, caspase inhibition, and macromolecular synthesis blockage, but not BCL-2 or BCL-XL proteins, protect SH-SY5Y cells from apoptosis triggered by two CDK inhibitory drugs. Exp. Cell Res. 2004;295:9–24. doi: 10.1016/j.yexcr.2003.12.019. [DOI] [PubMed] [Google Scholar]
- 49.Leost M, Schultz C, Link A, Wu Y-Z, Biernat J, Mandelkow E-M, Bibb JA, Snyder GL, Greengard P, Zaharevitz DW, Gussio R, Senderovitz A, Sausville EA, Kunick C, Meijer L. Paullones are potent inhibitors of glycogen synthase kinase - 3β and cyclin-dependent kinase 5/p25. Eur. J. Biochem. 2000;267:5983–5994. doi: 10.1046/j.1432-1327.2000.01673.x. [DOI] [PubMed] [Google Scholar]