

Abstract
A new nickel-catalyzed procedure for the [4+2+1] cycloaddition of (trimethylsilyl)diazomethane with alkynes tethered to dienes has been developed. A broad range of unsaturated substrates participates in the sequence, and stereoselectivities are generally excellent. Stereochemical studies provided evidence for a mechanism that involves the [3,3]-sigmatropic rearrangement of divinylcyclopropanes.
Keywords: nickel, cycloaddition, seven-membered rings
Introduction
Transition metal-catalyzed multicomponent cycloadditions have emerged as a powerful strategy for preparing complex ring systems. Particular utility in the construction of medium rings has been demonstrated since the corresponding thermal counterparts are often either inaccessible or inefficient. In recent years, the repertoire of available metal-catalyzed cycloaddition entries to medium rings has grown to include [4+4], [5+2], [6+2], [6+4], [4+2+2], [5+2+1], [2+2+2+1], and [5+1+2+1] processes.1 The development of an efficient [4+2+1] process has been the focus of a number of studies. In early work from Harvey, a molybdenum carbene-mediated process was developed in which dienynes underwent addition to the carbene unit in a [4+2+1] sense to afford seven-membered ring products.2 The mechanism for that class of reactions was proposed to involve an alkyne metathesis cascade to generate a divinylcyclopropane, which then underwent facile Cope rearrangement to afford the seven-membered ring adduct. While those studies were limited to stoichiometric processes, a related [4+3] cycloaddition process was extensively developed by Davies involving the rhodium catalyzed addition of unsaturated diazo species with dienes.3 Two isolated examples of the corresponding fully intramolecular [4+2+1] cycloaddition of a diazoalkane / alkyne / diene cycloaddition were also reported in studies from Padwa.4 While the developments involving rhodium catalysis provided elegant access to structurally-complex seven-membered rings, access to simpler seven membered ring systems by catalytic, partially-intermolecular [4+2+1] processes had remained elusive. Prior to our original report in the area,5 the only examples of partially intermolecular, catalytic [4+2+1] cycloadditions were from Wender.6 In their studies of [2+2+1] carbonylative cycloadditions of dienynes, the [4+2+1] cycloaddition product was observed as a minor component of the reaction mixture.
In surveying the literature on nickel carbene complexes, it became apparent that the involvement of nickel carbene species in synthetic applications was considerably underdeveloped. While a number of nickel carbene species had been characterized7 and nickel complexes of N-heterocyclic carbenes had been widely used in catalytic applications,8 the development of procedures in which the carbene unit was incorporated into an organic product structure had been little studied.9 An interesting report from Barluenga suggested that nickel catalyzes carbene transfer from a chromium carbene complex to an organic substrate,9a and a few studies involving olefin cyclopropanation with lithiated sulfones or diazoalkanes as the carbene precursor had appeared.9b-d Studies from Hillhouse had elegantly illustrated that reactive nickel carbene species could be generated from diazoalkanes by a pathway involving η2 coordination of the diazo unit prior to the extrusion of dinitrogen in the presence of Sm(OTf)3.7a,9d With this backdrop, it appeared to us that the development of new synthetic procedures involving the catalytic generation of nickel carbene intermediates from diazoalkane precursors was an area worthy of study. Considering the void in catalytic [4+2+1] cycloadditions and the void in synthetic applications of nickel carbene species, we chose the nickel-catalyzed [4+2+1] cycloaddition of (trimethylsilyl)diazomethane with dienynes as the initial target of our studies. Herein we report a full account of our work including reaction scope and mechanistic insight.
Results and Discussion
Development of a Catalytic [4+2+1] Cycloaddition Process
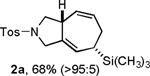
Our initial studies focused on the [4+2+1] cycloaddition of nitrogen-tethered substrate 1a (Table 1). A brief screen of conditions using Ni(COD)2 as catalyst and a 2:1 stoichiometry of (trimethylsilyl)diazomethane : substrate 1a illustrated that the desired cycloaddition was most effective in the absence of phosphine ligands. Both reaction rates and overall yields were lower in the presence of either monodentate or bidentate ligands (Table 1, entries 1-4). Variations in order of reagent addition were tolerated, and yields were highest in THF, although dioxane and toluene were also effective solvents (entries 5-6). Interestingly, diastereoselectivities improved as temperatures increased, with 60 °C being the optimum temperature to maximize both yield and diastereoselectivity (entries 7-8). The optimized procedure used throughout most of this study involved pre-mixing the diazoalkane and dienyne (2:1) in THF, followed by addition of 10 mol % Ni(COD)2, and stirring at 60 °C for 10-30 min (entry 8).
Table 1.
Reaction Optimizationa
| entry | ligand | solvent | temp (°C) | time (min) | % yield (dr) |
|---|---|---|---|---|---|
| 1 | dppe | THF | 60 | 30 | 42 |
| 2 | PCy3 | THF | 60 | 30 | 20 |
| 3 | P(t-Bu)3 | THF | 25 | 10 | 35 |
| 4 | none | THF | 0 | 10 | 64 (4.8:1) |
| 5 | none | dioxane | 90 | 30 | 54 (3.5:1) |
| 6 | none | toluene | 60 | 5 | 45 (>19:1) |
| 7 | none | THF | 25 | 5 | 72 (5.3:1) |
| 8 | none | THF | 60 | 5 | 68 (>19:1) |
Entries 1-4: 1a (1.0 equiv) was added to a premixed solution of TMSCHN2 (2.0 equiv) and Ni(COD)2 (10 mol %). Entries 5-8: Ni(COD)2 (10 mol %) was added to a premixed solution of 1a (1.0 equiv) and TMSCHN2 (2.0 equiv).
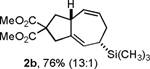
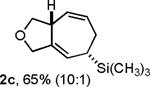
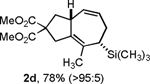
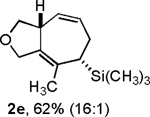
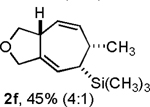
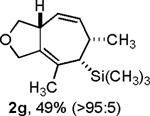
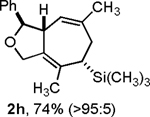
Following the optimized general procedure, a number of examples were carried out to illustrate the reaction scope (Table 2). In addition to the nitrogen-tethered substrate described above in our optimization studies (entry 1), we examined the preparation of carbocyclic and oxacyclic structures. Malonate-derived substrate 1b generated product 2b in 76% yield in 13:1 dr (entry 2), and oxygen-containing substrate 1c generated product 2c in 65% yield as a 10:1 ratio of diastereomers (entry 3). Internal alkynes were also effective participants, as evidenced by the generation of products 2d, 2e, 2g, and 2h (entries 4, 5, 7, 8). Substitution on either the diene terminus (entries 6 and 7) or an internal position of the diene (entry 8) was acceptable. Substitution within the tether chain was also tolerated with excellent resulting diastereoselectivity (entry 8). Notably, this latter example allowed the rapid preparation of densely functionalized product 2h with two rings, two sites of unsaturation, and three chiral centers in a 1,2,5 relationship in excellent yield and diastereoselectivity from relatively simple precursor 1h.
Table 2.
Reaction Scope.
| entry | substrate | product, yield (dr) |
|---|---|---|
| 1 | 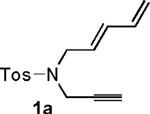 |
 |
| 2 | 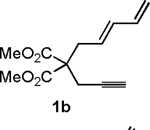 |
 |
| 3 | 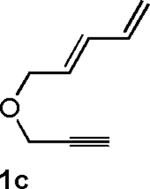 |
 |
| 4 | 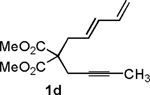 |
 |
| 5 | 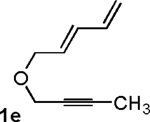 |
 |
| 6 | 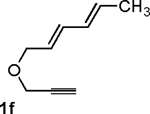 |
 |
| 7 | 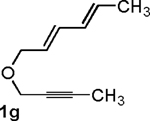 |
 |
| 8 | 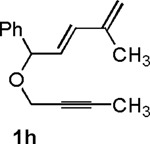 |
 |
The allyl silane handle installed in the cycloaddition process is potentially useful in a variety of postcycloaddition manipulations. However, in this study, we only examined simple desilylation. Interestingly, n-Bu4NF-mediated desilylation of 2b proceeds without alkene migration to generate product 3,10a whereas protodesilylation of 2b with BF3 / HOAc proceeds with allylic transposition to generate product 4 (eq 1).10b Thus, desilylated products 3 and 4 may be selectively prepared according to the reaction conditions chosen, although product 4 was prepared as a 3:1 ratio of isomers favoring the cis isomer. Unfortunately, readily available alkyl diazoacetates are unreactive in the protocol optimized for TMS diazomethane, although our studies to expand the scope of diazoacetates that participate are ongoing.
 |
(1) |
Possible Mechanisms
Based on a simple product analysis, we envisioned several mechanisms that may be operative in this new catalytic cycloaddition process (Scheme 1). As noted above, the stoichiometric behavior of molybdenum carbene complexes in [4+2+1] cycloadditions,2 as well as the related rhodium-catalyzed [4+2+1] processes3,4 were proposed to follow an alkyne metathesis / cyclopropanation / [3,3] rearrangement sequence. In analogy to these proposals, the nickel-catalyzed process, via mechanism A (Scheme 1), could involve formation of a nickel carbene intermediate 5, followed by alkyne metathesis to generate α,β-unsaturated carbene 7. Conversion of 7 to 8 and reductive elimination of 8 to produce 9, followed by [3,3] rearrangement, would afford product 2 via mechanism A (Scheme 1).11 As a second alternative (mechanism B, Scheme 1), ring expansion of 8 to 10a would allow direct production of 2 upon reductive elimination.12 In completely distinct mechanism C (Scheme 1), oxidative cyclization of nickel(0) with dienyne 1 could allow production of metallacycloheptadiene 11.13 Carbene insertion into either metal carbon bond of 11 to generate metallacycle 10a or 10b, followed by reductive elimination, would afford product 2. This type of oxidative cyclization, migratory insertion sequence is commonly invoked in carbonylative cycloadditions, and the insertion of a metal carbene ligand into a metal-carbon bond is well precedented in other contexts.14 Finally, a fourth alternative (mechanism D, Scheme 1) could involve ring contraction of metallacycle 11 to nickel bis-carbene 12. Carbene-carbene coupling of 12 could then generate divinylcyclopropane 9, which rearranges to 2 as previously described. The ring contraction of a metallacycle to a cyclopropane has been demonstrated in a number of contexts.15 Although mechanisms C and D have been depicted with the trimethylsilyl carbene unit present in the intermediate structures 11 and 12, the analogous structures that lack the trimethylsilyl carbene unit could also be involved, with diazoalkane incorporation occurring at a late stage of the reaction pathway. In this study, we have focused largely on the key question of involvement of divinylcyclopropanes (mechanisms A or D) vs. the direct kinetic production of a seven-membered ring (mechanisms B or C). A systematic examination of chemical reactivity and stereochemistry and a qualitative comparison of relative rates have provided significant insight into these intriguing mechanistic questions as described below.
Scheme 1.

Possible Mechanisms.
Evidence for the Involvement of Cyclopropane Intermediates
A key feature of mechanisms A and D is the formation of divinylcyclopropane 9 followed by its thermal conversion to 2. To gain evidence for the involvement of structures such as 9, we sought evidence that the nickel adduct generated from Ni(COD)2 and TMS diazomethane could directly add to a simple enyne. This observation would serve as a direct model for the portions of mechanisms A and D that correspond to the 1 to 9 conversion while avoiding the subsequent sigmatropic rearrangement. Notably, Hoye had previously demonstrated this mode of reactivity upon treatment of enynes to molybdenum carbene species,16 and the corresponding catalytic reaction was recently developed by Dixneuf upon exposure of enynes and TMS diazomethane to Cp*RuCl(COD).17 Both malonate-derived substrate 13a and nitrogen-tethered substrate 13b were examined in the catalytic reaction with TMS diazomethane and Ni(COD)2 (Table 3, entries 1-2). In both cases, the reactions proceeded smoothly at 60 °C to afford [3.1.0]-bicyclohexane ring systems 14a-b in analogy to the studies from Hoye and Dixneuf. An important observation is that the E-vinyl silane was preferentially generated in both cases. In contrast, the ruthenium-catalyzed method of Dixneuf, involving the same the diazoalkane, favored the Z-isomer.17 Examination of substrates 13c and 13d clearly illustrated that this addition step is stereospecific (Table 3, entries 3-4). Although the chemical yields were low with 1,2-disubstituted alkenes, substrate 13c afforded the cyclopropane 14c, whereas 13d afforded cyclopropane 14d. Diastereoselectivities were excellent and opposite for these two cases, and the E-vinyl silane was produced in both instances with high selectivity. Knowledge about the stereochemistry of the vinyl silane produced and about the relationship of starting material olefin stereochemistry to product cyclopropane stereochemistry proved to be important features in our stereochemical analysis of the [4+2+1] process (vide infra).
Table 3.
Enyne Cyclizations
| ||||
|---|---|---|---|---|
| entry (substrate) |
X | R1 | R2 | product, yield (dr, E/Z) |
| 1 (13a) | C(CO2Me)2 | H | H | 14a, 70% (E only) |
| 2 (13b) | NTs | H | H | 14b, 60% (10:1 E/Z) |
| 3 (13c) | O | Et | H | 14c, 36% (>95:5 dr, E only) |
| 4 (13d) | O | H | Et | 14d, 25%(>95:5 dr, 9:1 E/Z) |
Further Evidence for the Involvement of a [3.3] Rearrangement of Divinylcyclopropanes
A considerable amount is known about the mechanism of [3,3] rearrangements of divinylcyclopropanes. The reaction proceeds through a boat transition state, and the alkene stereochemistry controls both the relative stereochemistry of the newly formed chiral centers as well as the reaction rate.18 In particular, the development of A1,3 strain in conformation 15 (eq 2), via substitution of R1 or R3, results in a significant retardation of reaction rates.18f,g
 |
(2) |
Since the enyne cyclizations described above (Table 3) provide a clear predictor of the stereochemistry of the vinyl silane and cyclopropane stereocenters, it is useful to consider how these stereochemical features would be expected to impact the stereoselectivity and reaction rates of dienyne cyclizations. Accordingly, if dienyne 1 participates in mechanism A described above (Scheme 1), then the initially formed intermediate 7 should possess the E-vinyl silane configuration (Scheme 2). Addition of the nickel carbene of 7 to the proximal E-alkene of the tethered diene should proceed in a stereospecific sense to afford compound 9 with the stereochemistry shown (Scheme 2). A similar analysis of mechanism D also predicts the observed stereochemistry of product 2 if one assumes that the E-vinyl silane of 9 is generated from 12 (Scheme 3). The key features of 9 to note are the E-stereochemistry of the vinyl silane, the cis orientation of the two alkenes with respect to the cyclopropane, and the lack of a bridgehead substituent. All three of these features combine to provide a divinylcyclopropane structure that would be expected to undergo rapid [3,3] rearrangement to product 2, which is exactly the product stereochemistry observed in catalytic [4+2+1] cycloadditions. Failure to detect the unrearranged divinylcyclopropane in cyclizations of dienynes and observation of the stereochemical outcome predicted by the above analysis are thus consistent with mechanism A or D.
Scheme 2.

Stereochemical Analysis - Mechanism A.
Scheme 3.
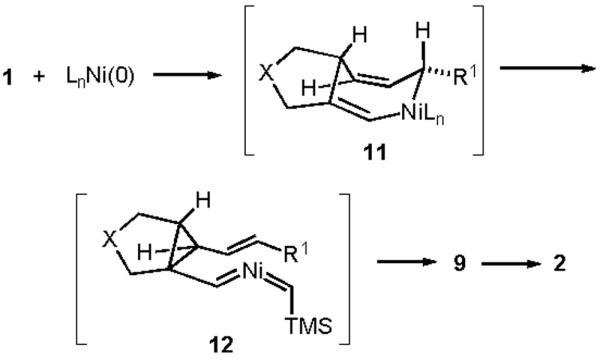
Stereochemical Analysis - Mechanism D.
To further probe the mechanistic features following this stereochemical analysis, we next installed functional groups into the starting dienyne that would introduce A1,3 strain in the intermediate divinylcyclopropanes produced via mechanisms A or D. We reasoned that mechanisms B and C would not be selectively and significantly impacted by these precise structural modifications. Installation of a bridgehead methyl in the intermediate divinylcyclopropane required the use of substrate 16 (Scheme 4). Treatment of 16 to the standard conditions at 60 °C in THF led to the selective formation of divinylcyclopropane 17 with the expected stereochemistry as shown. Significantly, the introduction of a bridgehead methyl substituent prevents the direct formation of a cycloheptadiene and instead allows divinylcyclopropane 17 to be isolated. Heating 17 to 110 °C in toluene led to a slow rearrangement of 17 to product 19, and conformation 18 depicts the A1,3 strain that slows the rate of this [3,3] sigmatropic rearrangement. Alternatively, carrying out the nickel-catalyzed reaction of 16 in toluene at 110 °C provided direct access to 19 in 50% yield without requiring isolation of cyclopropane 17. The rate of [3,3] rearrangement of 17 was unaffected by the presence of Ni(COD)2, suggesting that the rearrangement is a purely thermal, uncatalyzed process. On the basis of this experiment, the suggested involvement of mechanism A or D via a divinylcyclopropane rearrangement appears to be more secure.
Scheme 4.

Isolation of a Divinylcyclopropane via Quaternization.
A caveat in the above analysis is that installation of a quaternary center could potentially result in a change of mechanism. To avoid this significant structural change, we prepared both E,E-diene 1g and E,Z-diene 20 (Scheme 5). This comparison now allows two stereoisomers to be directly analyzed. By the proposed mechanism A, E,E-diene 1g will generate a divinylcyclopropane that lacks the A1,3 strain that will slow the [3,3] rearrangement as depicted in conformation 22a, whereas E,Z-diene 20 will generate divinylcyclopropane 21, which possesses the A1,3 strain that will slow the [3,3] rearrangement as depicted in confomation 22b. Indeed, these two stereoisomers 1g and 20 afforded completely different reaction outcomes at 60 °C in THF, with 1g exclusively producing rearrangement product 2g, while 20 exclusively produced divinylcyclopropane 21. Heating compound 21 at 105 °C in toluene resulted in the smooth conversion to cycloheptadiene 23, whereas carrying out the nickel-catalyzed reaction of substrate 20 in toluene at 110 °C provided direct access to 23 in 60% yield without requiring isolation of cyclopropane 21. The divergent behavior of these two stereoisomeric starting materials provides compelling evidence that a mechanism involving divinylcyclopropane formation and rearrangement is the operative pathway in [4+2+1] cycloadditions.
Scheme 5.

Isolation of a Divinylcyclopropane without Quaternization.
Mechanisms A and D (Scheme 1) cannot be distinguished by the above experiments. However, failure to observe [4+2] cycloaddition products seems unlikely if a structure related to 11 is involved in the reaction pathway.13 Furthermore, Wender has previously demonstrated that phosphine promotion is required to induce nickel-catalyzed [4+2] cycloadditions of dienyne substrates. Additionally, 1, 1-disubstituted dienes with an E internal alkene (related to structure 16, Scheme 4) failed to undergo cyclization in the Wender study. These observations suggest that fundamental differences exist between the [4+2] and [4+2+1] cycloaddition pathways. Finally, our prior demonstrations of ring contractions of nickel metallacycles related to the 11 to 12 conversion (Scheme 1) required oxidative promotion with molecular oxygen.15a Based upon these issues, mechanism A (alkyne metathesis, divinylcyclopropane formation, [3,3] rearrangement) is perhaps most consistent with the reactivity trends noted. The current study strongly suggests that divinylcyclopropanes, irrespective of the precise mechanism by which they are formed, are key intermediates in the nickel-catalyzed [4+2+1] reaction.
Conclusions
In summary, a new catalytic [4+2+1] cycloaddition of dienynes and (trimethylsilyl)diazomethane has been developed. A variety of carbocyclic and heterocyclic bicycles may be produced in a stereospecific fashion. Through a stereochemical analysis, strong evidence in favor of a mechanism involving formation and [3,3]-rearrangement of divinylcyclopropanes was presented.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge support for this research from NIH grant GM57014. Professor Jin Cha is thanked for many helpful discussions.
Footnotes
SUPPORTING INFORMATION. Full experimental details and copies of 1H NMR spectra of all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) For [4+4] reactions: Wender PA, Ihle NC. J. Am. Chem. Soc. 1986;15:4678. (b) For [5+2] reactions: Wender PA, Takahashi H, Witulski B. J. Am. Chem. Soc. 1995;117:4720. (c) For [6+2] reactions: Rigby JH, Henshilwood JA. J. Am. Chem. Soc. 1991;113:5122. (d) For [6+4] reactions: Rigby JH, Ateeq HS, Charles NR, Cuisiat SV, Ferguson MD, Henshilwood JA, Krueger AC, Ogbu CO, Short KM, Heeg MJ. J. Am. Chem. Soc. 1993;115:1382. (e) For [4+2+2] reactions: Evans PA, Robinson JE, Baum EW, Fazal AN. J. Am. Chem. Soc. 2002;124:8782. doi: 10.1021/ja026351q. (f) Gilbertson SR, DeBoef B. J. Am. Chem. Soc. 2002;124:8784. doi: 10.1021/ja026536x. (g) For [5+2+1] reactions: Wender PA, Gamber GG, Hubbard RD, Zhang L. J. Am. Chem. Soc. 2002;124:2876. doi: 10.1021/ja0176301. (h) Ojima I, Lee S-Y. J. Am. Chem. Soc. 2000;122:2385. (i) Wender PA, Gamber GG, Hubbard RD, Pham SM, Zhang L. J. Am. Chem. Soc. 2005;127:2836. doi: 10.1021/ja042728b.
- (2).(a) Harvey DF, Lund KP. J. Am. Chem. Soc. 1991;113:5066. (b) Harvey DF, Grenzer EM, Gantzel PK. J. Am. Chem. Soc. 1994;116:6719. (c) See also: Herndon JW, Chatterjee G, Patel PP, Matasi JJ, Tumer SU, Harp JJ, Reid MD. J. Am. Chem. Soc. 1991;113:7808.
- (3).(a) Davies HML, McAfee MJ, Oldenburg CEM. J. Org. Chem. 1989;54:930. (b) Davies HML, Doan BD. J. Org. Chem. 1999;64:8501. (c) Davies HML, Calvo RL, Townsend RJ, Ren P, Churchill RM. J. Org. Chem. 2000;65:4261. doi: 10.1021/jo991959b. (d) Davies HML, Stafford DG, Doan BD, Houser JH. J. Am. Chem. Soc. 1998;120:3326. (e) Deng L, Giessert AJ, Gerlitz OO, Dai X, Diver ST, Davies HML. J. Am. Chem. Soc. 2005;127:1342. doi: 10.1021/ja045173t.
- (4).Padwa A, Krumpe KE, Gareau Y, Chiacchio U. J. Org. Chem. 1991;56:2523. [Google Scholar]
- (5).For a preliminary communication of this work, see: Ni Y, Montgomery J. J. Am. Chem. Soc. 2004;126:11162. doi: 10.1021/ja046147y.
- (6).Wender PA, Deschamps NM, Gamber GG. Angew. Chem. Int. Ed. 2003;42:1853. doi: 10.1002/anie.200350949. [DOI] [PubMed] [Google Scholar]
- (7).(a) Mindiola DJ, Hillhouse GL. J. Am. Chem. Soc. 2002;124:9976. doi: 10.1021/ja0269183. (b) Gabor B, Krüger C, Marczinke B, Mynott R, Wilke G. Angew. Chem., Int. Ed. Engl. 1991;30:1666. (c) Arduengo AJ, Gamper SF, Calabrese JC, Davidson F. J. Am. Chem. Soc. 1994;116:4391. (d) Hou H, Gantzel PK, Kubiak CP. Organometallics. 2003;22:2817.
- (8).(a) Herrmann WA. Angew. Chem. Int. Ed. 2002;41:1290. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y. (b) Mahandru GM, Liu G, Montgomery J. J. Am. Chem. Soc. 2004;126:3698. doi: 10.1021/ja049644n. (c) Zuo G, Louie J. Angew. Chem. Int. Ed. 2004;43:2277. doi: 10.1002/anie.200353469. (d) Louie J, Gibby JE, Farnworth MV, Tekavec TN. J. Am. Chem. Soc. 2002;124:15188. doi: 10.1021/ja027438e. (e) Sato Y, Sawaki R, Mori M. Organometallics. 2001;20:5510. (f) Dible BR, Sigman MS. J. Am. Chem. Soc. 2003;125:872. doi: 10.1021/ja0286876. (g) Blakey SB, MacMillan DWC. J. Am. Chem. Soc. 2003;125:6046. doi: 10.1021/ja034908b.
- (9).(a) Barluenga J, Barrio P, López LA, Tomás M, García-Granda S, Alvarez-Rúa C. Angew. Chem. Int. Ed. 2003;42:3008. doi: 10.1002/anie.200351216. (b) Nakamura A, Yoshida T, Cowie M, Otsuka S, Ibers JA. J. Am. Chem. Soc. 1977;99:2108. (c) Gai Y, Julia M, Verpeaux J-N. Synlett. 1991:269. (d) Waterman R, Hillhouse GL. J. Am. Chem. Soc. 2003;125:13350. doi: 10.1021/ja0381914.
- (10).(a) Hulme AN, Henry SS, Meyers AI. J. Org. Chem. 1995;60:1265. (b) Knölker H-J, Baum E, Graf R, Jones PG, Spiess O. Angew. Chem. Int. Ed. 1999;38:2583. doi: 10.1002/(sici)1521-3773(19990903)38:17<2583::aid-anie2583>3.0.co;2-1. (c) For a discussion of the intermediacy of silanols in protodesilylations of C(sp3)-SiMe2Ph derivatives, see: Heitzman CL, Lambert WT, Mertz E, Shotwell B, Tinsley JM, Va P, Roush WR. Org. Lett. 2005;7:2405. doi: 10.1021/ol0506821.
- (11).For a discussion of cyclopropanation and metathesis pathways of enynes, see: Peppers BP, Diver ST. J. Am. Chem. Soc. 2005;126:9524. doi: 10.1021/ja049079o.
- (12).Involvement of ring-expanded nickel metallacycles in medium ring synthesis has precedent in Wender's [4+4] cycloaddition reactions. See reference 1a
- (13).Structure 11 has direct precedent in Wender's [4+2] alkyne / diene cycloadditions. See references: (a) Wender PA, Jenkins TE. J. Am. Chem. Soc. 1989;111:6432. (b) Wender PA, Smith TE. Tetrahedron. 1998;54:1255.
- (14).(a) Greenman KL, Van Vranken DL. Tetrahedron. 2005;61:6438. (b) Solé D, Vallverdú L, Solans X, Font-Bardia M, Bonjoch J. Organometallics. 2004;23:1438.
- (15).(a) Chowdhury SK, Amarasinghe KKD, Heeg MJ, Montgomery J. J. Am. Chem. Soc. 2000;122:6775. (b) Urabe H, Suzuki K, Sato F. J. Am. Chem. Soc. 1997;119:10014.
- (16).(a) Korkowski PF, Hoye TR, Rydberg DB. J. Am. Chem. Soc. 1988;110:2676. (b) Hoye TR, Rehberg GM. J. Am. Chem. Soc. 1990;112:2841.
- (17).Monnier F, Castillo D, Dérien S, Toupet L, Dixneuf PH. Angew. Chem. Int. Ed. 2003;42:5474. doi: 10.1002/anie.200352477. [DOI] [PubMed] [Google Scholar]
- (18).(a) Wender PA, Filosa MP. J. Org. Chem. 1976;41:3490. (b) Marino JP, Browne LJ. Tetrahedron Lett. 1976:3245. (c) Piers E, Nagakura I. Tetrahedron Lett. 1976:3237. (d) Lee J, Kim H, Cha JK. J. Am. Chem. Soc. 1995;117:9919. (e) Hudlicky T, Fan R, Reed JW, Gadamesetti KG. Org. React. 1992;41:1. (f) Rhoads SJ, Raulins NR. Org. React. 1975;22:1. (g) Ohloff G, Pickenhagen W. Helv. Chim. Acta. 1969;52:880.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.