

Abstract
Background
Clusterin expression in various types of human cancers may be higher or lower than in normal tissue, and clusterin may promote or inhibit apoptosis, cell motility, and inflammation. We investigated the role of clusterin in tumor development in mouse models of neuroblastoma.
Methods
We assessed expression of microRNAs in the miR-17-92 cluster by real-time reverse transcription–polymerase chain reaction in MYCN-transfected SH-SY5Y and SH-EP cells and inhibited expression by transfection with microRNA antisense oligonucleotides. Tumor development was studied in mice (n = 66) that were heterozygous or homozygous for the MYCN transgene and/or for the clusterin gene; these mice were from a cross between MYCN-transgenic mice, which develop neuroblastoma, and clusterin-knockout mice. Tumor growth and metastasis were studied in immunodeficient mice that were injected with human neuroblastoma cells that had enhanced (by clusterin transfection, four mice per group) or reduced (by clusterin short hairpin RNA [shRNA] transfection, eight mice per group) clusterin expression. All statistical tests were two-sided.
Results
Clusterin expression increased when expression of MYCN-induced miR-17-92 microRNA cluster in SH-SY5Y neuroblastoma cells was inhibited by transfection with antisense oligonucleotides compared with scrambled oligonucleotides. Statistically significantly more neuroblastoma-bearing MYCN-transgenic mice were found in groups with zero or one clusterin allele than in those with two clusterin alleles (eg, 12 tumor-bearing mice in the zero-allele group vs three in the two-allele group, n = 22 mice per group; relative risk for neuroblastoma development = 4.85, 95% confidence interval [CI] = 1.69 to 14.00; P = .005). Five weeks after injection, fewer clusterin-overexpressing LA-N-5 human neuroblastoma cells than control cells were found in mouse liver or bone marrow, but statistically significantly more clusterin shRNA-transfected HTLA230 cells (3.27%, with decreased clusterin expression) than control-transfected cells (1.53%) were found in the bone marrow (difference = 1.74%, 95% CI = 0.24% to 3.24%, P = .026).
Conclusions
We report, to our knowledge, the first genetic evidence that clusterin is a tumor and metastasis suppressor gene.
CONTEXT AND CAVEATS
Prior knowledge
The role of clusterin in cancer is unclear, and experimental results on its expression and activities are inconsistent across studies.
Study design
Cultured neuroblastoma cell lines, mouse models of human neuroblastoma, and human patient specimens were used. Expression of clusterin, MYCN, and miR-17-92 microRNAs in neuroblastoma cells was altered by various molecular biology techniques. The impact of such genetic alterations on tumor growth and metastasis was investigated.
Contribution
Clusterin appears to be a tumor and metastasis suppressor that is negatively regulated by MYCN and miR-17-92 microRNAs. Increased clusterin expression was associated with decreased tumor growth and decreased metastasis to mouse liver or bone marrow, whereas decreased clusterin expression was associated with increased tumor growth and metastasis.
Implications
More research into the role of clusterin in various cancers is warranted.
Limitations
Small numbers of mice and of patient specimens were used. Immunocompromised mice were used. The age of patients who contributed neuroblastoma samples was skewed to older ages, and the neuroblastoma stage was skewed to higher stage.
From the Editors
Neuroblastoma, which originates in the sympathetic nervous system, is the most common extracranial solid tumor in childhood, with approximately 90% occurring in children who are younger than 5 years (1,2). Neuroblastoma is prone to spontaneous differentiation and regression in some children but can manifest as a highly aggressive tumor in others. Patients with stage IV metastatic neuroblastoma show an initial response to chemotherapeutic treatment but typically relapse with an incurable form of the disease. Bone marrow is involved only in metastatic (stage IV or IVS), but not in localized (stages I–III), neuroblastoma stages in children (1). The most common cytogenetic alterations in metastatic neuroblastoma are the loss or rearrangement of the distal portion of the short arm of chromosome 1 and the amplification of the MYCN gene. Children carrying these alterations have a greatly increased chance of aggressive disease and fatal outcome (1).
Clusterin, also known as apolipoprotein J, is a heterodimeric sulfated glycoprotein that is expressed in most human tissues and body fluids and is associated with many biological activities, including regulation of apoptosis and cancer (3,4). Cytoplasmic clusterin, which originates from retrotranslocation via the endoplasmic reticulum or Golgi organelles (5), and secreted clusterin have been shown to cooperate with c-MYC during transformation, to promote tumor cell survival through inhibition of activated BAX at the mitochondria (6) and to protect cells from various death stimuli. For example, expression of clusterin decreases the cytotoxic effects of doxorubicin or cisplatin in neuroblastoma and osteosarcoma cells (7,8). However, a nuclear form of clusterin that originates from a differentially spliced mRNA or from an alternative translation start site has been shown to be proapoptotic in prostate and breast cells (9–11). Consequently, the relative expression of the two clusterin isoforms in a cell may dictate whether that cell survives or undergoes apoptosis and may explain the contradictory results obtained with different cell types and in different contexts.
In various cancers, the expression of clusterin has been reported to be either increased or decreased, compared with that in normal tissues. Alterations of clusterin expression patterns have been observed in prostate, mammary, esophageal, and pancreatic carcinomas and seminoma (12–17). Furthermore, Gleave and coworkers (18–21) have shown that neoplasias of the urinary tract have increased clusterin expression. On the basis of this evidence, a clinical trial was initiated in which prostate cancer patients were injected with a clusterin antisense oligonucleotide, called OGX-011, to ablate clusterin expression and to induce tumor regression (22,23). The results of this trial have not yet been reported.
In contrast with the hypothesis that clusterin is a tumor promoter, we have shown previously (15,24) that ectopic expression of clusterin suppresses proliferation of prostate cancer cells in vitro and that clusterin expression is reduced in human prostate cancer biopsy samples compared with normal tissue and in prostate cancer cells developing into tumors in transgenic adenocarcinoma mouse prostate (TRAMP) mice. In keeping with the hypothesis that clusterin is a suppressor rather than a promoter of tumorigenesis, we have shown previously (25) that adenovirus-mediated delivery of c-MYC ablates the expression of clusterin in mouse keratinocytes and that, in the absence of clusterin expression, the formation of chemically induced skin papillomas is accelerated. To reconcile these contrasting observations, one could hypothesize that the action of clusterin on tumorigenesis is context and signal dependent. This hypothesis is supported by our previous observations (7,26) in neuroblastoma cells that clusterin mediates resistance to the cytotoxic effects of doxorubicin and also suppresses in vitro invasion by inhibiting nuclear factor κB (NF-κB), whose expression has been associated with increased metastasis (27).
A mouse model of neuroblastoma was established by Weiss et al. (28) by directing ectopic expression of MYCN to the neuroectoderm. Genes in the MYC family modulate tumorigenesis by regulating the expression of various genes and microRNAs by recruiting chromatin remodeling enzymes and transcriptional cofactors (29–31). Tumors arising in these mice faithfully recapitulate the human disease, including the cytogenetic abnormalities typically found in children with advanced neuroblastoma. The purpose of this study was to investigate whether MYCN regulates clusterin expression, resulting in modification of neuroblastoma development, and, if so, to investigate the molecular mechanisms used.
Patients, Materials, and Methods
Immunodeficient Mice
The immunodeficient mice used were common cytokine gamma chain, RAG2 double-knockout mice or RAG2, complement C3, and common cytokine gamma chain triple-knockout mice (32). These mice were bred in-house and used for experiments at approximately 3 months of age. A 1:1 mixture of male and female mice was used in treatment and control groups. All experimental procedures involving transgenic mice were approved by the University College London (London) and were conducted under the Animal (Scientific Procedures) Act, 1986 (United Kingdom).
DNA Isolation and Genotyping
Genomic DNA was isolated from mouse tail tips harvested when MYCN-transgenic mice and mice with clusterin deletions (ie, clusterin-knockout mice) were 3 weeks of age by use of proteinase K digestion and phenol–chloroform extraction, as described previously (33). The genotype of MYCN-transgenic mice was determined by polymerase chain reaction (PCR) as described (28) and by real-time PCR, by use of oligonucleotide primers and probes specific for the human MYCN transgene (forward, 5′-CGACCACAAGGCCCTCAGTA-3′; reverse, 5′-CAGCCTTGGTGTTGGAGGAG-3′; probe, FAM-5′-CGCTTCTCCACAGTGACCACGTCG-3′) and for the murine β-actin gene (forward, 5′-TGCGTCTGGACTTGGCTG-3′; reverse, 5′-TAGCCACGCTCGGTCAGG-3′; probe, FAM-5′-CGGGACCTGACTGACTACCTCATGA-3′), as described previously (34). Briefly, the reaction conditions were as follows: incubation at 95°C for 10 minutes, followed by 40 cycles at 95°C for 15 seconds and 55°C for 1 minute. The data were analyzed with an ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA). To determine MYCN-transgene dosage, the comparative cycle threshold method (DDCt) was used. This method is a comparative technique that involves determining the comparative cycle threshold (35) value, defined as the cycle number at which a particular sample achieves when it crosses a set fluorescence threshold intensity, normalized to that of β-actin, and expressed relative to a calibrator (36). Genomic DNA from a MYCN heterozygous mouse was used as the calibrator sample. Mice with MYCN levels that were increased more than twofold, compared with the calibrator, were considered to be MYCN homozygous mice.
Antibodies
The primary antibodies used were anti-neural cell adhesion molecule, rabbit polyclonal (product AB5032; Millipore, Billerica, MA); anti-clusterin, mouse monoclonal (clone 41D, product 05-354; Millipore); anti-β-actin, goat polyclonal (clone I-19, product sc-1616; Santa Cruz Biotechnology, Santa Cruz, CA); anti-MYCN, mouse monoclonal (product sc-142; Santa Cruz Biotechnology); anti-clusterin-a, goat polyclonal (clone M-18, product sc-6420; Santa Cruz Biotechnology); anti-connective tissue growth factor (Santa Cruz Biotechnology); anti-glyceraldehyde-3-phosphate dehydrogenase, rabbit monoclonal (product 2118; Cell Signalling Technology, Danvers, MA); anti-vimentin mouse monoclonal (product VIM-13.2; Sigma-Aldrich, St Louis, MO); anti-fibronectin, mouse monoclonal (product 610077; BD Biosciences, Oxford, UK); anti-disialoganglioside GD2, mouse monoclonal (clone 14.2Ga; Millipore); and anti-NF-κB p65, mouse monoclonal (clone F-6, product sc-8008; Santa Cruz Biotechnology). The secondary antibodies used were biotinylated anti-mouse IgG (GE Healthcare, Little Chalfont, Buckinghamshire, UK), Alexa Fluor 568–conjugated goat anti-mouse IgG (product A21112; Invitrogen, Paisley, UK), fluorescein isothiocyanate (FITC)-conjugated donkey anti-rabbit IgG (H+L) (product 711-095-152; Jackson Immunoresearch, Newmarket, UK), FITC-conjugated goat anti-mouse IgG (Autogen Bioclear, Calne, UK), sheep anti-mouse IgG conjugated to horseradish peroxidase (HRP) (product RPN4201; GE Healthcare), HRP-conjugated sheep anti-rabbit IgG (product RPN4301; GE Healthcare), HRP-conjugated donkey anti-goat IgG (product sc-2033; Santa Cruz Biotechnology), and HRP-conjugated rabbit anti-goat IgG (Dako, Carpinteria, CA).
Immunocytochemistry
Cells were fixed for 10 minutes in paraformaldeyde or ice-cold methanol, washed with phosphate-buffered saline (PBS), and permeabilized with 0.5% Triton X-100 for 15 minutes. Nonspecific binding was blocked by incubation with 5% horse serum for 30 minutes. Antibodies against neural cell adhesion molecule (1:100 dilution) or clusterin (clone 41D, 1:150 dilution) were incubated with the fixed cells at 4°C overnight or at room temperature for 1 hour. Cells were washed with PBS and incubated with a secondary antibody, Alexa Fluor 568–conjugated anti-mouse IgG or FITC-conjugated anti-rabbit IgG. After several washes, the cells were incubated with 4′,6-diamidino-2-phenylindole to visualize nuclei and were embedded in Mowiol (Merk, Darmstadt, Germany). Cells were photographed with a camera connected to a Zeiss Axioplan 2 microscope.
Cell Culture
Human neuroblastoma cell lines HTLA230 and LA-N-5 were as described previously (37). They were confirmed to be neuroblastoma cell lines because both cell lines expressed GD2 (1). HEK-293 human embryonic kidney cells, Phoenix-A cells (a derivative of HEK-293 cells in which genes have been inserted), and IMR32 and SH-SY5Y human neuroblastoma cell lines were obtained from the American Type Culture Collection (Teddington, Middlesex, UK). SH-EP human neuroblastoma cells are diploid for MYCN and have no 1p deletion. SH-EP cells that had been transfected with the MYCN–estrogen receptor (ER) fusion gene (termed SH-EP MYCN-ER cells) express a conditionally active MYCN–ER fusion protein in which the full-length human MYCN sequence is fused in-frame with the ER-α ligand-binding domain, as described previously (38). The fusion protein is constitutively expressed but conformationally inactive unless the ER-binding site is bound by 4-hydroxytamoxifen.
HTLA230, LA-N-5, and IMR32, but not SH-SY5Y, cells contain amplified MYCN genes and express high levels of MYCN protein. Clusterin expression is higher in SH-SY5Y and HTLA230 cells than in LA-N-5 and IMR32 cells. All cell lines except SH-EP MYCN-ER cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum and 2 mM glutamine. SH-EP MYCN-ER cells were cultured in RPMI medium 1640 containing 10% charcoal-stripped fetal calf serum, 2 mM glutamine, penicillin (100 mg/mL), and streptomycin (100 mg/mL). MYCN was activated by adding ethanol-solubilized 4-hydroxytamoxifen (Sigma, Inc., St Louis, MO) to the culture medium at a final concentration of 400 nM.
The mouse neuroblastoma cell line IPR6-2 was obtained from a chest neuroblastoma tumor that developed in transgenic CBA–C57BL/6 mixed-background mice with homozygous MYCN expression and heterozygous clusterin deletion. IPR6-2 cells were cultured in RPMI-1640 medium supplemented with 2 mM l-glutamine, 10 μM 2-mercaptoethanol, 1 mM sodium pyruvate, 1× nonessential amino acids, and 20% heat-inactivated fetal calf serum.
Patient Samples
After institutional board approval, archived diagnostic samples from 100 patients were identified by searching the histopathology database of a single institution (Great Ormond Street Hospital, London, UK) with the search criteria neuroblastoma or ganglioneuroblastoma and identifying patients who were diagnosed from August 1, 1994, through August 31, 2005. Written consent was obtained from children's parents after September 1, 2006, as required by UK law. The hospital's ethics committee approved use of samples before this date. Data on patient stage, histology, and diagnosis were retrieved from hospital records. Data from three patients were not analyzed because these patients had been misclassified as having neuroblastoma. Among the remaining 97 patients, 51 were boys, the median age at diagnosis was 27 months, the mean age at diagnosis was 52 months, and the age range was 0–420 months. Among the 97 specimens, 84 were pretreatment biopsy specimens or specimens obtained from primary resections and 10 were from resections after chemotherapy; for three patients, the relation to chemotherapy could not be determined. For this analysis, tumors were classified as undifferentiated if diagnosed as undifferentiated neuroblastoma (Schwannian stroma poor, undifferentiated) and classified as differentiated if the diagnosis was differentiating neuroblastoma, ganglioneuroma, or ganglioneuroblastoma or if the patient had undergone extensive ganglionic differentiation after chemotherapy (four of the 10 samples obtained after chemotherapy). The MYCN gene was amplified in 15 specimens and nonamplified in 60 specimens; 22 were not tested for MYCN amplification (Table 1).
Table 1.
Characteristics of the 97 neuroblastoma patients analyzed in this study*
| Characteristics | Value |
| Mean age at diagnosis,† (±SD; range), mo | 51.6 (±66.7; 0–420) |
| Tissue sampling relative to chemotherapy, No. | |
| After | 10 |
| Before | 84 |
| ND | 3 |
| Stage or histology, No. | |
| Stage IV | 44 |
| Stage III | 7 |
| Stage IIA/B | 9 |
| Stage I | 3 |
| Stage IVS | 3 |
| Ganglioneuroblastoma | 5 |
| Ganglioneuroma | 1 |
| ND | 25 |
| MYCN amplification | |
| Yes | 15 |
| No | 60 |
| ND | 22 |
ND = not determined.
The age was unknown for 18 of the 97 patients.
Quantitative PCR
Expression of mature microRNAs from the miR-17-92 cluster was analyzed by quantitative real-time reverse transcription–PCR (qPCR) by use of the TaqMan miRNA assay system (Applied Biosystems), as described by the supplier. Total RNA from SH-SH5Y cells was isolated with Tri-Reagent (Sigma-Aldrich) by following the manufacturer's instructions. PCRs were performed on an ABI 7500 Fast Real-Time PCR System as described previously (39). The qPCRs for primary miR-17-92 transcripts and clusterin mRNAs were performed essentially as described previously (40). Primers for human clusterin were 5′-GACTCTGCTGCTGTTTGTG-3′ (forward) and 5′-GCTTTGTCTCTGATTCCCTG-3′ (reverse). Primers for human miR-17-92 were 5′-CTGTCGCCCAATCAAACTG-3′ (forward) and 5′-GTCACAATCCCCACCAAAC-3′ (reverse). The thermal cycling conditions were as follows: 50°C for 2 minutes, 95°C for 10 minutes, and 40 cycles of 95°C for 15 seconds and 60°C for 1 minute.
Antisense Inhibition of the miR-17-92 Cluster
SH-SY5Y cells overexpressing MYCN were transfected with a mixture of antisense oligoribonucleotides targeting six microRNAs from the miR-17-92 cluster (miR-17-5p, -18, -19a, -19b-1, -20, and -92-1), as described previously (40). Briefly, a mixture of 2′-O-methyl oligoribonucleotides (each at 100 pmol) targeting individual members of the cluster or 600 pmol of a nontargeting oligoribonucleotide was transfected by use of Lipofectamine 2000 (Invitrogen, Carlsbad, CA) into cells that had been plated 24 hours earlier in six-well plates in 2 mL of RPMI-1640 medium supplemented with 10% fetal bovine serum. Protein lysates from the cells were prepared 48 hours after transfection and analyzed by immunoblotting for clusterin, connective tissue growth factor, and β-actin. Connective tissue growth factor was included to monitor the efficacy of antisense inhibition, as we previously had shown that oligonucleotides targeting miR-17-92 members relieve repression of connective tissue growth factor by this microRNA cluster (40). Bands on the scanned blots were quantified with NIH ImageJ software. We confirmed transfection efficiency (>90%; data not shown) by transfecting cells in a separate well with an FITC-labeled oligonucleotide (BLOCK-iT; Invitrogen, Carlsbad, CA) and identifying transfected cells by flow cytometry.
Transfection With Clusterin Small Interfering RNA
Two small interfering RNAs (siRNAs) corresponding to human clusterin mRNA sequences 1 (5′-GGUUUAUAUGAUCUUCAUA-3′, nucleotide positions 2472–2490 in clusterin mRNA) and 2 (5′-AGGAAGAACCCUAAAUUUA-3′, nucleotide positions 1578–1596 in clusterin mRNA) were purchased from Qiagen, Valencia, CA and transfected into the neuroblastoma cell lines HTLA230, IMR32, SH-SY5Y, and LA-N-5. After transfection, we verified that sequence 2 but not sequence 1 decreased the expression of clusterin in each cell line, compared with nontransfected cells, by western blotting with a clusterin antibody. We therefore used sequence 1 as a control oligonucleotide in transfection experiments. In some experiments, we used another clusterin siRNAs purchased from Ambion, Austin, TX (named Clu 2 = 5′-CCGGAGGCCUCACUUCUUC-3′, nucleotide positions 824–842 in clusterin mRNA), and a siRNA that is guaranteed not to target any gene product (Ambion Silencer Negative Control, product AM4611, identification number 114526), which was used as a negative control.
The day before siRNA transfections, cells were seeded at a density of 200 000 cells per well in 1.5 mL of DMEM supplemented with 10% fetal calf serum in 12-well plates. For the transfection mixture, 1 μg of siRNA duplex was used. The RNA duplex was diluted in Opti-MEM I serum-free medium (Invitrogen, Carlsbad, CA), and a ratio of 1 μL of Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) to 2 μg of siRNAs was added by following the manufacturer's instructions. Cells were harvested after 24 hours for further assays, including western blotting and in vitro invasion assays.
In Vitro Invasion and Cell Viability Assays
The invasion assay was carried out as described previously (26). Briefly, IMR32, HTLA230, LA-N-5, or SH-SY5Y cells were plated at a density of 2.5 × 104 cells per chamber in a 24-well plate in serum-free DMEM in the top invasion assay chamber by following manufacturer's instructions (BD Biosciences). DMEM containing 10% fetal calf serum was placed in the bottom chamber as the chemoattractant. After 24 hours, cells migrating into the bottom chambers were stained with crystal violet and counted. If cells were metastatic, they could invade the Matrigel layer, which separates the top and bottom wells. The number of cells recovered in the bottom well was used as a measure of the invasive potential of that cell line.
Western Blot Analysis
For this analysis, 0.5 × 106 IMR32, HTLA230, LA-N-5, or SH-SY5Y cells were lysed in modified radio immuno precipitation buffer (RIPA) buffer containing 50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 0.1% sodium deoxycholate, 1% Triton X-100, 0.1% sodium dodecyl sulfate, and a mixture of protease inhibitors (Complete, protease inhibitor cocktail tablets; Roche Diagnostics, Indianapolis, IN) at 4°C for 30 minutes. Proteins were quantified by the dye-fixation Bradford method (Sigma-Aldrich), with bovine serum albumin as standard, and protein bands were resolved by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes (GE Healthcare). The primary antibodies against clusterin (1:1000 dilution), clusterin-α chain (1:500 dilution), β-actin (1:500 dilution), MYCN (1:200 dilution), connective tissue growth factor (1:200 dilution), glyceraldehyde-3-phosphate dehydrogenase (1:200 dilution), vimentin (1:200 dilution), and fibronectin (1:1000 dilution) were used. The membranes were then incubated with a secondary antibody–HRP-conjugated anti-mouse IgG (1:10 000 dilution), HRP-conjugated anti-rabbit IgG (1:10 000 dilution), or HRP-conjugated anti-goat IgG (1:5000 dilution). Antibody binding was detected by enhanced chemiluminesence (GE Healthcare).
Immunohistochemical Analysis
Immunohistochemical staining was performed as reported previously (24). Briefly, 4-mm sections from formalin-fixed, paraffin-embedded samples from neuroblastoma tumors were mounted onto slides, treated with xylene and graded alcohol, and equilibrated in PBS. Antigen retrieval was performed by microwaving the slides for three 5-minute periods at 400 W in a buffer containing 10 mM sodium citrate (pH 6). Endogenous peroxidase was blocked by incubating the slides in methanol containing 3% H2O2 at room temperature for 10 minutes. Nonspecific binding was blocked by incubation with normal goat serum (Pierce, Rockford, IL) for 30 minutes at room temperature. Anti-clusterin antibody (clone 41D) was used at a dilution of 1:50. The antibody was incubated for 1 hour at room temperature. Slides were washed several times in PBS and incubated with biotinylated anti-mouse IgG (dilution 1:100) for 30 minutes at room temperature, followed by peroxidase-labeled streptavidin (dilution 1:200, product RPN4401; GE Healthcare) for 30 minutes at room temperature. The antigen was visualized by incubation for 10–20 minutes at room temperature with diaminobenzidine tetrahydrochloride by following manufacturer's instruction (Sigma-Aldrich). Negative controls were treated similarly, except that monoclonal antibodies were excluded from the reaction mixture. Slides were counterstained with hematoxylin (Sigma-Aldrich), and coverslips were mounted with Eukitt (O. Kindler GmbH, Freiburg im Breisgau, Germany). The cut point for clusterin staining was 30% immunostained cells (either cytoplasmic or nuclear) in neuroblastic or ganglionic cells in four independent fields, with a positive result being 30% or more and a negative result being fewer than 30%, which we defined a priori (41). Slides were independently evaluated by two investigators (A. E. Caccamo and A. Sala). Positivity was detected in the nuclei, cytoplasm, or extracellular spaces of specimen examined.
Plasmid Construction and Generation of Stable Clones
The short hairpin oligonucleotides containing siRNA sequences 1 and 2 were annealed and ligated to the pSUPER.gfp+neo vector (OligoEngine, Seattle, WA) to produce pSUPER.gfp+neo.Clu and pSUPER.gfp+neo.Scr plasmids, respectively. Plasmids were transfected into HTLA230 cells by use of Lipofectamine 2000 (Invitrogen, Paisley, UK), as described in the manufacturer's protocol. Cells were maintained in DMEM containing G418 at 1.0 mg/mL for 3 weeks to select stable clones. We analyzed several G418-resistant clones for clusterin expression by Western blot analysis. We named the clone with the lowest expression of clusterin protein HTLA230-19b and the clone with the same clusterin expression as parental cells HTLA230-16, which was used as the control. These clones were used in the luciferase assay and xenotransplant experiments.
We generated the MYCN expression vector by excising a MYCN-containing fragment from pSP64-CRNX-3-MYCN vector (42) with XbaI and EcoRI enzymes and ligating it into a pcDNA3.1(+)/His vector (Invitrogen, Carlsbad, CA) to generate the pcDNA3.1(+)/His-MYCN vector. SH-SY5Y cells were transfected with pcDNA3.1(+)/His-MYCN or pcDNA3.1(+)/His, as a control, with Lipofectamine 2000 (Invitrogen, Carlsbad, CA), as described in the manufacturer's protocol. After 3 weeks of culture in DMEM medium with heat-inactivated 10% fetal calf serum containing G418 at 0.8 mg/mL, we selected the cell lines SH-SY5Y-MYCN, which express MYCN, and SH-SY5Y-Empty, as the control. These mixed cell population–stable clones were used to investigate the effect of MYCN overexpression on clusterin expression.
Luciferase Assays and BAY 11-7082 Treatment to Inhibit NF-κB
HTLA-16, HTLA-19b, and IPR6-2 cells were cultured in 12-well plate at a density of 200 000 cells per well in 1.5 mL of DMEM containing 10% fetal calf serum per well. Cells were transfected with 0.5 μg of the pNF-κB-LUC reporter plasmid (Clontech, Mountain View, CA) by use of Lipofectamine 2000 (Invitrogen, Carlsbad, CA), according the manufacturer's protocol. After 18 hours, cells were harvested and monitored for luciferase activity with a Dual-Luciferase Assay Kit (Promega, Madison, WI) by following the manufacturer's instructions. Light emission was measured with a luminometer and expressed as light units, which were normalized with a Renilla luciferase reference plasmid. In some experiments, 18 hours after transfection, IPR6-2 cells were exposed to 5 μM BAY 11-7082 (Merck KgaA, Darmstadt, Germany) for 22 hours, harvested, and monitored for luciferase activity.
Retroviral Infections and In Vivo Experiments
We investigated the role of clusterin in neuroblastoma growth in vivo by use of LA-N-5 cells and two retroviral vectors that express green fluorescent protein (26), MIGR1 and MIGR1-ApoJ. MIGR1-ApoJalso carries the clusterin sequence. Briefly, the amphotropic-packaging cell line Phoenix-A was transfected with MIGR1 or MIGR1-ApoJ by the calcium phosphate method according to the manufacturer's protocol (Promega). Viral particles were collected in DMEM after 48 hours of culture, filtered through a 0.45-μm (pore size) filter to remove cell debris, and then used to infect LA-N-5 cells. Cells were collected 48 hours after infection and subjected to Western blot analysis to monitor clusterin expression. Cells were sorted for green fluorescent protein expression by flow cytometry, and 5 × 106 LAN-5-MIGR1-ApoJ- or LAN-5-MIGR1-infected cells expressing green fluorescent protein were injected in the tail vein of common gamma chain, RAG double-knockout immunodeficient mice, which are permissive to human neuroblastoma cell growth. After 5 weeks, mice were killed by CO2 inhalation, cells in the dissected organs (livers, lungs, kidneys, and bone marrows) were mechanically dispersed, and tumor cells were recovered. The number of neuroblastoma cells, which were identified by their expression of green fluorescent protein, was quantified by flow cytometry. Neuroblastoma cells were detected consistently only in livers and bone marrows.
To investigate the role of endogenous clusterin in neuroblastoma cell growth, we generated stable HTLA230 clones with normal or suppressed clusterin expression. We injected 5 × 106 HTLA-19b cells (suppressed clusterin expression) or HTLA-16 cells (normal clusterin expression) in the tail vein of immunodeficient (ie, RAG, complement C3, and common gamma chain triple-knockout) mice. After 5 weeks, mice were killed by CO2 asphyxiation and the dissected organs were mechanically dispersed to recover tumor cells. The number of neuroblastoma cells in the liver or bone marrow was quantified by flow cytometry with an anti-disialoganglioside GD2 antibody. GD2 is a glycosphingolipid typically expressed on the surface of human neuroblastoma cells, including HTLA230. Briefly, cells were washed with PBS and incubated with 2 μg of GD2 antibody on ice for 30 minutes. Cells were then washed twice with PBS containing 3% fetal calf serum, resuspended in ice-cold 3% bovine serum albumin in PBS, and incubated for 30 minutes at room temperature with FITC-coupled anti-mouse secondary antibody (dilution 1:100). Cells were washed three times with PBS containing 3% fetal calf serum, and then, GD2-positive cells were counted by flow cytometry.
Genetically Modified Mice
Human MYCN-transgenic mice in the CBA mouse background (that were obtained from M. S. Jackson, University of Newcastle, UK) and clusterin-knockout mice (43) were maintained at University College London animal facility. The MYCN mice are the only available mouse model of neuroblastoma. The formation of tumors was monitored by inspection every other day until mice were 8 months old. The mice were killed by cervical dislocation or CO2 asphyxiation when a palpable tumor was detected or at the first signs of discomfort or stress, which is indicative of disease. Tumors were excised and snap-frozen in liquid nitrogen for subsequent characterization and protein analysis. All experimental procedures involving transgenic mice were approved by the University College London and were conducted under the Animal (Scientific Procedures) Act, 1986 (United Kingdom).
Electrophoretic Mobility Shift Assay
Tumor cells were lysed in modified RIPA buffer (50 mM Tris–HCl at pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% sodium deoxycholate, 1% Triton X-100, 0.1% sodium dodecyl sulfate, and a mixture of protease inhibitors [Complete, Roche Diagnostics]). Double-stranded DNA oligomers containing a wild-type (5′-AGTTGAGGGGACTTTCCCAGGC-3′) or mutant (5′-AGTTGAGGCGACTTTCCCAGGC-3′) NF-κB–binding site (underlined sequence) were labeled with [32P]dCTP (44) and used as probes for gel shift analysis. Relevant complexes were identified by incubation with 1 μg of antibody against p65-NF-κB for 5 minutes before adding the probe (supershift assay). Whole tumor cell extract (10 μg) and 1 ng of probes were mixed for 30 minutes at room temperature in 25 μL of binding buffer [10 mM Tris–HCl at pH 8.0, 130 mM KCl, 0.5 mM EDTA, 0.1% Triton X-100, 12.5% glycerol, 1 mM dithiothreitol, and 1.5 μg of poly(dI-dC)]. Shifted protein complexes were resolved by electrophoresis in 5% native polyacrylamide gel.
MicroRNA Target Prediction
Several computational methods have been developed to predict microRNA targets. MicroRNA target prediction for clusterin was performed by use of the miRanda algorithm (www.microrna.org).
Statistical Analysis
The χ2 test, a nonparametric test of statistical significance for bivariate analysis, with Yates correction was used to assess the statistical significance of the differences found in the immunohistochemistry analysis. The statistical significance in the cell invasion, xenotransplant, and luciferase experiments was calculated by Student t test or Welch t test, when applicable. Normal distribution was verified by observing that the data were symmetrically distributed around the mean. Kaplan–Meier survival curves and log-rank test were used to compare the groups of mice with different MYCN and/or ApoJ genotypes. A P value of less than .05 was interpreted to represent a statistically significant difference. All statistical tests were two-sided.
Results
MYCN and Regulation of Clusterin Expression
During a screening of Affymetrix datasets for the expression of clusterin, which are available on the Oncomine (45) Web site (www.oncomine.org), we observed that expression of the clusterin mRNA was lower in virtually all types of human cancer than in the corresponding normal tissues (Supplementary Figure 1, available online). In particular, the expression of clusterin in neuroblastoma was statistically significantly lower in specimens in which the MYCN proto-oncogene was amplified (the median value of normalized expression units was 1.9 in MYCN-negative neuroblastomas and 1.0 in MYCN-positive neuroblastomas; difference = 0.9 unit, 95% confidence interval [CI] = 0.82 to 1.14 units, P < .001) (Figure 1, A). This result led us to assess whether clusterin is negatively regulated by MYCN in neuroblastoma by stably transfecting SH-SY5Y neuroblastoma cells, in which MYCN is not amplified, with an expression vector containing the full-length MYCN cDNA [ie, pcDNA3.1(+)/His-MYCN]. We found that expression of endogenous clusterin mRNA and protein in these transfected cells was strongly inhibited by ectopic expression of MYCN (Figure 1, B and C). In addition, transient overexpression of MYCN in the human embryonic kidney cell line HEK-293 decreased the expression of clusterin compared with cells transfected with empty vector, implying that the mechanism of clusterin repression is not tissue specific (Figure 1, B).
Figure 1.

Expression of clusterin and MYCN in neuroblastoma. A) Box plot of the expression of clusterin mRNA in primary human neuroblastomas with MYCN amplification (Positive) or without MYCN amplification (Negative). The figure was generated with tools found in the Oncomine Web site (www.oncomine.org), where the information regarding the neuroblastoma samples can also be found. Statistical significance was assessed by Student t test. In the Oncomine Web site, all mRNA datasets are normalized by being log2-transformed, with the median set to 0 and SD set to 1. All statistical tests were two-sided. B) WB analysis. Whole-cell lysates were prepared from SH-SY5Y and HEK-293 cells that were stably (SH-SY5Y cells) or transiently (HEK-293 cells) transfected with an MYCN (pcMYCN) or control (Empty) vector and subjected to western blot analysis with antibodies specific for MYCN, the precursor form of clusterin (pCLU), or actin (as a loading control). C) Semiquantitative reverse transcription–PCR. Clusterin mRNA expression was assessed in SH-SY5Y cells stably transfected with MYCN (pcMYCN) or with control (Empty) vector. H2O indicates PCR amplification without reverse transcription (negative control). Samples were analyzed after completion of the indicated number of PCR cycles. Amplification of glyceraldehyde-3-phosphate dehydrogenase served as a loading control. M indicates molecular weight marker. PCR = polymerase chain reaction; WB = Western blot.
c-MYC has been shown to regulate gene expression through activation of microRNAs (40). To investigate whether microRNAs mediate the regulation of clusterin by MYCN, we performed a search for microRNAs that were predicted to target clusterin by use of miRanda, a web-based algorithm for finding genomic targets for microRNAs (46). The results predicted that clusterin mRNA was a target for four of the six microRNAs in the miR-17-92 locus (Figure 2, A; and the alignment for the closely related miR-19b; data not shown), which is known to be induced by c-MYC in a human B-cell line (47). We hypothesized that the expression of this microRNA cluster might also be increased in the presence of MYCN. Indeed, we found by use of real-time PCR that the expression of two of the six miR-17-92 microRNAs, miR-18a and miR-19a, was higher in MYCN-transfected SH-SY5Y cells than in nontransfected SH-SY5Y cells (Figure 2, B, left panel). To determine whether the increased expression of these microRNAs is a direct effect of MYCN, we selected SH-EP MYCN-ER neuroblastoma cells that express a conditionally active MYCN–ER fusion protein (38). Transient activation of MYCN with 4-hydroxytamoxifen resulted in an approximately fourfold increase in the level of the primary miR-17-92 transcript compared with that observed in untreated cells (miR-17-92 expression in treated cells = 263.90 relative units, 95% CI = 206.77 to 321.03 relative units; and miR-17-92 expression in untreated cells = 51.53 relative units, 95% CI = 45.6 to 57.47 relative units) and a concomitant approximately fourfold decrease in the level of clusterin mRNA (clusterin mRNA in treated cells = 83.66 relative units, 95% CI = 55.54 to 111.8 relative units; and in untreated cells = 271.81 relative units, 95% CI = 247.82 to 295.8 relative units) (Figure 2, B, right panel). We then investigated whether increased miR-17-92 levels in SH-SY5Y MYCN–transduced neuroblastoma cell lines were partially responsible for the decreased expression of clusterin mRNA by inhibiting microRNA expression by transfecting SH-SY5Y neuroblastoma cells with sequence-specific 2′-O-methyl oligoribonucleotides against the six microRNAs in the miR-17-92 cluster, as described previously (48).
Figure 2.

MicroRNAs of the 17-92 cluster and clusterin in neuroblastoma cells that overexpress MYCN. A) miRanda algorithm–predicted alignment of miR-17a, -18a, and -19a RNAs with clusterin (CLU) mRNA. The closely related miR-19b aligns with the same sequence as miR-19a. Alignment scores reflect sequence complementarity by using a position-weighted local alignment algorithm. Phastcons scores are a measure of evolutionary conservation of sequence blocks across multiple vertebrate species. Energy is an estimate of the free energy of formation of the microRNA–mRNA duplex. Perfect base pairing and G:U wobble base pairing are indicated by the vertical bars and colons, respectively. The numbers 1146, 764, and 2 are nucleotide positions in clusterin mRNA. B) Real-time polymerase chain reaction analysis. Left) Mature miR-17-92 cluster members in SH-SY5Y cells transfected with MYCN or control (vector) plasmids. The small nucleolar RNA U18 was used as control for RNA quality and/or input. Values on the y-axis equal , where Ct is the threshold cycle (ie, the cycle at which fluorescence rises statistically significantly above the baseline). The ABI sequence detection system software was set to automatically generate baseline and threshold values. Error bars = 95% confidence intervals. Right) miR-17-92 and clusterin transcripts in SH-EP cells expressing the MYCN–estrogen receptor fusion protein before and 48 hours after treatment with 4-hydroxytamoxifen (4OHT). A 24-hour treatment yielded essentially identical results. Expression levels were adjusted for β-actin. C) Western blot analysis. Blots were probed with antibodies against precursor (pCLU) or secreted (sCLU) clusterin, connective tissue growth factor (CTGF), and β-actin in MYCN-expressing SH-SY5Y cells transfected with anti-microRNA 17-92 (α-miR-17-92) or scrambled 2′-O-methyl oligoribonucleotides. Numbers refer to expression levels relative to those observed in mock-transfected cells. For clusterin, values represent the sum of the precursor and secreted forms under each condition. This assay was repeated twice with similar results.
We found an approximately twofold increase in the expression of connective tissue growth factor protein, a known miR-17-92 target, in the transfected cells compared with mock-transfected cells and cells transfected with scrambled oligonucleotides. In addition, levels of the precursor and secreted isoforms of clusterin were increased two- and ninefold, respectively, compared with those of mock-transfected cells or cells transfected with scrambled oligonucleotides; actin levels, used as loading control, in all cells were similar (Figure 2, C). We conclude that MYCN is a negative regulator of clusterin expression via activation of members of the miR-17-92 cluster of microRNAs.
Genetic Inactivation of Clusterin and Neuroblastoma Development in MYCN-Transgenic Mice
To test whether MYCN tumorigenic activity requires inactivation of clusterin expression in vivo, we used a transgenic MYCN mouse model in which targeted overexpression of MYCN in the neural crest leads to neuroblastoma. We crossed MYCN-transgenic mice with clusterin-knockout mice to obtain mice transgenic for MYCN in a clusterin-null background. Clusterin-knockout mice had been generated previously and shown to develop normally and to reach adulthood without overt physiological alterations (43). We backcrossed clusterin-knockout mice to the C57BL/6 strain, which is less susceptible to MYCN-induced neuroblastoma than the original FVB/N strain (28,49), to investigate whether clusterin functions as a tumor suppressor. We observed that the tumor-free survival among all MYCN-transgenic mice with two normal clusterin alleles was statistically significantly higher than the survival of mice with one (relative risk [RR] = 3.85, 95% CI = 1.26 to 11.81; P = .023) or no (RR = 4.87, 95% CI = 1.69 to 14.00; P = .005) normal clusterin alleles (Figure 3, A). Because tumor incidence is proportional to the dosage of the MYCN transgene (28), we also examined tumor-free survival by clusterin level when mice were divided into two main groups: those homozygous for the MYCN transgene and those heterozygous for the MYCN transgene. Tumor-free survival among mice homozygous for the MYCN transgene carrying two clusterin alleles was statistically significantly higher than that of mice with deleted clusterin alleles (RR = 3.76, 95% CI = 0.89 to 15.98; P = .049) (Figure 3, B). The same trend was observed in MYCN-transgene heterozygous mice, although the difference was not statistically significant (RR = 5.42, 95% CI = 1.07 to 27.36; P = .080) (Figure 3, C).
Figure 3.
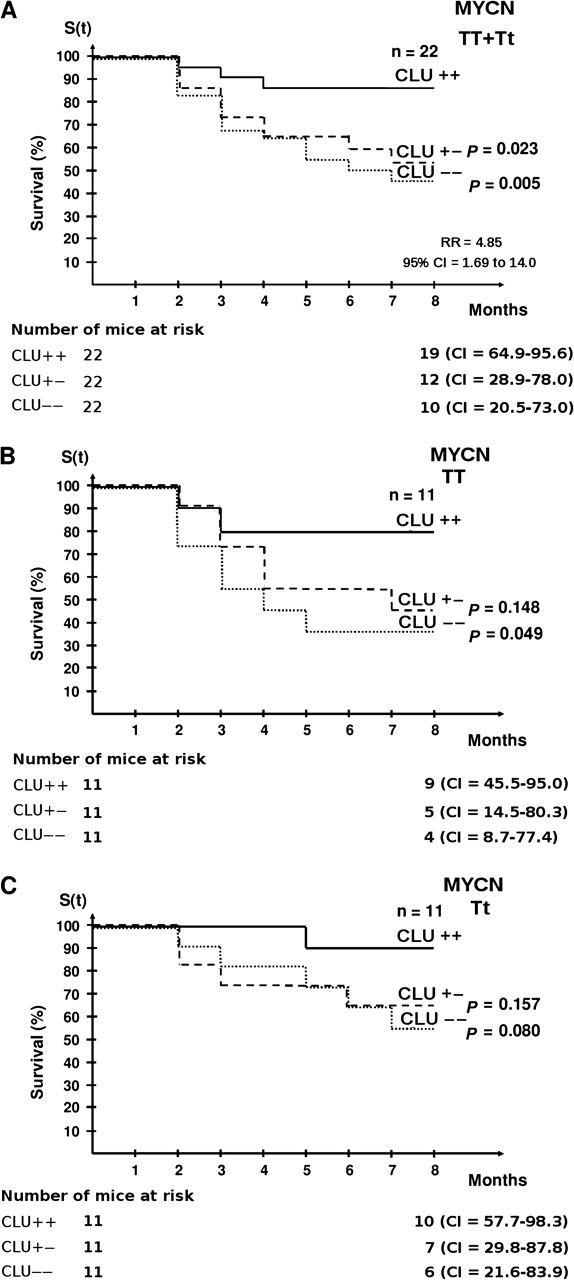
Tumor-free survival in MYCN-transgenic mice with different dosages of clusterin alleles. Kaplan–Meier survival curves of mice with different MYCN-transgene dosages (homozygous = TT or heterozygous = Tt) and clusterin genotypes (homozygously deleted = − −; heterozygously deleted = + −; wild type = + +) are shown. A) Cumulative tumor-free survival of the entire population of homozygous (MYCN TT) plus heterozygous MYCN-transgenic mice (MYCN Tt) by clusterin expression. There were 22 mice in each MYCN-transgenic group. B) Tumor-free survival of homozygous MYCN-transgenic mice (MYCN TT) by clusterin expression. There were 11 mice in each MYCN-transgenic group. C) Tumor-free survival of hemizygous MYCN-transgenic mice (MYCN Tt) by clusterin expression. There were 11 mice in each MYCN-transgenic group. The log-rank test was used to assess whether the difference in cumulative tumor-free survival between groups was statistically significant (RR for neuroblastoma development in clusterin +/+ vs clusterin −/− for all MYCN-transgenic mice = 4.85, 95% CI = 1.69 to 14.00). The number of mice at risk at 0 and 8 months and the 95% CI for the survival at 8 months are also shown. All statistical tests were two-sided. CI = confidence interval; RR = relative risk.
The Role of Clusterin in NF-κB Activity and Metastatic Progression in MYCN-Initiated Neuroblastomas
We have shown previously that the full-length clusterin protein is a potent repressor of NF-κB activity and that its overexpression inhibits the in vitro invasion of the human neuroblastoma cell line LA-N-5 (26). We extended these results by investigating whether genetic inactivation of clusterin leads to the activation of NF-κB in neuroblastomas developing in MYCN-transgenic mice. First, we monitored clusterin protein levels in tumors developing in MYCN-transgenic mice. We detected virtually no expression of clusterin protein in MYCN-transgenic homozygous tumors, regardless of the clusterin genotype, supporting the hypothesis that MYCN is a suppressor of clusterin (Figure 4, A). Normal adrenal glands expressed high levels of endogenous clusterin protein (Figure 4, A), whereas levels of clusterin protein in the neuroblastoma tumors or adrenal glands were lower than expected in mice with only one allele of the clusterin gene, which may explain why there is similar tumor incidence in mice with deletion of one or two alleles of clusterin.
Figure 4.

MYCN overexpression, clusterin expression, NF-κB activation, and the epithelial-to-mesenchymal transition. A) Western blot (WB) analysis of lysates of samples obtained from neuroblastoma tumors, normal adrenal glands, or embryonic fibroblasts from mice homozygous (MYCN TT) or hemizygous (MYCN Tt) for the MYCN transgene with wild-type or heterozygously or homozygously deleted clusterin alleles (indicated by + +, + −, or − −, respectively). Blots were incubated with antibodies against clusterin, vimentin, fibronectin, or glyceraldehyde-3-phosphate dehydrogenase (as a loading control), as indicated. Nonspecific bands in some of the lanes are indicated by an asterisk. B) EMSA of tumor protein lysates obtained from MYCN-transgenic mice with various MYCN and clusterin genotypes. Tumor extracts were incubated with a 32P-labeled oligonucleotide probe containing a wild-type (wt; lanes 3, 6, and 9) or mutated (mt; lanes 4, 7, and 10) NF-κB consensus motif. Lanes 1 and 2 contain free wt and mt probes, respectively. Supershifted complexes of the oligonucleotide and p65-NF-κB were identified by use of anti-p65-NF-κB antibody (lanes 5, 8, 11, and 13). Nuclear extract of Jurkat cells that had been stimulated with 12-O-tetradecanoylphorbol-13-acetate (Jurkat NE) was used as a positive control for NF-κB activation. The mouse genotypes for MYCN and clusterin are indicated at the bottom. pClu = precursor clusterin; sClu = secreted clusterin; NS = nonspecific DNA–protein complex; NF-κB = nuclear factor κB; EMSA = electrophoretic mobility shift assay.
To assess whether the p65 subunit of NF-κB was activated in primary neuroblastoma tumor isolated from MYCN-transgenic mice, we carried out a supershift analysis of MYCN-transgenic heterozygous tumors, in which NF-κB activation is indicated by a band-shift caused by the binding of the p65 subunit to the probe. The identity of the complex was validated by using the p65 antibody, and the band-shift was more pronounced in clusterin-knockout tumors than in tumors expressing normal levels of clusterin, suggesting that NF-κB is activated in parallel with the deletion of clusterin (Figure 4, B). Furthermore, expression of vimentin and fibronectin, two markers of mesenchymal tissue and NF-κB target genes, was generally higher in tumors with low or absent expression of clusterin than in normal adrenal glands or tumors expressing high levels of clusterin (Figure 4, A).
We investigated whether NF-κB is required for the metastatic activity of MYCN tumors by generating the IPR6-2 cell line from a mouse visceral neuroblastoma. We confirmed that this cell line was a homogeneous population of neuronal cells by staining these cells with CD56 antibody specific for neural cell adhesion molecule, a homophilic binding glycoprotein that is expressed on the surface of neurons (data not shown). We first assessed whether a classic inhibitor of NF-κB, BAY 11-7082, could inhibit an NF-κB–responsive promoter linked to the luciferase gene transiently transfected into IPR6-2 cells. Indeed, treatment of IPR6-2 cells with 5 μM BAY 11-7082 for 22 hours inhibited binding activity of NF-κB protein to its consensus binding sites, as shown by a drop in the activity of the NF-κB luciferase reporter of approximately twofold (from 0.88 to 0.47 relative luciferase units, 95% CI = 0.14 to 0.68; P = .002). Treatment of cells with BAY 11-7082 did not change the rate of apoptosis or the cell cycle distribution of neuroblastoma cells, as indicated by propidium iodide staining for DNA and flow cytometry analysis (Supplementary Figure 2, available online). Next, we examined the effect of BAY 11-7082 treatment on the metastatic activity of IPR6-2 cells with in vitro invasion assays. Statistically significantly fewer IPR6-2 cells treated with BAY 11-7082 (mean = 109 cells) than untreated IPR6-2 cells (mean = 270 cells) invaded the Matrigel layer in the invasion assays (difference = 161 cells, 95% CI = 100 to 224; P = .004). Thus, NF-κB may be required for metastatic activity of MYCN-transformed neuroblastoma cells.
Expression of Clusterin Protein in Primary Human Neuroblastomas
The findings of increased neuroblastoma tumor incidence and activated NF-κB in mice with zero or one normal clusterin alleles indicated that clusterin expression might be associated with a more benign phenotype in spontaneous human neuroblastomas. To investigate this possibility, we examined clusterin expression patterns in 97 primary human neuroblastomas from archival diagnostic biopsy examinations or surgical resections by use of a clusterin antibody that recognizes both nuclear and secreted clusterin. Specimens could have been obtained before or after chemotherapeutic treatments. We observed clusterin immunostaining in the cytoplasm, nucleus, and the extracellular space. In approximately 50% of the specimens, we observed strong or moderate extracellular and cytoplasmic clusterin immunostaining in the neuropil, a proteinaceous matrix typically associated with neuroblastic cells (Figure 5, A). Nuclear staining of clusterin was observed only in morphologically necrotic or apoptotic cells (Figure 5, B and D), a finding that supports nuclear clusterin as being proapoptotic, as reported previously (9–11). Unequivocal cytoplasmic staining was detected in ganglion cells, which are differentiated neuroblasts found most commonly in localized disease or neuroblastoma after chemotherapy (Figure 5, C). Ganglion cells are much larger than neuroblastsand so the identification of cytoplasmic clusterin was more accurate in these cells than in neuroblasts. It has been reported that the mature 60-kDa clusterin isoform is localized cytoplasmically and extracellularly, whereas the 54-kDa clusterin variant, which is derived from an alternatively spliced mRNA, is localized in the nucleus (3,4,50). For the purpose of determining the association between clusterin expression and biological or clinical tissue features, we merged extracellular and cytoplasmic clusterin into one category because we could not easily distinguish between them. Expression of the extracellular and cytoplasmic clusterin isoform was associated with differentiated or localized neuroblastomas, that is, 17 (74%) clusterin-positive samples were identified among 23 differentiated neuroblastomas and 17 (65%) were identified among 26 localized tumors (P < .001 for differentiated vs undifferentiated neuroblastomas and P = .002 for metastatic vs localized neuroblastomas, respectively) (Supplementary Table 1, available online). Nuclear clusterin expression was not associated with neuroblastoma metastasis or differentiation (data not shown). Only 10 (24.3%) of 41 metastatic neuroblastoma samples were positive for cytoplasmic and secreted clusterin.
Figure 5.

Immunohistochemical staining for clusterin expression in sections from neuroblastoma tumors. Representative histological sections stained with hematoxylin and anti-clusterin (CLU) antibodies (brown color). A) Extracellular or cytoplasmic staining for clusterin. Clusterin staining in the neuropil = arrows. Original magnification was ×200. Scale bar = 0.05 mm. B) Necrotic tissue. Thin arrow = clusterin staining in necrotic tissue; thick arrow = nuclear clusterin staining. Original magnification was ×100. Scale bar = 0.1 mm. C) Ganglion cells. Thick arrow = differentiated ganglion cells; thin arrow = immature neuroblasts. Original magnification was ×200. Scale bar = 0.05 mm. D) Apoptotic cells. Thick arrow = normal nucleus; thin arrow = pyknotic nucleus. Note that only cells with fragmented or pyknotic, but not normal, nuclei are stained by the antibody against clusterin. Original magnification was ×400. Scale bar = 0.001 mm.
Clusterin Expression and Invasion of Human Neuroblastoma Cell Lines In Vitro
The low percentage of metastatic neuroblastoma samples that were positive for cytoplasmic and secreted clusterin prompted us to further investigate the association between clusterin expression and metastatic activity in human neuroblastoma cells by transfecting siRNAs against clusterin into MYCN-amplified HTLA230 and IMR32 neuroblastoma cells and SH-SY5Y neuroblastoma cells, which lack MYCN amplification. Transfection of cells with clusterin siRNAs, but not control siRNAs, reduced expression of both precursor and mature (secreted) clusterin proteins (Figure 6, A) but did not change the survival or proliferation of these neuroblastoma cells within 48 hours of transfection (data not shown). We conducted in vitro invasion assays 24 hours after transfection by placing neuroblastoma cells in the top wells of invasion assay chambers. We found that more cells transfected with clusterin siRNA invaded the bottom chamber than cells transfected with the control siRNA (Figure 6, B). Thus, endogenous levels of clusterin appeared to suppress the metastatic behavior of both MYCN-amplified and MYCN-nonamplified neuroblastoma cell lines. These results were confirmed by use of other clusterin and control siRNAs (Figure 6, C).
Figure 6.

Clusterin and in vitro invasion of neuroblastoma. A) WB analysis of clusterin expression in neuroblastoma cells after transfection with siRNA against clusterin. Cell lysates were collected 24 hours after transfection of control sequence 1 (Seq 1) or clusterin sequence 2 (Seq 2) and then subjected to WB analysis with antibodies against clusterin to detect precursor (pClu) and secreted (sClu) clusterin. Expression of housekeeping genes, as a loading control, was monitored by WB with actin (HTLA cells) or keratin antibodies (IMR32 and SH-SY5Y cells). B) In vitro invasion assay with cells transfected with siRNAs against clusterin. The numbers of IMR32, HTLA230, and SH-SY5Y cells that migrated to the bottom chamber after mock transfection (wt), transfection of control sequence 1 (Seq1), or clusterin sequence 2 (Seq2) siRNAs are indicated. C) In vitro invasion assay with cells transfected with other siRNAs against clusterin. Left) WB analysis with a clusterin antibody of IMR-32 cells transfected with clusterin (Clu2) or control (Ctr) siRNAs. Middle) Crystal violet staining of invading IMR32 cells. Scale bars = 0.05 mm. Right) In vitro invasion assay results with cells transfected with the control (Ctr) or clusterin (Clu2) siRNAs. Statistical significance was calculated with a Student t test (n = number of independent assays). Error bars = 95% confidence intervals. All statistical tests were two-sided. siRNA = small interfering RNA; WB = Western blot.
Clusterin and Metastatic Growth of Xenotransplanted Human Neuroblastoma Cells
We observed by Western blot analysis that LA-N-5 cells expressed low levels of the precursor and secreted clusterin protein isoforms and HTLA230 cells expressed high levels of both (Figure 7, A). To assess whether increasing clusterin levels in LA-N-5 cells were associated with a change in their metastatic behavior in vivo, we infected neuroblastoma cells with a bicistronic retroviral vector, termed MIGR1/ApoJ, that carries full-length cDNAs for clusterin and for green fluorescent protein and expresses high levels of clusterin (26). After retroviral infection, we observed that the overexpressed clusterin protein was localized in the cytoplasm (Supplementary Figure 3, available online).
Figure 7.

Clusterin and an experimental model of metastasis in mice bearing xenotransplanted human neuroblastoma. A) Western blot analysis of clusterin expression in human neuroblastoma cell lines LA-N-5 and HTLA230. Cells were lysed and subjected to Western blot analysis with a clusterin antibody. Positions of the human 60-kDa clusterin precursor (pClu) and the mature 36-kDa secreted (sClu) clusterin are shown. Actin was used as the loading control. B) Quantification of neuroblastoma cells recovered from BM or liver of mice after 5 weeks that had been injected with LA-N-5 cells transduced with empty or clusterin MIGR1 (Clusterin) vectors (n = 4 mice per group). Tumor cells which were positive for green fluorescent protein expression from the MIGR1 vector were counted by flow cytometry. The statistical significance between the clusterin-transduced and untransduced groups was assessed with Student t test. C) Western blot analysis of clusterin expression in HTLA clones. HTLA cells were stably transfected with control vector (Empty), a plasmid containing short hairpin RNA control sequence 1 (termed HTLA-16 cells), or a plasmid containing clusterin short hairpin RNA sequence 2 (termed HTLA-19b cells), and then subjected to Western blot analysis for clusterin with a clusterin antibody. Actin was used as the loading control. D) Luciferase assays and the activity of NF-κB in HTLA cells with (HTLA-16 cells) or without (HTLA-19b cells) clusterin expression. A NF-κB luciferase reporter construct was transiently transfected into HTLA-16 and -19b cells. Firefly luciferase activity was measured and normalized to that of Renilla luciferase. Statistical significance was assessed with the two-sample Welch t test (n = 3 independent experiments). Error bars = 95% confidence intervals. E) Quantification of neuroblastoma cells recovered from BM of mice injected with HTLA clones HTLA-16 or -19b (n = 8 mice per group). Grey diamonds = HTLA-16 cells; black diamonds = HTLA-19b cells. Statistical significance was assessed with Student t test. All statistical tests were two-sided. NF-κB = nuclear factor κB; BM = bone marrow.
LA-N-5 cells that had been transduced with clusterin or control vectors were injected in the tail vein of immunodeficient mice. These mice were killed 5 weeks later by asphyxiation with carbon dioxide, and their livers and bone marrow were harvested. Large metastatic nodules were found by visual inspection in the livers of mice injected with control vector–infected neuroblastoma cells, but no metastatic nodules were observed in the livers of mice injected with clusterin-expressing cells. Flow cytometry analysis of cells isolated from liver or bone marrow found few or no cells that were positive for green fluorescent protein in mice injected with clusterin-expressing cells but found many cells that were positive for green fluorescent protein in mice injected with control cells (P = .049) (Figure 7, B). Inhibition of experimental metastasis in LA-N-5 cells was not caused by toxic effects or reduced cell proliferation because ectopic clusterin did not induce apoptosis or modify the cell cycle (data not shown).
We next transfected HTLA230 cells, which express high levels of clusterin, with a clusterin short hairpin RNA construct to observe the biological effects of ablating the expression of clusterin in human neuroblastoma cells. After selection with G418, we picked one clone with low clusterin expression (named HTLA-19b) and one clone with unchanged clusterin expression (named HTLA-16) as a control (Figure 7, C). Increased NF-κB activity was detected in HTLA-19b cells, the cells with low clusterin expression, but not in HTLA-16 cells, the cells with normal clusterin expression, further supporting the hypothesis that endogenous clusterin is a repressor of NF-κB in neuroblastoma cells (Figure 7, D). Next, HTLA-19b or -16 cells were injected in the tail vein of immunodeficient mice, these mice were killed after 6 weeks, and their livers were examined for metastatic nodules. We found that livers in four of the eight mice injected with clusterin-negative HTLA-19b cells had large nodules but that livers in only three of the seven mice injected with clusterin-positive HTLA-16 cells had small nodules. In addition, four of the eight mice injected with HTLA-19b cells and one of the seven mice injected with HTLA-16 cells had enlarged spleens (data not shown). Statistically significantly more clusterin-deficient HTLA-19b cells (3.27%) invaded the bone marrow than clusterin-positive HTLA-16 control cells (1.53%) (difference = 1.74%, 95% CI = 0.24 to 3.24, P = .026) (Figure 7, E). Thus, clusterin appears to be a suppressor of the metastatic behavior of human neuroblastoma cells.
Discussion
We have provided, to our knowledge, the first evidence to show that clusterin is a tumor suppressor gene that is negatively regulated by the proto-oncogene MYCN. Mice with a disrupted clusterin gene developed more neuroblastomas than mice with a normal clusterin gene (Figure 3). We have also shown that MYCN protein inhibited the expression of clusterin by inducing transcription of the miR-17-92 microRNA cluster. In support to the hypothesis that clusterin is a suppressor rather than a promoter of human tumorigenesis, two research teams (51,52) have found decreased levels of clusterin in specimens from patients with von Hippel–Lindau disease and proposed that clusterin mediates a von Hippel–Lindau tumor suppressor function that is independent of the transcription factor HIF, the primary target of the von Hippel–Lindau tumor suppressor protein. In addition, by analyzing data from available expression profiling datasets, we observed that clusterin mRNA levels were reduced in virtually all types of human cancers compared with normal tissues (Supplementary Figure 1, available online).
Our observations in neuroblastoma are consistent with the hypothesis that clusterin is a tumor suppressor gene or a tumor modifier. Although no mutations of the clusterin gene have been reported so far, its expression is often silenced by epigenetic mechanisms in human neoplasia. Furthermore, we have observed that clusterin-knockout mice spontaneously develop prostate carcinomas with high incidence, supporting the hypothesis that clusterin is a tumor suppressor gene (S. Bettuzzi, P. Davalli, S. Davoli, O. Chayka, F. Rizzi, L. Belloni, D. Pellacani, G. Fregni, S. Astancolle, M. Fassan, A. Corti, R. Baffa, and A. Sala, unpublished results).
Evaluation of the biological role of clusterin is complicated by the existence of two protein variants. Expression of the nuclear form of clusterin is restricted to apoptotic and/or necrotic cells (10,11). In neuroblastoma, we observed nuclear clusterin only in dying cells that were within or near areas of necrotic or apoptotic tissue (Figure 5). The cytoplasm location of clusterin is functionally relevant because a research group (6) has claimed that cytoplasmic clusterin binds to activated BAX in the mitochondria and thus blocks apoptosis.
We have shown previously (26) that clusterin inhibits the proteosomal degradation of the inhibitors of kappa B protein and, consequently, the activation of NF-κB and the in vitro invasion of human neuroblastoma cells. Other research teams (35,53) have shown that clusterin inhibits NF-κB activity in other pathophysiological contexts, including rheumatoid arthritis and apoptosis of tubular cells. In epithelial cancers, activation of NF-κB and inflammation are thought to induce the epithelial-to-mesenchymal transition and a metastatic phenotype (27,54–56). The results presented in this manuscript are consistent with this model because suppression of clusterin elicits activation of NF-κB and markers of the epithelial-to-mesenchymal transition in MYCN-induced neuroblastomas. Suppression of the invasive potential of a neuroblastoma cell line derived from a MYCN-induced tumor by treatment with the NF-κB inhibitor, BAY 11-7082, demonstrated that NF-κB may have an important role in the aggressive behavior of neuroblastoma. This concept is not without a precedent because Farina and coworkers (57) have showed previously that MYCN-transfected neuroblastoma cells have activated NF-κB, are resistant to differentiation induced by retinoic acid, and have higher metastatic potential than the corresponding parental cells. Thus, the oncogenic activity of MYCN appears, at least in part, to be mediated by NF-κB, which explains why overcoming the tumor suppressive function of clusterin appears to be important in neuroblastoma tumorigenesis. In spite of the fact that NF-κB promotes metastatic activity, the cytotoxic effects of chemotherapeutic drugs on neuroblastoma cells also require NF-κB activity (58). Thus, it is not surprising that endogenous clusterin protects neuroblastoma cells from the cytotoxic effects of doxorubicin (7). A potentially relevant clinical implication is that clusterin-negative tumors, although intrinsically more aggressive, may respond better to chemotherapy.
This study had several limitations. We demonstrated the requirement of NF-κB for the metastatic activity in only one neuroblastoma cell line in vitro. In future investigations, it will be important to study whether pharmacological inhibition of NF-κB causes inhibition of the growth of other neurblastoma cell lines in mouse models of neuroblastoma and, hopefully, in neuroblastoma patients. The sample sizes were small in our experiments in mice with retroviral expression of clusterin, with groups of four mice injected with LA-N-5 neuroblastoma cells infected with control or clusterin vectors. Specimens from only 97 neuroblastoma tumors were analyzed, 10 of which were from postchemotherapy patients and could not be used for the analysis of the association of clusterin expression with biological features. The population was skewed toward an older median age, and patients were diagnosed more frequently with stage IV disease than with disease at other stages.
Our results with the miR-17-92 cluster are supported by a recent report (59) that the expression of two microRNAs in the miR-17-92 cluster, miR-17-5p and miR-92, is increased by MYCN expression in SH-EP neuroblastoma cells. We cannot rule out, however, that MYCN represses clusterin expression by other mechanisms in addition to RNA interference. Indeed, we identified two putative MYC-binding sites (E-boxes) in the clusterin promoter sequence. A retarded complex was detected by gel shift analysis with one of the two putative MYC-binding sites in cells overexpressing exogenous MYCN (O. Chayka, D. Corvetta, and A. Sala, unpublished data). One could speculate that to achieve complete silencing of clusterin, MYCN may have to act via a combination of direct and indirect mechanisms. Other MYC family members, like c-MYC, have been shown to bind to the promoter of growth suppressor genes, such as p21, and to inhibit their transcription by recruiting chromatin modifiers, including Dnmt3a (29). Interestingly, the clusterin gene is repressed by epigenetic mechanisms in endothelial tumor cells and in neuronal tumor cell lines, including neuroblastoma (60,61). MYCN, in addition to inducing microRNAs that suppress clusterin expression, could promote recruitment of DNA methylases or histone deacethylases to the clusterin promoter, resulting in silencing of transcription. Because histone deacetylase and DNA methylation inhibitors are being used to treat some forms of adult human cancers, it is possible to envisage that these compounds and/or molecules targeting the miR-17-92 cluster could reactivate clusterin expression and induce tumor regression in children with neuroblastoma.
Funding
Sport Aiding Medical Research for Kids (SPARKS), Great Ormond Street Hospital/National Health Service to A.S.; RO1 (CA 122334) from the National Cancer Institute to A.T.-T; FIL 2008, University of Parma to S.B.
Supplementary Material
Footnotes
Sponsors had no role in the collection of the data, the analysis and interpretation of the data, the decision to submit the manuscript for publication, and the writing of the manuscript.
References
- 1.Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003;3(3):203–216. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- 2.Lonergan GJ, Schwab CM, Suarez ES, Carlson CL. Neuroblastoma, ganglioneuroblastoma, and ganglioneuroma: radiologic-pathologic correlation. Radiographics. 2002;22(4):911–934. doi: 10.1148/radiographics.22.4.g02jl15911. [DOI] [PubMed] [Google Scholar]
- 3.Shannan B, Seifert M, Leskov K, et al. Challenge and promise: roles for clusterin in pathogenesis, progression and therapy of cancer. Cell Death Differ. 2006;13(1):12–19. doi: 10.1038/sj.cdd.4401779. [DOI] [PubMed] [Google Scholar]
- 4.Trougakos IP, Gonos ES. Clusterin/apolipoprotein J in human aging and cancer. Int J Biochem Cell Biol. 2002;34(11):1430–1448. doi: 10.1016/s1357-2725(02)00041-9. [DOI] [PubMed] [Google Scholar]
- 5.Nizard P, Tetley S, Le Drean Y, et al. Stress-induced retrotranslocation of clusterin/ApoJ into the cytosol. Traffic. 2007;8(5):554–565. doi: 10.1111/j.1600-0854.2007.00549.x. [DOI] [PubMed] [Google Scholar]
- 6.Zhang H, Kim JK, Edwards CA, Xu Z, Taichman R, Wang CY. Clusterin inhibits apoptosis by interacting with activated Bax. Nat Cell Biol. 2005;7(9):909–915. doi: 10.1038/ncb1291. [DOI] [PubMed] [Google Scholar]
- 7.Cervellera M, Raschella G, Santilli G, et al. Direct transactivation of the anti-apoptotic gene apolipoprotein J (clusterin) by B-MYB. J Biol Chem. 2000;275(28):21055–21060. doi: 10.1074/jbc.M002055200. [DOI] [PubMed] [Google Scholar]
- 8.Lourda M, Trougakos IP, Gonos ES. Development of resistance to chemotherapeutic drugs in human osteosarcoma cell lines largely depends on up-regulation of clusterin/apolipoprotein J. Int J Cancer. 2007;120(3):611–622. doi: 10.1002/ijc.22327. [DOI] [PubMed] [Google Scholar]
- 9.Caccamo AE, Scaltriti M, Caporali A, et al. Ca2+ depletion induces nuclear clusterin, a novel effector of apoptosis in immortalized human prostate cells. Cell Death Differ. 2005;12(1):101–104. doi: 10.1038/sj.cdd.4401491. [DOI] [PubMed] [Google Scholar]
- 10.Leskov KS, Klokov DY, Li J, Kinsella TJ, Boothman DA. Synthesis and functional analyses of nuclear clusterin, a cell death protein. J Biol Chem. 2003;278(13):11590–11600. doi: 10.1074/jbc.M209233200. [DOI] [PubMed] [Google Scholar]
- 11.O’Sullivan J, Whyte L, Drake J, Tenniswood M. Alterations in the post-translational modification and intracellular trafficking of clusterin in MCF-7 cells during apoptosis. Cell Death Differ. 2003;10(8):914–927. doi: 10.1038/sj.cdd.4401254. [DOI] [PubMed] [Google Scholar]
- 12.Behrens P, Jeske W, Wernert N, Wellmann A. Downregulation of clusterin expression in testicular germ cell tumours. Pathobiology. 2001;69(1):19–23. doi: 10.1159/000048753. [DOI] [PubMed] [Google Scholar]
- 13.He HZ, Song ZM, Wang K, et al. Alterations in expression, proteolysis and intracellular localizations of clusterin in esophageal squamous cell carcinoma. World J Gastroenterol. 2004;10(10):1387–1391. doi: 10.3748/wjg.v10.i10.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Redondo M, Villar E, Torres-Munoz J, Tellez T, Morell M, Petito CK. Overexpression of clusterin in human breast carcinoma. Am J Pathol. 2000;157(2):393–399. doi: 10.1016/S0002-9440(10)64552-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scaltriti M, Brausi M, Amorosi A, et al. Clusterin (SGP-2, ApoJ) expression is downregulated in low- and high-grade human prostate cancer. Int J Cancer. 2004;108(1):23–30. doi: 10.1002/ijc.11496. [DOI] [PubMed] [Google Scholar]
- 16.Steinberg J, Oyasu R, Lang S, et al. Intracellular levels of SGP-2 (Clusterin) correlate with tumor grade in prostate cancer. Clin Cancer Res. 1997;3(10):1707–1711. [PubMed] [Google Scholar]
- 17.Xie MJ, Motoo Y, Su SB, et al. Expression of clusterin in human pancreatic cancer. Pancreas. 2002;25(3):234–238. doi: 10.1097/00006676-200210000-00004. [DOI] [PubMed] [Google Scholar]
- 18.Hadaschik BA, Sowery RD, Gleave ME. Novel targets and approaches in advanced prostate cancer. Curr Opin Urol. 2007;17(3):182–187. doi: 10.1097/MOU.0b013e3280dd8a4f. [DOI] [PubMed] [Google Scholar]
- 19.Miyake H, Gleave M, Kamidono S, Hara I. Overexpression of clusterin in transitional cell carcinoma of the bladder is related to disease progression and recurrence. Urology. 2002;59(1):150–154. doi: 10.1016/s0090-4295(01)01484-4. [DOI] [PubMed] [Google Scholar]
- 20.Miyake H, Nelson C, Rennie PS, Gleave ME. Testosterone-repressed prostate message-2 is an antiapoptotic gene involved in progression to androgen independence in prostate cancer. Cancer Res. 2000;60(1):170–176. [PubMed] [Google Scholar]
- 21.Zellweger T, Miyake H, July LV, Akbari M, Kiyama S, Gleave ME. Chemosensitization of human renal cell cancer using antisense oligonucleotides targeting the antiapoptotic gene clusterin. Neoplasia. 2001;3(4):360–367. doi: 10.1038/sj.neo.7900174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chi KN, Siu LL, Hirte H, et al. A phase I study of OGX-011, a 2′-methoxyethyl phosphorothioate antisense to clusterin, in combination with docetaxel in patients with advanced cancer. Clin Cancer Res. 2008;14(3):833–839. doi: 10.1158/1078-0432.CCR-07-1310. [DOI] [PubMed] [Google Scholar]
- 23.Schmitz G. Drug evaluation: OGX-011, a clusterin-inhibiting antisense oligonucleotide. Curr Opin Mol Ther. 2006;8(6):547–554. [PubMed] [Google Scholar]
- 24.Caporali A, Davalli P, Astancolle S, et al. The chemopreventive action of catechins in the TRAMP mouse model of prostate carcinogenesis is accompanied by clusterin over-expression. Carcinogenesis. 2004;25(11):2217–2224. doi: 10.1093/carcin/bgh235. [DOI] [PubMed] [Google Scholar]
- 25.Thomas-Tikhonenko A, Viard-Leveugle I, Dews M, et al. Myc-transformed epithelial cells down-regulate clusterin, which inhibits their growth in vitro and carcinogenesis in vivo. Cancer Res. 2004;64(9):3126–3136. doi: 10.1158/0008-5472.can-03-1953. [DOI] [PubMed] [Google Scholar]
- 26.Santilli G, Aronow BJ, Sala A. Essential requirement of apolipoprotein J (clusterin) signaling for IkappaB expression and regulation of NF-kappaB activity. J Biol Chem. 2003;278(40):38214–38219. doi: 10.1074/jbc.C300252200. [DOI] [PubMed] [Google Scholar]
- 27.Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18(1):19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weiss WA, Aldape K, Miohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16(11):2985–2995. doi: 10.1093/emboj/16.11.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brenner C, Deplus R, Didelot C, et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 2005;24(2):336–346. doi: 10.1038/sj.emboj.7600509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, Eisenman RN. Myc influences global chromatin structure. EMBO J. 2006;25(12):2723–2734. doi: 10.1038/sj.emboj.7601152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wanzel M, Herold S, Eilers M. Transcriptional repression by Myc. Trends Cell Biol. 2003;13(3):146–150. doi: 10.1016/s0962-8924(03)00003-5. [DOI] [PubMed] [Google Scholar]
- 32.Goldman JP, Blundell MP, Lopes L, Kinnon C, Di Santo JP, Thrasher AJ. Enhanced human cell engraftment in mice deficient in RAG2 and the common cytokine receptor gamma chain. Br J Haematol. 1998;103(2):335–342. doi: 10.1046/j.1365-2141.1998.00980.x. [DOI] [PubMed] [Google Scholar]
- 33.Sambrook J, Maniatis T, Fritsch EF. Molecular Cloning: A Laboratory Manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 34.Norris MD, Burkhart CA, Marshall GM, Weiss WA, Haber M. Expression of N-myc and MRP genes and their relationship to N-myc gene dosage and tumor formation in a murine neuroblastoma model. Med Pediatr Oncol. 2000;35(6):585–589. doi: 10.1002/1096-911x(20001201)35:6<585::aid-mpo20>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 35.Devauchelle V, Essabbani A, De Piinieux G, et al. Characterization and functional consequences of underexpression of clusterin in rheumatoid arthritis. J Immunol. 2006;177(9):6471–6479. doi: 10.4049/jimmunol.177.9.6471. [DOI] [PubMed] [Google Scholar]
- 36.Winer J, Jung CK, Shackel I, Williams PM. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem. 1999;270(1):41–49. doi: 10.1006/abio.1999.4085. [DOI] [PubMed] [Google Scholar]
- 37.Brignole C, Marimpietri D, Pastorino F, et al. Effect of bortezomib on human neuroblastoma cell growth, apoptosis, and angiogenesis. J Natl Cancer Inst. 2006;98(16):1142–1157. doi: 10.1093/jnci/djj309. [DOI] [PubMed] [Google Scholar]
- 38.Ushmorov A, Hogarty MD, Liu X, Knauss H, Debatin KM, Beltinger C. N-myc augments death and attenuates protective effects of Bcl-2 in trophically stressed neuroblastoma cells. Oncogene. 2008;27(24):3424–3434. doi: 10.1038/sj.onc.1211017. [DOI] [PubMed] [Google Scholar]
- 39.Chung EY, Dews M, Cozma D, et al. c-Myb oncoprotein is an essential target of the dleu2 tumor suppressor microRNA cluster. Cancer Biol Ther. 2008;7(11):1758–1764. doi: 10.4161/cbt.7.11.6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dews M, Homayouni A, Yu D, et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38(9):1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wolff AC, Hammond ME, Schwartz JN, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch Pathol Lab Med. 2007;131(1):18. doi: 10.5858/2007-131-18-ASOCCO. [DOI] [PubMed] [Google Scholar]
- 42.Wittke I, Wiedemeyer R, Pillmann A, Savelyeva L, Westermann F, Schwab M. Neuroblastoma-derived sulfhydryl oxidase, a new member of the sulfhydryl oxidase/Quiescin6 family, regulates sensitization to interferon gamma-induced cell death in human neuroblastoma cells. Cancer Res. 2003;63(22):7742–7752. [PubMed] [Google Scholar]
- 43.McLaughlin L, Zhu G, Mistry M, et al. Apolipoprotein J/clusterin limits the severity of murine autoimmune myocarditis. J Clin Invest. 2000;106(9):1105–1113. doi: 10.1172/JCI9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Urban MB, Schreck R, Baeuerle PA. NF-kappa B contacts DNA by a heterodimer of the p50 and p65 subunit. Embo J. 1991;10(7):1817–1825. doi: 10.1002/j.1460-2075.1991.tb07707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rhodes DR, Yu J, Shanker K, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6(1):1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;(11):2, e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV. Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435(7043):839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 48.Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10(3):544–550. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hackett CS, Hodgson JG, Law ME, et al. Genome-wide array CGH analysis of murine neuroblastoma reveals distinct genomic aberrations which parallel those in human tumors. Cancer Res. 2003;63(17):5266–5273. [PubMed] [Google Scholar]
- 50.Klokov D, Criswell T, Leskov KS, Araki S, Mayo L, Boothman DA. IR-inducible clusterin gene expression: a protein with potential roles in ionizing radiation-induced adaptive responses, genomic instability, and bystander effects. Mutat Res. 2004;568(1):97–110. doi: 10.1016/j.mrfmmm.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 51.Nakamura E, Abreu-e-Lima P, Awakura Y, et al. Clusterin is a secreted marker for a hypoxia-inducible factor-independent function of the von Hippel-Lindau tumor suppressor protein. Am J Pathol. 2006;168(2):574–584. doi: 10.2353/ajpath.2006.050867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou M, Shen D, Head JE, et al. Ocular clusterin expression in von Hippel-Lindau disease. Mol Vis. 2007;13:2129–2136. [PMC free article] [PubMed] [Google Scholar]
- 53.Takase O, Minto AW, Puri TS, et al. Inhibition of NF-kappaB-dependent Bcl-xL expression by clusterin promotes albumin-induced tubular cell apoptosis. Kidney Int. 2008;73(5):567–577. doi: 10.1038/sj.ki.5002563. [DOI] [PubMed] [Google Scholar]
- 54.Grund EM, Kagan D, Tran CA, et al. TNF-{alpha} regulates inflammatory and mesenchymal responses via MEK, p38, NF-{kappa}B in human endometriotic epithelial cells [published online ahead of print] Mol Pharmacol. 2008;73(5):1394–1404. doi: 10.1124/mol.107.042176. [DOI] [PubMed] [Google Scholar]
- 55.Julien S, Puig I, Caretti E, et al. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26(53):7445–7456. doi: 10.1038/sj.onc.1210546. [DOI] [PubMed] [Google Scholar]
- 56.Radisky DC, Bissell MJ. NF-kappaB links oestrogen receptor signalling and EMT. Nat Cell Biol. 2007;9(4):361–363. doi: 10.1038/ncb0407-361. [DOI] [PubMed] [Google Scholar]
- 57.Farina AR, Masciulli MP, Tacconelli A, et al. All-trans-retinoic acid induces nuclear factor kappaB activation and matrix metalloproteinase-9 expression and enhances basement membrane invasivity of differentiation-resistant human SK-N-BE 9N neuroblastoma Cells. Cell Growth Differ. 2002;13(8):343–354. [PubMed] [Google Scholar]
- 58.Bian X, McAllister-Lucas LM, Shao F, et al. NF-kappa B activation mediates doxorubicin-induced cell death in N-type neuroblastoma cells. J Biol Chem. 2001;276(52):48921–48929. doi: 10.1074/jbc.M108674200. [DOI] [PubMed] [Google Scholar]
- 59.Schulte JH, Horn S, Otto T, et al. MYCN regulates oncogenic MicroRNAs in neuroblastoma. Int J Cancer. 2008;122(3):699–704. doi: 10.1002/ijc.23153. [DOI] [PubMed] [Google Scholar]
- 60.Hellebrekers DM, Melotte V, Vire E, et al. Identification of epigenetically silenced genes in tumor endothelial cells. Cancer Res. 2007;67(9):4138–4148. doi: 10.1158/0008-5472.CAN-06-3032. [DOI] [PubMed] [Google Scholar]
- 61.Nuutinen T, Suuronen T, Kyrylenko S, Huuskonen J, Salminen A. Induction of clusterin/apoJ expression by histone deacetylase inhibitors in neural cells. Neurochem Int. 2005;47(8):528–538. doi: 10.1016/j.neuint.2005.07.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.