

Abstract
The engineering of functional skeletal muscle tissue substitutes holds promise for the treatment of various muscular diseases and injuries. However, no tissue fabrication technology currently exists for the generation of a relatively large and thick bioartificial muscle made of densely packed, uniformly aligned, and differentiated myofibers. In this study, we describe a versatile cell/hydrogel micromolding approach where polydimethylsiloxane (PDMS) molds containing an array of elongated posts were used to fabricate relatively large neonatal rat skeletal muscle tissue networks with reproducible and controllable architecture. By combining cell-mediated fibrin gel compaction and precise microfabrication of mold dimensions including the length and height of the PDMS posts, we were able to simultaneously support high cell viability, guide cell alignment along the microfabricated tissue pores, and reproducibly control the overall tissue porosity, size, and thickness. The interconnected muscle bundles within the porous tissue networks were composed of densely packed, aligned, and highly differentiated myofibers. The formed myofibers expressed myogenin, developed abundant cross-striations, and generated spontaneous tissue contractions at the macroscopic spatial scale. The proliferation of non-muscle cells was significantly reduced compared to monolayer cultures. The more complex muscle tissue architectures were fabricated by controlling the spatial distribution and direction of the PDMS posts.
Keywords: Skeletal myoblasts, myotube alignment, rapid prototyping, myogenesis, reproducibility
1. Introduction
The reconstruction of the lost skeletal muscle tissue function caused by congenital defects, tumor ablation, prolonged denervation, traumatic injury, or different myopathies [1] is hampered by the lack of functional tissue substitutes. Current surgical treatments including autologous muscle transplantation and transposition only yield a limited degree of success [2]. An alternative approach is the transplantation of exogenous myogenic cells (satellite cells and myoblasts) in the site of injury. However, previous clinical trials with intramuscular injection of skeletal myoblasts yielded poor outcomes due to inadequate distribution and low retention and survival of the injected cells [1]. The implantation of an engineered skeletal muscle tissue, although surgically more complex, holds potential to improve retention and survival of the transplanted cells [3]. In addition, the shape and architecture of the engineered muscle could be pre-designed for the precise structural repair at the site of damage (e.g., during craniofacial reconstruction). The engineered muscle could also be preconditioned for specific mechanically or metabolically demanding host environments (e.g., sarcopenic muscle in the elderly or at the site of traumatic injury). Upon implantation, the tissue graft is expected to incorporate into the host neuromuscular system and directly augment the compromised muscle function while potentially being designed to exert additional regenerative effects via localized delivery of angiogenic and anti-apoptotic paracrine factors.
From a biomimetic perspective, an ideal engineered skeletal muscle tissue needs to: 1) be adequately large and thick, 2) consist of densely packed and highly differentiated myofibers, and 3) mimic the aligned architecture of native muscle in order to generate sufficient and appropriately distributed active forces for the effective restoration of the impaired muscle function. However, the current methods to align skeletal muscle cells in 3 dimensions rely on the constraint of cell growth within single, few hundred μm thick muscle bundles that generate active forces more than an order of magnitude lower than normal adult muscle [4, 5]. Recent attempts to grow larger muscle tissues with aligned cells have included successive layering of collagen matrix and the seeding of myoblasts in a Petri dish to produce dense, 100-150 μm thick muscle sheets [6] and the use of relatively thick (2-2.5mm) polymer scaffolds with oriented pores seeded with C2C12 cells [7]. Nevertheless, the inability to provide sufficient oxygen and nutrient supply to match the high metabolic demand of differentiated engineered muscle [8] and the lack of reliable and reproducible methods to control muscle cell density and alignment within a relatively large tissue volume still remain as the main obstacles to the in vitro fabrication of clinically relevant bioengineered muscle.
Recently, we utilized sucrose leaching to produce porous, oriented poly(lactic-co-glycolic) acid scaffolds that supported 3-D cardiac cell alignment and anisotropic electrical propagation over a relatively large area (∼1-2 cm2) [9]. However, the mechanical rigidity of the scaffold, which prevented macroscopic tissue contractions and the inability to control the tissue anisotropy by controlling the scaffold structure were the major drawbacks of this approach. Bioactive hydrogels, on the other hand, are attractive scaffold materials for the engineering of skeletal muscle, because they allow for spatially uniform cell entrapment, high ultimate cell density due to significant cell-mediated gel compaction [10, 11], control of cell alignment by application of specific geometrical constraints [12], and macroscopic tissue contractions. Despite being difficult to be chemically manipulated and having significant batch-to-batch variability, natural hydrogels (e.g., collagen [5, 13], matrigel [5, 13] and fibrin [4]) still appear to be superior to synthetic hydrogels for muscle tissue engineering primarily due to the higher density of cell adhesion sites required for the 3D cell spreading. Recently, methods for photo- [14] and soft-lithographic [15] micropatterning of hydrogels have been applied to the engineering of complex hepatic [14] and vascular tissue structures [16]. These rapid prototyping techniques enable reproducible design of scaffold geometry [17], systematic control of porosity and pore interconnectivity [18], precise positioning of one or more cell types in the desired 3D configuration [15], and layered assembly of 3D objects [14], all of which may facilitate the reproducible engineering of customized muscle tissue architectures.
In this study, we developed a cell/hydrogel micromolding approach to fabricate relatively large (0.5-2 cm2) and thick (127-384 μm) skeletal muscle tissue networks with dense, aligned and highly differentiated muscle fibers. Specifically, a cell/hydrogel mixture was cast inside microfabricated polydimethylsiloxane (PDMS) molds with staggered elongated posts to create porous tissue networks. We hypothesized that the control of the network pore size and elongation will enable: 1) increased cell viability due to improved nutrient and oxygen transport through the network pores and 2) effective and uniform cell alignment along the repetitive pore boundaries [12, 15]. Using primary skeletal myoblasts from neonatal rats and the mouse C2C12 myoblast cell line, we demonstrated the high versatility of this technique including the ability to accurately and reproducibly vary the engineered tissue size, thickness, porosity and the spatial distribution of cell alignment.
2. Materials and Methods
2.1 Fabrication of Tissue Molds
Tissue molds made of polydimethylsiloxane (PDMS, Dow Corning, Midland, MI) were created by casting against patterned master templates (Figure 1.A). We compared two approaches for master fabrication: 1) standard photolithography with SU-8 photoresist (Microchem, Newton, MA) and 2) rapid photopatterning with a thiolene-based optical adhesive, Norland 81 (Norland Products, Cranbury, NJ) [19]. Photomasks were designed using Postcript language and printed at high resolution (6.35 μm/pixel) on transparencies (Advance reproductions, North Andover, MA). For standard photolithography, silicon wafers (Wafer World Inc., West Palm Beach, FL) were cleaned in H2O2/ H2SO4 (1:3, v/v) solution and treated with UV-generated ozone (PSD-UVT system, Novascan Technologies Inc, Ames, IO). The cleaned wafers were coated with a 1-2 mm thick layer of SU-8 100 photoresist, prebaked overnight, cooled, and exposed to UV through a photomask using a vacuum mask aligner (Suss MicroTec, Garching, Germany). Exposed masters were postbaked, cooled, developed in propylene-glycol-methyl-ether-acetate (PGMEA, Sigma, St. Louis, MO) solution, rinsed with isopropyl alcohol, dried and silanized overnight to facilitate PDMS removal. For rapid photopatterning with Norland 81, the liquid adhesive was poured within a ∼1mm thick PDMS spacer on a glass slide and covered by the photomask coated with a thin layer of PDMS. After a short UV exposure, the photomask and the PDMS spacer were removed and the glass slide with the patterned adhesive was immersed in acetone to dissolve the uncrosslinked adhesive residuals. The slide was dried with nitrogen, exposed to UV to solidify the adhesive and baked overnight at 50°C. Dip-coating of Novec™ EGC-1700 reagent (3M, St. Paul, MN) was utilized to prevent PDMS adhesion. The dimensions of the fabricated master templates were visualized and measured by a 3D optical profiler (ZYGO, Middlefield, CT). The PDMS prepolymer mixture was degassed, poured onto the master templates and baked overnight at 80°C. The polymerized PDMS was carefully peeled off the master template to yield a replica of the tissue mold. The dimensions, shape, and orientation of the PDMS posts within the mold were varied by the use of different photomasks to microfabricate the corresponding masters. A PDMS mold without the posts and otherwise with identical dimensions was used for the fabrication of non-porous tissue sheets.
Figure 1. Engineering of porous skeletal muscle tissue networks.
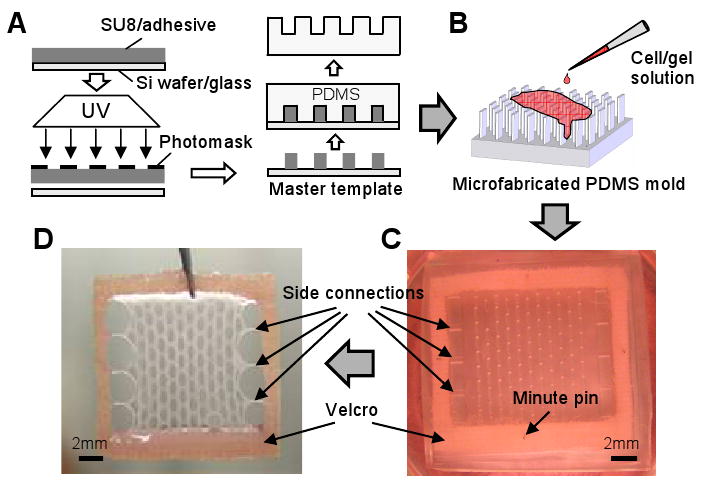
(A) Microfabrication of PDMS molds using soft lithography. (B) Injection of cell/hydrogel mixture into the PDMS mold. (C) Polymerized cell/gel mixture within a PDMS mold submerged in culture medium. The gels are anchored to a pinned Velcro® frame. (D) A representative tissue network shown after removal from the PDMS mold. The tissue network is attached fully on two opposite sides and via 3 side connections on perpendicular sides to the Velcro frame and readily transferred using a pair of forceps.
2.2 Culture of C2C12 myoblasts
Murine C2C12 myoblasts (< 4 passages; American Type Culture Collection, Manassas, VA) were maintained at below 70% confluence in growth medium containing DMEM (American Type Culture Collection), 10% fetal bovine serum (Gibco, Carlsbad, CA) and 1% penicillin-streptomycin (Gibco).
2.3 Isolation of primary skeletal myoblasts
Primary rat skeletal myoblasts (rSKM) were isolated as described by De Deyne [20]. Briefly, muscle tissue from the lower hindlimbs of 2∼3 day old Sprague-Dawley rats was dissected, separated from bones and connective tissue, minced into a coarse slurry, pooled in a 60mm dish and treated with 1mg/ml collagenase (Worthington, Lakewood, NJ) in Wyles solution (137mM NaCl, 5mM KCl, 21mM HEPES, 0.7mM Na2HPO4, 100mM glucose, and 0.1 mg/ml BSA) for 2 hrs at 37°C. The slurry was passed through a 40μm strainer (BD), centrifuged, resuspended in growth medium (DMEM (Gibco),10% fetal bovine serum, 0.5% penicillin-streptomycin and 0.1% gentamicin (Gibco)) and preplated for 30 min at 37°C to reduce the portion of fibroblasts and obtain an enriched myoblast population. The average cell yield was 12.8±3.4 million cells/pup. Myoblasts identified as MyoD positive cells comprised 79.9±8.0% of the total cell population. The freshly isolated cells were either plated on coverslips at a density of 0.6×106 cells/cm2 for monolayer cultures or mixed with hydrogel for micromolding experiments. All experiments involving animals were conformed to the protocols in the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, Revised 1996) and the animal protocol was approved by Duke Animal Care and Use Committee.
2.4 Micromolding of cell/hydrogel networks
The PDMS molds were treated with air plasma (March Instruments, Concord, CA) to increase the surface hydrophilicity, sterilized with ethanol, coated with 0.2% (w/v) pluronic F-127 (Molecular probes, Eugene, OR) to prevent gel adhesion, rinsed and dried with nitrogen. The cell/gel mixture consisted of different volume ratios of fibrinogen (2mg/ml) (Sigma) and collagen type I (1mg/ml) (BD, Franklin Lakes, NJ) [11]. Polymerization in gels containing fibrinogen was initiated by the addition of 0.4U thrombin (Sigma) per mg of fibrinogen. NaOH was used to adjust the pH in collagen containing gels. Matrigel (BD) was added to all gels at 10% (v/v) to support faster cell spreading, Cells were added to the cold hydrogel solution to yield final concentrations of 5×106 cells/ml for C2C12 myoblasts and 10×106 cells/ml for rSKMs.
The 100-600μl of cell/gel mixture was injected into the PDMS molds and allowed to gel in the incubator for 1hr (Figure 1.B). A rectangular Velcro® frame pinned to the PDMS mold provided an anchor for the gels which were fully attached to the frame in one direction and via three thin connections in the perpendicular direction (Figure 1.C and D). The frame served not only to facilitate the handling and transfer of the gel networks during analysis but also to provide additional alignment cues by exerting uniaxial passive tension on the cells [12]. After 3 days, growth medium in the monolayers and in the cell/hydrogel cultures was switched to a differentiation medium (DMEM, 3% horse serum (Hyclone, Logan, UT), 0.5% penicillin-streptomycin and 0.1% gentamicin) to induce myotube formation. For fibrin containing gels, medium was supplemented with 1mg/ml ε-aminocaproic acid (Sigma) to prevent fibrin degradation by serum plasmin.
2.5 Analysis of Gel Compaction
Two hundred μL of gel solution (with collagen:fibrin volume ratios of 1:0, 3:1, 1:1, 1:3, or 0:1) containing 5×106 Cell Tracker Green (Molecular probes) labeled C2C12 myoblasts per ml of gel were allowed to polymerize inside the wells of a 24-well plate previously coated with 0.2% pluronic F-127 and rinsed with PBS. The diameters of the resulting free-floating discs (relative to the well size) were measured daily starting from culture day 0 (7hrs after gelation) to day 10 using a fluorescence microscope (Eclipse TE 2000E, Nikon, Melville, NY), a cooled CCD camera (Sensicam QE, Cooke Corp., Auburn Hills, MI) and IPLab software (BD Biosciences, Rockville, MD). Note that throughout the text, these gels are referred to as either “pure” (1:0, 0:1) or collagen/fibrin composites, with understanding that 10% v/v matrigel was also included in all of the pure and composite gels.
2.6 Morphometric Assessment of Tissue Networks
Three-dimensional video-rate optical coherence tomography (OCT) [21] was used to non-invasively acquire volume images of the same tissue network on culture days 1, 4, 6, 9, 11 and 15. Tissue thickness was measured by averaging multiple OCT cross-sections of the sample using ImageJ software (NIH, Bethesda, MD). The obtained values were expressed relative to the height of the PDMS mold to quantify the decrease in tissue thickness with culture time due to cell-mediated gel compaction. The thicknesses of the tissue networks fabricated using 0.8mm, 1.2mm and 1.8mm tall PDMS molds were compared on culture day 15 (12 days after the onset of differentiation).
The maximum length and width of individual network pores and the minimum width of the tissue bundles were also measured on culture day 9 (6 days after the onset of differentiation) using phase contrast microscopy and ImageJ software. Because the entire tissue network is composed of repeating identical rectangular subunits (Figure 2.A1), the overall tissue porosity was determined by averaging the ratio of the acellular area to the total area in 3 representative subunits of each tissue network. The dependence of the resulting tissue network pore dimensions and the total porosity as a function of the length of the hexagonal PDMS posts was studied for three different network types fabricated using post lengths of 1120μm, 1330μm and 1613μm. Obtained images were analyzed using Matlab (Mathworks, Natick, MA) and measured values were expressed relative to the initial values prior to gel compaction.
Figure 2. Fabrication of PDMS molds with different dimensions.
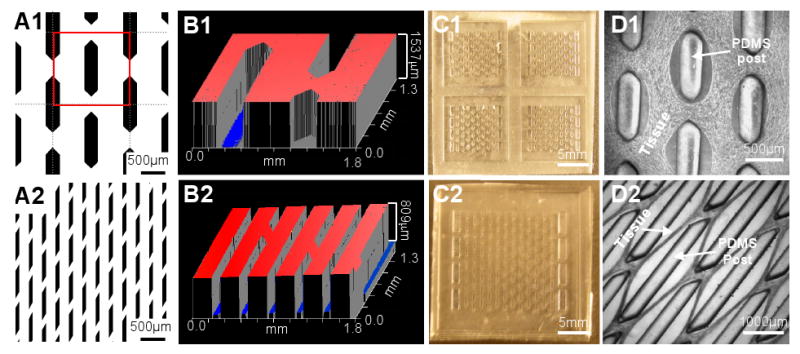
(A1-2) Examples of photomasks with different post (black) shapes and sizes. The red frame in A1 indicates one of the repeating subunits used for morphometric analyses. (B1-2) Examples of optical profiles (ZYGO) of master templates with different thicknesses. (C1-2) Representative PDMS molds of different sizes (C1, four 7×7mm molds; C2, a 14×14mm mold). (D1-2) The resulting tissue network morphology depends on the post size and shape.
2.7 Quantification of cell alignment
The degree of cell alignment was quantified from phase contrast microscopic images taken by a CCD camera (Sensicam QE) at 7× magnification using an image intensity gradient algorithm adapted from Karlon et al. [22]. Local cell orientation vectors were obtained in each 50μm×50μm subregion and the deviation of the vector angles from the mean angle of all the vectors was used as a measure of the degree of cell alignment (with 0° and 45° deviations respectively representing perfect and random alignments). Cell alignments in the repeating rectangular subunits of the tissue networks (i.e. around the tissue pores) were measured from composite microscopic images and averaged to obtain the global cell alignment within the network on culture day 13 (10 days after the onset of differentiation). Average alignment in the network bundles formed between the tissue pores was obtained by analyzing 4 different bundles per tissue network. The results were compared with those obtained in the non-porous tissue sheets on the same culture day.
2.8 Cell viability
The 5 day old tissue networks and non-porous sheets made of mouse C2C12 myoblasts were washed with PBS and incubated with agitation for 30min at 37°C in DMEM medium containing 5μM SYTO13 green nuclear stain (Molecular probes) to label all cells and 4μM ethidium homodimer-1 (EthD-1, Molecular probes) red nuclear stain to label dead cells. Confocal microscopic images of the stained cell nuclei at a tissue depth of 40μm were analyzed using a nuclei counting algorithm written in Matlab to determine the cell viability (i.e., (total number - number of dead)/total number of cells).
2.9 Histological assessment
For conventional histology, tissue networks were fixed in formalin at 4°C overnight, dehydrated in ethanol, embedded in paraffin, sectioned into 10μm sections and stained with hematoxylin and eosin (H&E) [23]. Alternatively, tissue networks were fixed with 4% formaldehyde for 20 min at room temperature with agitation, soaked in sucrose solutions of increasing concentration, embedded in O.C.T. medium (Tissue-Tek) on dry ice, cryosectioned into 20μm sections and immunostained with FITC-conjugated phalloidin (Sigma) and DAPI (Sigma).
2.10 Immunostaining
Cells were fixed with 4% formaldehyde for 10 min (monolayers) or 20 min with agitation (tissue networks and sheets) at room temperature, permeabilized with 0.5% Triton-X, blocked with 20% chicken serum in 1% bovine serum albumin (BSA) and incubated with primary antibodies (sarcomeric α-actinin, Sigma; MyoD, BD; myogenin, SantaCruz Biotech, Santa Cruz, CA) overnight at 4°C. Secondary antibodies (Alexa488 and Alexa594, Molecular Probes) were then applied together with a nuclear dye (DAPI or PI) or FITC-conjugated phalloidin for 1hr (monolayers) or 2∼3 hrs with agitation (tissue networks) at room temperature [23]. Images were acquired using a fluorescence microscope (Eclipse TE 2000E) or a Zeiss confocal microscope (LSM510, Carl Zeiss MicroImaging Inc., Thornwood, NY).
2.11 Statistics
Data are expressed as mean±SD. Statistical significance was determined by student t test between two groups and one-way ANOVA test among three and more groups. Differences were considered to be significant when p <0.05. Correlation was assessed using linear regression analysis.
3. Results
3.1 Mold fabrication
We compared two microfabrication methods to create the master templates, i.e., standard photolithography with SU8 photoresist and rapid photopatterning with Norland 81 adhesive. Standard photolithography required a longer processing time (>30hrs) but provided feature heights of up to 2mm with feature dimensions down to 200μm (i.e., up to a 10:1 aspect ratios), compared with rapid photopatterning, which had processing times of less than 12hrs but yielded feature heights of up to 1mm with sizes down to only 400μm (i.e., ∼2.5:1 ratios). The silanized SU8 photoresist templates could also be repeatedly used to cast PDMS molds (tested >5 times) while solidified Norland 81 adhesive patterns were limited to 1-2 times of usage because of the damage to the master template caused by the stress encountered during removal of the molded PDMS. For both methods, the obtained PDMS molds could be reused for several tissue cultures.
The PDMS molds were designed to contain an array of staggered PDMS posts positioned in the centers of the repeating rectangular subunits (Figure 2.A1). The shape and size of the posts and interpost distances could be systematically varied by modifying the design of the photomask (Figure 2.A1&2) and the resulting master templates (Figure 2.B1&2). The height of the photoresist determined the height of the posts and could be varied within a given range of aspect ratios (up to 10:1) while maintaining the high accuracy of fabrication (i.e., sharp features, Figure 2.B1&2). The overall size of the PDMS molds (Figure 2.C1&2) determined the size of the tissue constructs. Different initial dimensions of the microfabricated PDMS posts combined with cell-mediated gel compaction were used to obtain a variety of the tissue network topologies (Figure 2.D1&2).
3.2 Cell mediated gel compaction
To understand the factors that control the network geometry and cell alignment, we first undertook a series of measurements to characterize skeletal myoblast-mediated compaction of two naturally derived hydrogels, collagen and fibrin, as they have been previously reported to support high viability and spreading of skeletal myoblasts [4, 5]. A simple gel disk assay (Figure 3.A1) was conducted using C2C12 myoblasts to characterize the time course of the cell/gel compaction and examine the influence of gel composition (i.e., the mixing ratio of collagen and fibrin gels) on the degree of compaction. The compaction largely occurred in the first 2 days after gelation while the disk diameter remained unchanged after day 6 (Figure 3.A2). The “pure” collagen gel compacted significantly less than the “pure” fibrin gel (69.5±3.7% vs. 80.5±2.1% decrease in the disc diameter after 10 days, n = 3). The composite gels compacted to a similar level independent of the fraction of fibrin (collagen:fibrin 3:1, 77.5±0.4%; 1:1, 77.3±1.1%; 1:3, 81.5±2.2% decrease in the disc diameter after 10 days, n = 3 for each group). The increased compaction of fibrin containing disks appeared to occur due to the increased volume loss immediately upon gelation (Figure 3.A2).
Figure 3. Skeletal myoblast-mediated gel compaction.
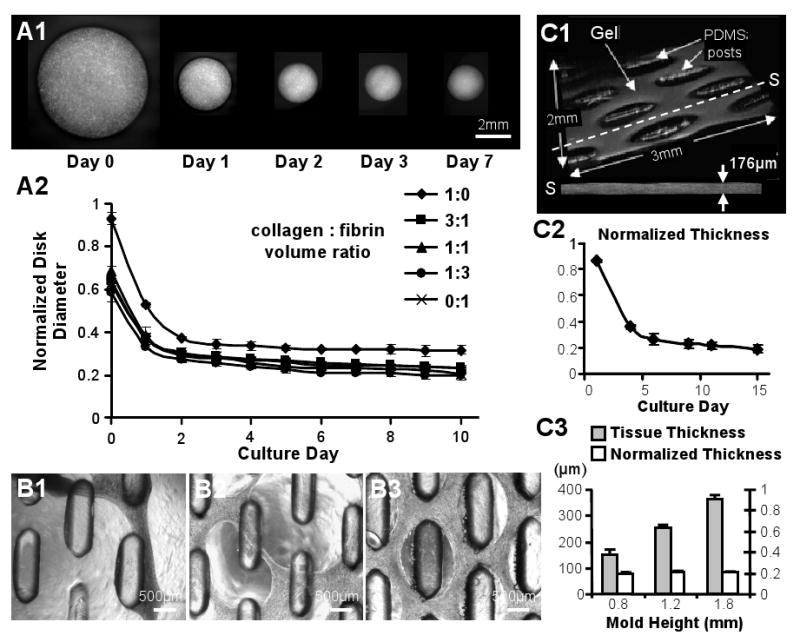
(A1) Serial images of gel disks with C2C12 myoblasts. (A2) Time course of compaction for free-floating C2C12 gel disks made with different ratios of collagen and fibrin. Disk diameter is normalized to that before gelation. (B1-3) Primary rSKM tissue networks made of (B1) “pure” collagen gel after 3 days of differentiation, (B2) 1:1 collagen:fibrin composite gel after 10 days of differentiation, and (B3) “pure” fibrin gel after 17 days of differentiation. Note that only rSKM/fibrin gel networks remained intact during long-term culture. (C1) Example of the volume OCT images used for non-invasive monitoring of tissue thickness. The dashed line indicates the position of a cross-sectional slice (S) with an average thickness of 176μm. (C2) Thickness of the rSKM/fibrin tissue networks as a function of culture time, normalized by the height of the PDMS mold. (C3) Final thicknesses of the 15 day old networks made using PDMS molds with different heights (and other dimensions the same). Note the linear dependence of the final tissue thickness on the mold height with a slope (average normalized thickness) of ∼0.2. Small error bars in C2 and C3 indicate highly reproducible tissue network geometry.
The compaction of gels with different collagen:fibrin ratios was also tested in tissue networks made of the primary rSKMs. Interestingly, after the rSKM differentiation was initiated, tissue bundles in the networks containing collagen gel exhibited significant thinning and eventually ruptured. In particular, the rSKM tissue networks made of “pure” collagen gel ruptured as early as 3-4 days after switching to the differentiation medium (Figure 3.B1), concomitant with the myotube fusion and the onset of spontaneous contractions, while the networks made of composite gel with a 1:1 collagen:fibrin ratio ruptured on differentiation days 10-11 (Figure 3.B2) when the tissue contractions were the most vigorous. In contrast, tissue networks composed of only sporadically contracting C2C12 myotubes in collagen containing gels remained intact even after 14 days of differentiation. Importantly, despite significant thinning of the tissue bundles and continuous contractions, the tissue networks made of “pure” fibrin gel remained intact during the entire course of study, i.e., up to 17 days of differentiation (Figure 3.B3), and therefore, the fibrin based tissue constructs were used for all the remaining experiments.
Based on non-invasive 3D OCT measurements (Figure 3.C1), the final decrease in the thickness of rSKM fibrin gel networks was found to be similar to that of the diameter of the C2C12 myoblast disks (compare Figure 3.C2 and 3.A2). Simultaneously, the time course of gel compaction for the primary rSKMs was slower than for C2C12 myoblasts, possibly due to a longer time required by the rSKMs to recover from enzymatic isolation. By varying the height of the PDMS molds, the final thickness of the formed tissue networks was varied in a linear fashion (Figure 3.C3), from 153.0±17.1μm for 0.8 mm tall molds (n = 6) and 255.6±11.1μm for 1.2 mm molds (n = 6) to 366.1±12.2μm for 1.8 mm molds (n = 4). The final percent reduction of tissue thickness due to gel compaction remained consistent for different mold heights averaging to 79.8±1.3% after 12 days of differentiation (Figure 3.C3).
3.3 Control of pore dimensions and overall tissue porosity
The initial pores in the skeletal muscle tissue networks were created by gel formation around the PDMS posts. Subsequently, the pore area was gradually enlarged during the process of cell-mediated gel compaction. The spatial distribution and dimensions of the microfabricated posts directly determined the shape and size of the resulting pores. Specifically, the elongated hexagonal shape and the staggered arrangement of PDMS posts directed the gel anchoring against the longitudinal post ends during the process of gel compaction, yielding the formation of symmetric elliptical pores with lengths equal to the post length (Figure 4.A1-3). Simultaneously, as gel compacted, the width of the pores and the width of the tissue bundles formed between the pores respectively increased and decreased relative to the values before gel compaction (set by the mold dimensions). Quantitatively, for increasing post lengths of 1120±20μm, 1330±13μm, and 1613±17μm (n = 5 for each group) and initial pore and bundle width of 418±30μm and 402±17μm, respectively, the resulting pore widths in 9 day old networks were found to be 1.34±0.02, 1.71±0.07, and 2.07±0.07 times larger than the initial pore width,(Figure 4.B1) while the resulting tissue bundle widths were 1.21±0.05, 1.30±0.03, and 1.49±0.04 times smaller than the initial bundle width (Figure 4.B2). As a result, longer posts also yielded increased tissue porosity (percent of void area within the network) relative to that before the gel compaction (Figure 4.B3). Notably, the length of the posts appeared linearly related to gel compaction-induced changes in pore and bundle width as well as tissue porosity (Figure 4.B1-3). This simple relationship was not found for the quadrangular posts (Figure 2.A2). Therefore, controlling the dimensions of the hexagonal posts (at least within the studied limits) allowed predictable and independent control of both the height and the porosity of the resulting tissue networks.
Figure 4. Effect of PDMS post length on the skeletal muscle tissue network dimensions and porosity.
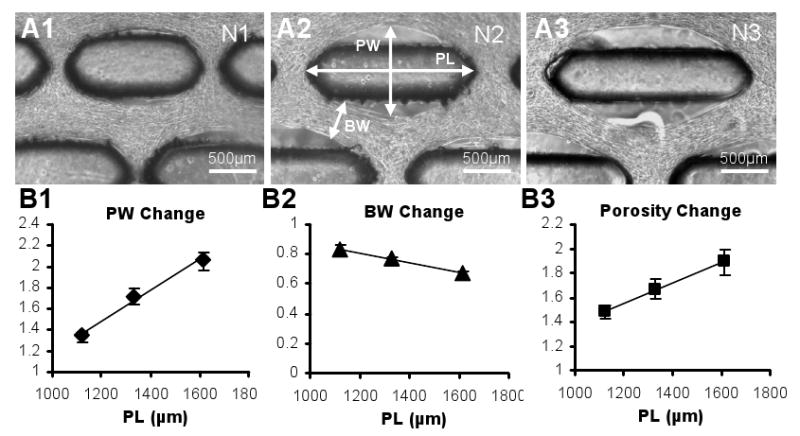
(A1-3) Representative phase contrast images of three tissue networks (N1-3) after 9 days of culture, fabricated using molds with post lengths of 1120μm (N1), 1330μm (N2), and 1613μm (N3). PL, maximum pore length; PW, maximum pore width; BW, minimum tissue bundle width. (B1-3) Change of PW, BW and porosity (void/total network area) relative to the respective values before gel compaction as a function of PL. For each PL, the shown relative value represents an average of individual relative values from all networks in the group. Note that due to gel anchoring at the longitudinal ends of the posts, the resulting PL is equal to the post length. Small error bars in B1-3 indicate the high reproducibility of the tissue network formation. Data for different PLs differ significantly. For all linear fits, R2>0.95.
3.4 Uniformity of cell alignment in tissue networks
The use of elongated hexagonal posts to control the spatial pattern of gel compaction not only allowed the precise control of the pore dimensions, but also served to locally guide the cell alignment within the resulting tissue bundles between the pores. Considering that tissue networks consisted of repeating rectangular subunits with similar tissue structure (Figure 2.A1), we compared the global cell alignments (Figure 5.A1-3) in networks, as well as the alignments within the tissue bundles (Figure 5.C1-2), by studying representative network subunits. For all the three studied post lengths, the mean (global) cell alignment in the network was directed along the long axis of the posts (Figure 5.A1-3). The deviations from the global alignment (22.8°±1.1°, 28.8°±1.9° and 25.3°±0.6°, Figure 5.B1) but not the deviations within tissue bundles (12.5°±3.5°, 12.1±3.9° and 11.8±3.7°, Figure 5.C3) significantly differed for the three post lengths, suggesting pore shape as one of the factors that determined the cell alignment in the network. In particular, the cells were expected to better align along more elongated pores and at locations closer to the pore boundary. Therefore, the deviation from the unidirectional cell alignment in a subunit was correlated with pore elongation (i.e., pore length divided by pore width) normalized by the tissue area in the subunit (Figure 5.B2). A linear correlation (r = 0.87) was found for a range of cell alignments suggesting that normalized pore elongation could be used as a geometrical parameter predictive of global cell alignment within the network. Finally, both the global and bundle cell alignments in engineered tissue networks were superior to virtually random cell orientation found in non-porous tissue sheets (deviation of ∼40°, Figure 5.B1 and 5.C3).
Figure 5. Cell alignment in skeletal muscle tissue networks as a function of PDMS post length.
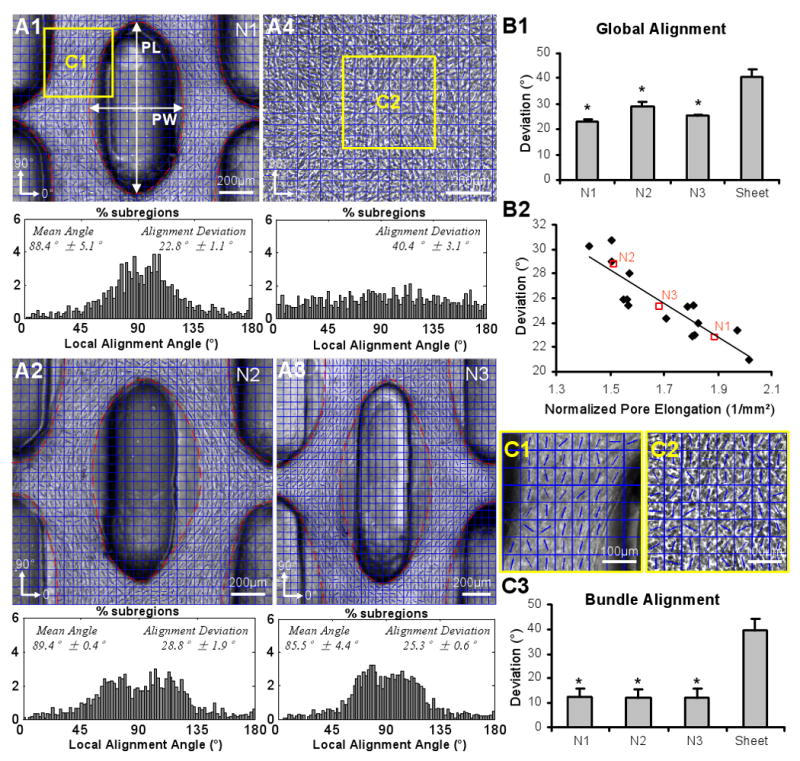
(A1-4) Phase contrast images of the repeating subunits for three types of tissue networks (N1-3) described in Figure 4 and a non-porous tissue sheet (A4). Blue grids denote square tissue subregions used to construct local cell alignment maps. Direction of cell alignment in each subregion is denoted by a blue line. Corresponding average histograms of local alignment angles with mean and standard deviation (alignment deviation) are shown underneath the phase contrast images. 0° and 180° denote the horizontal direction while 90° denotes the vertical direction. Note bimodal shape of angle distribution in tissue networks due to the divergence and convergence of the muscle fibers around the vertically oriented pore (i.e. around ∼90°). (B1) Global cell alignment (average alignment deviation) in tissue networks and non-porous tissue sheets. Small error bars reveal high reproducibility of the tissue network geometry. Deviations significantly differ among the three types of tissue networks (N1-3). *, significantly different from the tissue sheets. (B2) Linear correlation (r = 0.87) between global alignment and pore elongation (PL/PW in A1) normalized by tissue area in the subunit (outlined by red dashed lines in A1). Open red squares denote the mean deviations for each network type (N1-3). (C1-2) Representative cell alignment angle maps in tissue bundles (C1) and non-porous tissue sheets (C2) from areas denoted by yellow frames in A1&4. (C3) Cells in the bundles are highly aligned compared to those in the sheets. *, significantly different from the tissue sheets.
3.5 Cell viability, distribution and differentiation in tissue networks
The microfabricated tissue pores were expected to promote cell viability within tissue constructs by improving the nutrient and oxygen transfer to the cells. The cell viability after 5 days of culture at a tissue depth of 40 μm was significantly higher in the porous tissue networks (94.3±1.7%) than in the corresponding non-porous tissue sheets (90.6±1.7%) fabricated using the molds of the same height (Figure 6). The spatially uniform cell seeding within the hydrogel and the initial gel compaction resulted in the even distribution and uniform alignment of cells throughout the tissue depth as early as 3 days after seeding (Figure 7.A1&2), when the cell differentiation was initiated using a low serum medium. With further gel compaction, cell density and alignment significantly increased through culture day 5 (Figure 7.B1). After 4-6 days of differentiation, the bundle and pore dimensions within the network were stabilized (Figure 7.C1) and the networks anchored to the Velcro frames could be transferred by forceps without any damage (Figure 1.D).
Figure 6. Cell viability in porous skeletal muscle tissue networks and non-porous tissue sheets.
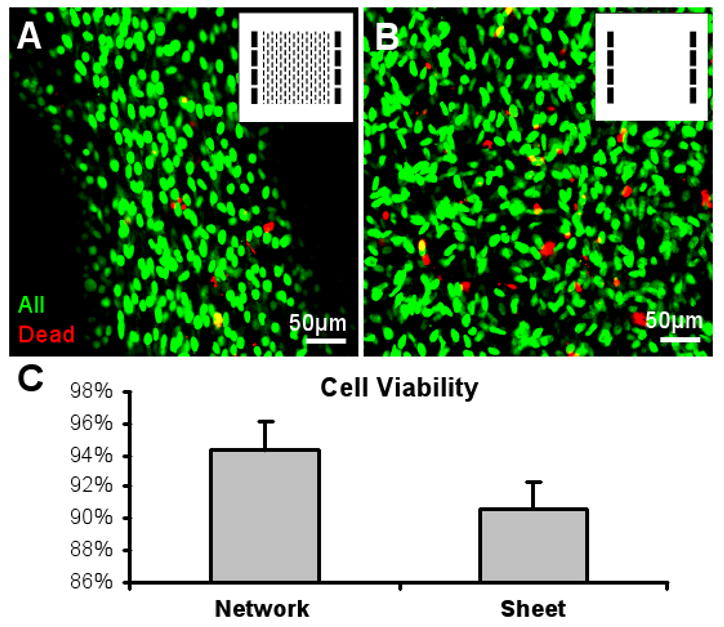
Representative live/dead images of a tissue network (A) and a tissue sheet (B) after 5 days of culture. Inserts, photomask patterns used for the fabrication of PDMS molds. The two molds had the same heights. Confocal images were taken at a tissue depth of 40 μm. (C) Cell viability is significantly higher in the porous networks than in the non-porous sheets (n = 5 per group).
Figure 7. Distribution and differentiation of skeletal muscle cells in engineered tissue networks.
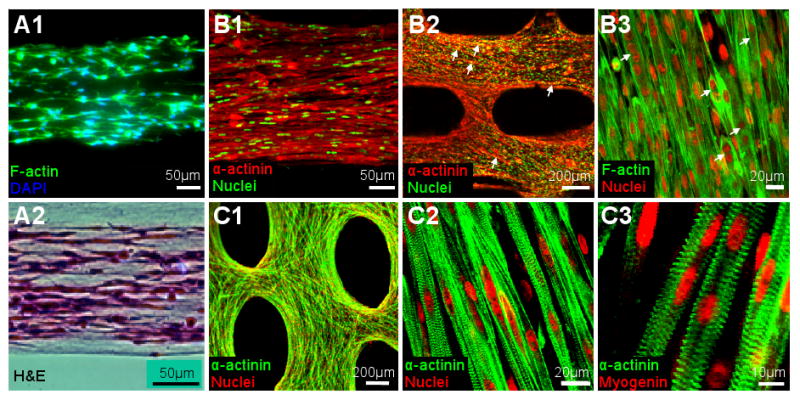
(A1-2) Longitudinal cross-sections of 3 day old tissue bundles showing evenly distributed cells throughout the tissue thickness. (B1-3) Representative immunofluorescence images revealing densely packed and highly aligned skeletal muscle cells in the tissue networks after 4-5 days of culture. Arrows in B2 and B3 denote the fusing myotubes. (C1-3) Representative immunofluorescence images showing the ubiquitous differentiation of primary rSKMs into striated myotubes after 10 days of culture. Virtually all fused nuclei were myogenin positive.
Fusion of myoblasts into myotubes was evident within 1-2 days after the onset of differentiation (Figure 7.B2&3) followed by the occurrence of spontaneous myotube contractions on differentiation day 4 (see supplemental movie). This spontaneous activity became more synchronized and stronger by differentiation day 10 and then slowly ceased by days 12-14. The onset of spontaneous contractions coincided with the occurrence of cross-striated myotubes that expressed myogenin (a regulatory transcription factor involved in the myogenesis). By differentiation day 7, almost all of the myogenin-positive nuclei resided in the cross-striated myotubes (Figure 7.C2&3). The percent of myotubes exhibiting cross-striations was already 88±5% at differentiation day 7 (estimated from multiple confocal sections) with no further increase observed between days 7 and 14 (not shown).
To quantify the fraction of cells that underwent fusion and myogenesis we counted the percent of myogenin-positive nuclei in the 2D monolayer cultures (Figure 8.A1), the non-porous tissue sheets (Figure 8.A2) and the porous tissue networks (Figure 8.A3) after 4 days of differentiation. The fraction of myogenin-positive nuclei was higher in the tissue networks (70.5±4.7% of the total nuclear count, n = 7) than in the tissue sheets (56.6±7.1%, n = 5) and monolayers (32.7±7.1%, n = 8) (Figure 8.B). Therefore, the 3D culture environment of the tissue networks and sheets prevented significant proliferation of non-myogenic cells compared to the 2D environment of the monolayers, yielding a substantially higher fraction of myogenin-positive cells. Furthermore, virtually all myoD-positive myoblasts (initially 80% of cells) in the tissue networks fused into myogenin-positive myotubes and underwent further maturation into aligned striated myofibers. In contrast, the same age non-porous 3D tissue sheets with randomly oriented cells still contained myoD-positive nuclei (data not shown) and exhibited lower percent of myogenin-positive cells (Figure 8.B), indicating that the local myoblast alignment, specific strain patterns, and/or improved mass transfer through the tissue pores may all have contributed to superior myoblast fusion and maturation within the network vs. sheet 3D environments.
Figure 8. Comparison of myogenesis in 2D monolayers, tissue sheets and tissue networks.
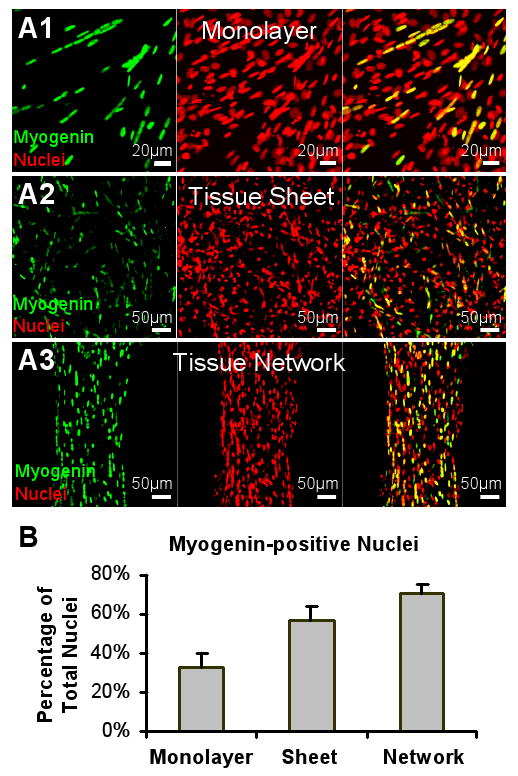
Representative images of myogenin-positive nuclei in a monolayer (A1), a non-porous tissue sheet (A2) and a porous tissue network (A3) after 4 days of differentiation. (B) The proportions of myogenin-positive nuclei significantly differ among the three groups.
3.6 Control of the regional myofiber alignment
The ability to precisely control local myofiber alignments within the relatively large engineered muscle tissues may allow for the more efficient structural and functional repair of complex muscle injuries (e.g. during aesthetic reconstructions). To demonstrate the ability to locally control myofiber directions in the tissue networks, we fabricated an abrupt change in the orientation of the hexagonal posts in adjacent regions of the PDMS mold ranging from 30° to 90° (Figure 9.A1-4). The tissue bundles that formed between the pores rapidly changed orientation within the 500μm wide border between the two regions. When traversing the border, long myotubes curved to adapt to changes in the pore and bundle orientation (Figure 9.B1&2). Conceivably, by controlling the shape and orientation of the individual posts within the mold, the local directions of the muscle bundles and myofibers could be arranged into customized tissue structures with potential to better “blend in” with the host muscle upon implantation.
Figure 9. Control of cell alignment in skeletal muscle tissue networks.

(A1-4) Three-dimensional OCT images showing the border between the two adjacent tissue network regions with a sharp change in pore orientation of 30° (A1), 45° (A2), 60° (A3) and 90° (A4). (B1) Immunofluorescence images of long curved myotubes (denoted by white arrows) traversing the border of the network shown in A4. B2 is the region denoted by yellow frame in B1.
4. Discussion
We developed a cell/hydrogel micromolding approach for the reproducible fabrication of relatively large skeletal muscle tissue networks made of viable, densely packed, highly aligned, cross-striated, and spontaneously contractile myofibers. The precise design of PDMS molds containing an array of staggered, pore-generating posts allowed us to systematically control engineered tissue size, thickness, and porosity. The presence of microfabricated tissue pores facilitated nutrient and oxygen transport within the relatively thick (127 - 384 μm) and metabolically active muscle constructs, thereby increasing the cell viability compared to that of non-porous muscle sheets and enabling the uniformly high cell density throughout the entire tissue volume. Simultaneously, the shape, distribution and orientation of the elongated PDMS posts governed the local cell-mediated gel compaction and enabled control of cell alignment over a relatively large area (0.5 - 2 cm2). The 3D muscle tissue network environment limited the proliferation of non-myogenic cells and promoted the myoblast fusion and formation of myogenin-positive myofibers compared to both 2D myoblast monolayers and 3D non-porous muscle sheets.
Rapid 3D prototyping techniques have been recently employed for the production of solid polymer scaffolds with controlled pore size and geometry [24]. However, these scaffolds are unsuitable for the engineering of dense and highly ordered muscle tissues due to: 1) the discontinuities in tissue structure created by the polymer scaffold and 2) the random structural and biochemical changes that would result from the polymer degradation. Alternatively, photopatterning and stereolithographic techniques have been used to directly fabricate soft tissue constructs in the form of reproducible, cell-laden hydrogel structures [25]. In particular, layer-by-layer assembly of photopatterned poly-ethylene glycol (PEG) hydrogels yielded the formation of functional 3D hepatic tissue constructs with complex internal architecture and controllable porosity [14]. Spatiotemporal 3D patterning of ligands, growth factors, extracellular matrix components and controlled release particles in microfabricated PEG hydrogels was also demonstrated using similar methods [26]. However, the ability of modified and unmodified PEG hydrogels to support cell proliferation, adhesion and spreading is, in general, inferior to the naturally derived gels, such as fibrin and collagen. Importantly, unlike fibrin and collagen gels, PEG hydrogel does not undergo significant cell-mediated compaction that allows for the: 1) high ultimate tissue density and 2) induction and control of cell alignment by applying specific geometrical constraints, both of which are required for the engineering of functional muscle tissues.
Therefore, for the fabrication of skeletal muscle tissue networks in this study, we relied on the ability of skeletal myoblasts to exhibit stress fibers and undergo alignment when collagen and fibrin gel compaction is restricted by immobilization at one or more surfaces or points [5, 13]. Theoretical models predict that this cell alignment arises from the imposed strains on the gel [27]. Compaction of the gel is critically dependent on the engagement of integrins to the cytoskeleton and contraction of the cytoskeleton. The stiffness of the 3D reconstituted tissue is dependent upon actin polymerization inside the cells [28] and the spatial rearrangement of gel fibers and has been shown to increase with gel compaction [29]. Following compaction, the mechanical strength increases further as collagen and other extracellular matrix proteins are synthesized by cells in a proper 3D orientation [30]. This phenomenon has been previously utilized to induce alignment of muscle cells in engineered blood vessels [30], skeletal and cardiac muscle bundles [5, 13, 31] and heart valves [32]. Nevertheless, as the specific geometrical constraints [12] were only applied at the outer boundary of the gels, these methods were limited to achieving uniform cell alignment in tissues with simple geometry (e.g., thin muscle bundles [5, 13] or rings [33]) or a rough control of bulk cell alignment within a relative large tissue area (e.g., heart valves [32]). In the current study, gel compaction between the boundaries of staggered hexagonal posts was utilized to create a large number of interconnected muscle bundles containing highly aligned cells. Importantly, control of the post geometry, orientation and distribution allowed for the precise control of the local myofiber alignment throughout the entire tissue area.
In the specific setting of the micromolded muscle tissue networks, the Velcro® frame was used to control (constrain) the total network area while enabling the unconstrained compaction of the tissue thickness. Unconstrained gel compaction to 20∼30% of initial gel dimensions exerted by the primary neonatal rat and mouse C2C12 skeletal myoblasts in this study (Figure 3.A2&C2) is similar to those previously shown for C2C12 myoblasts and human fetal and adult skeletal muscle cells [5, 13], but lower than the compactions induced by neonatal rat cardiomyocytes [31], rat aortic smooth muscle cells [11] or bovine chordal fibroblasts [34], confirming the dependence of the degree of gel compaction on the specific cell type. Furthermore, the higher skeletal myoblast-mediated compaction of fibrin versus collagen gels found in this study was consistent with previous findings for rat aortic smooth muscle cells [11]. In contrast to the unconstrained gel compaction (Figure 3.A1), where the initial tissue area decreased ∼25 times (∼5 times for each dimension), constrained gel compaction in the tissue network plane yielded only a 1.4-1.7 times decrease in the total gel area (i.e., a 1.5-1.9 times increase in the tissue porosity, Figure 4.B3). Notably, this increase in the tissue porosity and the decrease in the bundle width were linearly related to the length of the hexagonal posts (Figure 4.B2&3). While cell alignment within the tissue bundles was not affected by the post length, the overall (global) cell alignment in the tissue network could be predicted by measuring a geometrical parameter related to the pore length-to-width ratio (elongation) per tissue area (Figure 5.B2). Taken together, the described micromolding approach allowed the independent control of the engineered muscle thickness, porosity, and cell alignment in a predictable and reproducible fashion, by controlling the thickness of the PDMS mold, the length and width of the hexagonal post and the area of the tissue network subunit (Figure 2.A1).
An interesting finding of this study was that the vigorous contractions of differentiated skeletal myofibers resulted in the rupture of collagen but not fibrin gel based tissue networks. Higher mechanical stability of the contracting cell-laden fibrin gels in this study may stem from the improved endogenous synthesis of extracellular matrix proteins (e.g. elastin, collagen I) that was previously shown to occur in smooth muscle cells and dermal fibroblasts embedded in fibrin compared with collagen gels [35, 36]. In addition, fibrin fibers have been shown to exhibit two orders of magnitude lower stiffness (Young's modulus, ∼15 MPa) than collagen fibrils (2 - 5 GPa) [37, 38] and, similarly, pure fibrin gel based smooth muscle tissue constructs were found to be more compliant than those made of a mixture of collagen and fibrin gels or collagen gel alone [11]. Therefore, the relatively thin network bundles with aligned, densely packed and spontaneously contractile myofibers may be less prone to rupture when made of more elastic fibrin gel than stiffer collagen gel. However, it is also possible that the lower myofiber density and/or larger bundle diameter, previously used for the engineering of single collagen gel based muscle bundles, would yield lower contractile forces and therefore allow for the long-term culture of intact muscle tissue networks made of the collagen gel.
The potential of bioartificial skeletal muscle to generate contractile forces comparable to those of the native muscle is critically dependent on the ability to engineer dense, uniformly aligned, and differentiated myofibers throughout the entire tissue volume. The superior force generating capability of the muscle tissue networks is expected to arise from high myoblast density and alignment that both mimic native tissue structure and facilitate muscle fusion and maturation (Figures 7&8). On the other hand, the necessity to introduce pores (i.e., space void of muscle fibers) within the engineered muscle in order to facilitate metabolic supply to the embedded cells and fabricate thicker tissue constructs is expected to adversely affect the tissue tensile strength and generated contractile force. Therefore, in order to maximize the force generating capability of the engineered skeletal muscle tissue networks, the tissue porosity should be minimized under conditions of high cell viability, density and alignment. This goal could be achieved by creating PDMS molds with tall and thin posts via the use of high aspect ratio microfabrication. While our current ability to fabricate relatively thick (several hundreds of microns to millimeters) features with 10:1 aspect ratio is already approaching the limits of the UV photolithography, other techniques such as X-ray lithography [39] and high resolution stereolithography may offer room for further improvements. In addition, minimizing the deviation from the global and local unidirectional myofiber alignment within the engineered muscle tissue is expected to further augment the generated contractile force (in the direction of the cell alignment). While it is apparent that the elongated pore shape and uniform pore distribution are needed to induce cell alignment throughout the tissue network, a particular post geometry (e.g., hexagon, rectangle, diamond) and subunit size that would yield maximum local and global cell alignment are still to be determined. Future computational and experimental studies are thus warranted to investigate the role of muscle tissue network topology in the generation of contractile force. Finally, stacking the multiple networks (either in vitro or during implantation), while maintaining the tissue porosity and viability, may further augment the force generation capacity of the engineered muscle tissue networks.
It is important to note that similar to other skeletal and cardiac muscle tissue engineering approaches with hydrogel scaffolds [5, 13, 33], Matrigel was also used in this study to promote muscle cell spreading and differentiation. It is well recognized, however, that Matrigel can not be used in human studies due to its murine origin and potential tumorigenicity. Instead, a defined combination of extracellular matrix proteins and/or growth factors similar to those identified for engineered cardiac tissues [40], or a reconstituted human derived basement membrane matrix [41] will need to be developed to enable potential translation of this and similar approaches to clinics. Furthermore, a Velcro frame was used in this study (and studies by others [5, 13, 31]) to anchor the cell/gel mixture and, by acting as a surrogate for the native tendon, facilitated the generation of passive tension and cell alignment. Although maintaining the tension of engineered muscle for a certain period after implantation will likely be necessary to prevent its atrophy, Velcro will not be used for this purpose because of its potential to cause tissue inflammation. Instead, tissue engineered tendons or segments of native tendon [42] or porous anchors made of biocompatible materials (e.g. polyurethane, polycaprolactone, BioVyon [43], etc.) will be used to facilitate the surgical attachment of the engineered muscle to the host tendon or bone.
5. Conclusion
This study describes a cell/hydrogel micromolding approach to fabricate porous skeletal muscle tissue networks composed of densely packed, uniformly aligned and highly differentiated muscle fibers. Specifically, control of the size and elongation of microfabricated network pores in this system enabled independent control of the tissue porosity (i.e., metabolic supply) and 3D cell alignment. The reproducibility and versatility of the proposed approach rely on the use of CAD techniques to generate arbitrary photomask drawings and in turn yield the fabrication of tissue-specific PDMS molds, as well as the ability to repeatedly cast and reuse different PDMS molds to generate muscle tissues with highly reproducible and controllable 3D architecture. In the future, the versatility of the described methodology is expected to enable the engineering of customized functional skeletal muscle tissue substitutes for the reconstruction of the damaged muscle in a variety of pathological conditions.
Supplementary Material
Acknowledgments
We are grateful to Dr. Joseph Izatt and Hansford Hendargo for their assistance with OCT imaging, Dr. George Truskey and Caroline Rhim for their assistance with C2C12 culture, and Brian Liau for his assistance with high aspect microfabrication of PDMS molds. This work is supported by NIH grants HL080469 from National Heart Lung and Blood Institute and AR055226 from National Institute of Arthritis and Musculoskeletal and Skin Diseases to N.B.
Footnotes
Supplemental Data: Movie legend: A real-time movie showing the spontaneous macroscopic contraction of a skeletal muscle tissue network after 5 days of differentiation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mouly V, Aamiri A, Perie S, Mamchaoui K, Barani A, Bigot A, et al. Myoblast transfer therapy: is there any light at the end of the tunnel? Acta Myol. 2005;24(2):128–133. [PubMed] [Google Scholar]
- 2.Bach AD, Beier JP, Stern-Staeter J, Horch RE. Skeletal muscle tissue engineering. J Cell Mol Med. 2004;8(4):413–422. doi: 10.1111/j.1582-4934.2004.tb00466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kondoh H, Sawa Y, Miyagawa S, Sakakida-Kitagawa S, Memon IA, Kawaguchi N, et al. Longer preservation of cardiac performance by sheet-shaped myoblast implantation in dilated cardiomyopathic hamsters. Cardiovasc Res. 2006;69(2):466–475. doi: 10.1016/j.cardiores.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Huang YC, Dennis RG, Larkin L, Baar K. Rapid formation of functional muscle in vitro using fibrin gels. J Appl Physiol. 2005;98(2):706–713. doi: 10.1152/japplphysiol.00273.2004. [DOI] [PubMed] [Google Scholar]
- 5.Powell CA, Smiley BL, Mills J, Vandenburgh HH. Mechanical stimulation improves tissue-engineered human skeletal muscle. Am J Physiol Cell Physiol. 2002;283(5):C1557–1565. doi: 10.1152/ajpcell.00595.2001. [DOI] [PubMed] [Google Scholar]
- 6.Yan W, George S, Fotadar U, Tyhovych N, Kamer A, Yost MJ, et al. Tissue engineering of skeletal muscle. Tissue Eng. 2007;13(11):2781–2790. doi: 10.1089/ten.2006.0408. [DOI] [PubMed] [Google Scholar]
- 7.Kroehne V, Heschel I, Schugner F, Lasrich D, Bartsch JW, Jockusch H. Use of a novel collagen matrix with oriented pore structure for muscle cell differentiation in cell culture and in grafts. J Cell Mol Med. 2008;12:1582–1838. doi: 10.1111/j.1582-4934.2008.00238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis BH, Schroeder T, Yarmolenko PS, Guilak F, Dewhirst MW, Taylor DA. An in vitro system to evaluate the effects of ischemia on survival of cells used for cell therapy. Ann Biomed Eng. 2007;35(8):1414–1424. doi: 10.1007/s10439-007-9301-2. [DOI] [PubMed] [Google Scholar]
- 9.Bursac N, Loo Y, Leong K, Tung L. Novel anisotropic engineered cardiac tissues: studies of electrical propagation. Biochem Biophys Res Commun. 2007;361(4):847–853. doi: 10.1016/j.bbrc.2007.07.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi Y, Rittman L, Vesely I. Novel geometries for tissue-engineered tendonous collagen constructs. Tissue Eng. 2006;12(9):2601–2609. doi: 10.1089/ten.2006.12.2601. [DOI] [PubMed] [Google Scholar]
- 11.Cummings CL, Gawlitta D, Nerem RM, Stegemann JP. Properties of engineered vascular constructs made from collagen, fibrin, and collagen-fibrin mixtures. Biomaterials. 2004;(17):3699–3706. doi: 10.1016/j.biomaterials.2003.10.073. [DOI] [PubMed] [Google Scholar]
- 12.Costa KD, Lee EJ, Holmes JW. Creating alignment and anisotropy in engineered heart tissue: role of boundary conditions in a model three-dimensional culture system. Tissue Eng. 2003;9(4):567–577. doi: 10.1089/107632703768247278. [DOI] [PubMed] [Google Scholar]
- 13.Rhim C, Lowell DA, Reedy MC, Slentz DH, Zhang SJ, Kraus WE, et al. Morphology and ultrastructure of differentiating three-dimensional mammalian skeletal muscle in a collagen gel. Muscle Nerve. 2007;36(1):71–80. doi: 10.1002/mus.20788. [DOI] [PubMed] [Google Scholar]
- 14.Liu Tsang V, Chen AA, Cho LM, Jadin KD, Sah RL, DeLong S, et al. Fabrication of 3D hepatic tissues by additive photopatterning of cellular hydrogels. FASEB J. 2007;21(3):790–801. doi: 10.1096/fj.06-7117com. [DOI] [PubMed] [Google Scholar]
- 15.Norman JJ, Desai TA. Control of cellular organization in three dimensions using a microfabricated polydimethylsiloxane-collagen composite tissue scaffold. Tissue Eng. 2005;11(34):378–386. doi: 10.1089/ten.2005.11.378. [DOI] [PubMed] [Google Scholar]
- 16.Chrobak KM, Potter DR, Tien J. Formation of perfused, functional microvascular tubes in vitro. Microvasc Res. 2006;71(3):185–196. doi: 10.1016/j.mvr.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 17.Fedorovich NE, Alblas J, de Wijn JR, Hennink WE, Verbout AJ, Dhert WJ. Hydrogels as extracellular matrices for skeletal tissue engineering: state-of-the-art and novel application in organ printing. Tissue Eng. 2007;13(8):1905–1925. doi: 10.1089/ten.2006.0175. [DOI] [PubMed] [Google Scholar]
- 18.Bryant SJ, Cuy JL, Hauch KD, Ratner BD. Photo-patterning of porous hydrogels for tissue engineering. Biomaterials. 2007;28(19):2978–2986. doi: 10.1016/j.biomaterials.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harrison C, Cabral JT, Stafford CM, Karim A, Amis EJ. A rapid prototyping technique for the fabrication of solvent-resistant structures. J Micromech Microeng. 2004;14:153–158. [Google Scholar]
- 20.De Deyne PG. Formation of sarcomeres in developing myotubes: role of mechanical stretch and contractile activation. Am J Physiol Cell Physiol. 2000;279(6):C1801–1811. doi: 10.1152/ajpcell.2000.279.6.C1801. [DOI] [PubMed] [Google Scholar]
- 21.Yelbuz TM, Choma MA, Thrane L, Kirby ML, Izatt JA. Optical coherence tomography: a new high-resolution imaging technology to study cardiac development in chick embryos. Circulation. 2002;106(22):2771–2774. doi: 10.1161/01.cir.0000042672.51054.7b. [DOI] [PubMed] [Google Scholar]
- 22.Karlon WJ, Covell JW, McCulloch AD, Hunter JJ, Omens JH. Automated measurement of myofiber disarray in transgenic mice with ventricular expression of ras. Anat Rec. 1998;252(4):612–625. doi: 10.1002/(SICI)1097-0185(199812)252:4<612::AID-AR12>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 23.Bursac N, Papadaki M, White JA, Eisenberg SR, Vunjak-Novakovic G, Freed LE. Cultivation in rotating bioreactors promotes maintenance of cardiac myocyte electrophysiology and molecular properties. Tissue Eng. 2003;9(6):1243–1253. doi: 10.1089/10763270360728152. [DOI] [PubMed] [Google Scholar]
- 24.Zein I, Hutmacher DW, Tan KC, Teoh SH. Fused deposition modeling of novel scaffold architectures for tissue engineering applications. Biomaterials. 2002;23(4):1169–1185. doi: 10.1016/s0142-9612(01)00232-0. [DOI] [PubMed] [Google Scholar]
- 25.Du Y, Lo E, Ali S, Khademhosseini A. Directed assembly of cell-laden microgels for fabrication of 3D tissue constructs. Proc Natl Acad Sci U S A. 2008;105(28):9522–9527. doi: 10.1073/pnas.0801866105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mapili G, Lu Y, Chen S, Roy K. Laser-layered microfabrication of spatially patterned functionalized tissue-engineering scaffolds. J Biomed Mater Res B Appl Biomater. 2005;75(2):414–424. doi: 10.1002/jbm.b.30325. [DOI] [PubMed] [Google Scholar]
- 27.Ohsumi TK, Flaherty JE, Evans MC, Barocas VH. Three-dimensional simulation of anisotropic cell-driven collagen gel compaction. Biomech Model Mechanobiol. 2008;7(1):53–62. doi: 10.1007/s10237-007-0075-0. [DOI] [PubMed] [Google Scholar]
- 28.Wakatsuki T, Kolodney MS, Zahalak GI, Elson EL. Cell mechanics studied by a reconstituted model tissue. Biophys J. 2000;79(5):2353–2368. doi: 10.1016/S0006-3495(00)76481-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wille JJ, Elson EL, Okamoto RJ. Cellular and matrix mechanics of bioartificial tissues during continuous cyclic stretch. Ann Biomed Eng. 2006;34(11):1678–1690. doi: 10.1007/s10439-006-9153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Isenberg BC, Tranquillo RT. Long-term cyclic distention enhances the mechanical properties of collagen-based media-equivalents. Ann Biomed Eng. 2003;31(8):937–949. doi: 10.1114/1.1590662. [DOI] [PubMed] [Google Scholar]
- 31.Zimmermann WH, Fink C, Kralisch D, Remmers U, Weil J, Eschenhagen T. Three-dimensional engineered heart tissue from neonatal rat cardiac myocytes. Biotechnol Bioeng. 2000;68(1):106–114. [PubMed] [Google Scholar]
- 32.Robinson PS, Johnson SL, Evans MC, Barocas VH, Tranquillo RT. Functional Tissue-Engineered Valves from Cell-Remodeled Fibrin with Commissural Alignment of Cell-Produced Collagen. Tissue Eng Part A. 2007;14(1):83–95. doi: 10.1089/ten.a.2007.0148. [DOI] [PubMed] [Google Scholar]
- 33.Zimmermann WH, Schneiderbanger K, Schubert P, Didie M, Munzel F, Heubach JF, et al. Tissue engineering of a differentiated cardiac muscle construct. Circ Res. 2002;90(2):223–230. doi: 10.1161/hh0202.103644. [DOI] [PubMed] [Google Scholar]
- 34.Shi Y, Vesely I. Fabrication of mitral valve chordae by directed collagen gel shrinkage. Tissue Eng. 2003;9(6):1233–1242. doi: 10.1089/10763270360728143. [DOI] [PubMed] [Google Scholar]
- 35.Long JL, Tranquillo RT. Elastic fiber production in cardiovascular tissue-equivalents. Matrix Biol. 2003;22(4):339–350. doi: 10.1016/s0945-053x(03)00052-0. [DOI] [PubMed] [Google Scholar]
- 36.Clark RA, Nielsen LD, Welch MP, McPherson JM. Collagen matrices attenuate the collagen-synthetic response of cultured fibroblasts to TGF-beta. J Cell Sci. 1995;108(Pt 3):1251–1261. doi: 10.1242/jcs.108.3.1251. [DOI] [PubMed] [Google Scholar]
- 37.Yang L, van der Werf KO, Koopman BF, Subramaniam V, Bennink ML, Dijkstra PJ, et al. Micromechanical bending of single collagen fibrils using atomic force microscopy. J Biomed Mater Res A. 2007;82(1):160–168. doi: 10.1002/jbm.a.31127. [DOI] [PubMed] [Google Scholar]
- 38.Collet JP, Shuman H, Ledger RE, Lee S, Weisel JW. The elasticity of an individual fibrin fiber in a clot. Proc Natl Acad Sci U S A. 2005;102(26):9133–9137. doi: 10.1073/pnas.0504120102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perennes F, Marmiroli B, Tormen M, Matteucci M, Di Fabrizio E. Replication of deep x-ray lithography fabricated microstructures through casting of soft material. J Microlith Microfab Microsyst. 2006;5(1):011007. [Google Scholar]
- 40.Naito H, Melnychenko I, Didie M, Schneiderbanger K, Schubert P, Rosenkranz S, et al. Optimizing engineered heart tissue for therapeutic applications as surrogate heart muscle. Circulation. 2006;114(1 Suppl):I72–78. doi: 10.1161/CIRCULATIONAHA.105.001560. [DOI] [PubMed] [Google Scholar]
- 41.Siegal GP, Wang MH, Rinehart CA, Jr, Kennedy JW, Goodly LJ, Miller Y, et al. Development of a novel human extracellular matrix for quantitation of the invasiveness of human cells. Cancer Lett. 1993;69(2):123–132. doi: 10.1016/0304-3835(93)90164-5. [DOI] [PubMed] [Google Scholar]
- 42.Larkin LM, Calve S, Kostrominova TY, Arruda EM. Structure and functional evaluation of tendon-skeletal muscle constructs engineered in vitro. Tissue Eng. 2006;12(11):3149–3158. doi: 10.1089/ten.2006.12.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eastwood M, McGrouther DA, Brown RA. A culture force monitor for measurement of contraction forces generated in human dermal fibroblast cultures: evidence for cell-matrix mechanical signalling. Biochim Biophys Acta. 1994;1201(2):186–192. doi: 10.1016/0304-4165(94)90040-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.